

Abstract
The sideroblastic anemias (SAs) are a group of inherited and acquired bone marrow disorders defined by pathological iron accumulation in the mitochondria of erythroid precursors. Like most hematological diseases, the molecular genetic basis of the SAs has ridden the wave of technology advancement. Within the last 30 years, with the advent of positional cloning, the human genome project, solid-state genotyping technologies, and next-generation sequencing have evolved to the point where more than two-thirds of congenital SA cases, and an even greater proportion of cases of acquired clonal disease, can be attributed to mutations in a specific gene or genes. This review focuses on an analysis of the genetics of these diseases and how understanding these defects may contribute to the design and implementation of rational therapies.
Introduction
The sideroblastic anemias (SAs) are a group of inherited and acquired bone marrow disorders defined by pathological iron accumulation in the mitochondria of erythroid precursors (Figure 1).1-3 Abnormal, iron-laden mitochondria appear to encircle erythroblast nuclei, giving rise to the characteristic morphological feature of SAs, the ring (or ringed) sideroblast. Originally recognized in the 1940s,4,5 and codified as a class of anemia in the 1960s,6 like most hematological diseases, the molecular genetic basis of the SAs has ridden the wave of technology advancement. Within the last 30 years, with the advent of positional cloning, the human genome project, solid-state genotyping technologies, and next-generation sequencing (NGS) have evolved to the point where more than two-thirds of congenital SA (CSA) cases, and an even greater proportion of cases of acquired clonal disease, can be attributed to mutations in a specific gene or genes (Table 1). This review focuses on an analysis of the genetics of these diseases and how understanding these defects may contribute to the design and implementation of rational therapies. It does not take into consideration the numerous acquired SAs due to toxic and metabolic conditions not thought to have a genetic component, even though many have been linked to pathways common to the CSAs.
![Figure 1. Morphological features of SA. (A) May-Gruenwald-Giemsa (MGG)-stained peripheral blood smear of a man with mild XLSA demonstrating hypochromia, anisocytosis, and microcytosis. (B) Iron-stained peripheral blood smear from the same patient highlighting a siderocyte (arrow). (C) MGG-stained peripheral blood smear from the patient’s mother demonstrating the dimorphic red blood cell population, including hypochromic microcytes containing Pappenheimer bodies (arrow). (D) Iron-stained bone marrow aspirate smear from a man with XLSA demonstrating iron granules (blue) ringing around late erythroblast nuclei. (E) Transmission electron micrograph from a patient with RARS demonstrating electron densities (black) within degenerating mitochondria (pale vacuoles, indicated by an arrow) ringing around erythroblast nuclei (photograph courtesy of Marcel Seiler, Boston Veterans Affairs Medical Center [VAMC], Boston, MA). (A-D) Equally scaled; original magnification ×1000. (E) Original magnification ∼×8000.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/1/10.1182_blood-2018-08-815951/9/m_blood815951f1.png?Expires=1768957545&Signature=U5~yocZZ~jONlx4v4vvbWmg2Sha7XpfKwNvEQ2RZq5zN3Pkja5YXgW0p3DnWmDl~VfJCq9yYqhWCs-yL6w1TfXvnusHHz1xitfdEm-7aUd9KbO4JdEarPC~EvW0VzCmlaEoLP2hw1AndtwM5N8YrAw7y5zVPJc8~y9SmVz6P2OtvBAIrzQUWs8~SUbMNMBJoktg~05DzYIFD5NQxHYikretHThZ9Vf4sGr-E2IR8qK1Y6wOJ~kdUsHTxDUsRTZS~XRrEayky1zuYGEwcGZOLrL~CiDIs66JJtZNEiUOPpt6IdIKzh-xuCDC-a4rWisRruYj8Gp66380dEi93VUSMQw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Morphological features of SA. (A) May-Gruenwald-Giemsa (MGG)-stained peripheral blood smear of a man with mild XLSA demonstrating hypochromia, anisocytosis, and microcytosis. (B) Iron-stained peripheral blood smear from the same patient highlighting a siderocyte (arrow). (C) MGG-stained peripheral blood smear from the patient’s mother demonstrating the dimorphic red blood cell population, including hypochromic microcytes containing Pappenheimer bodies (arrow). (D) Iron-stained bone marrow aspirate smear from a man with XLSA demonstrating iron granules (blue) ringing around late erythroblast nuclei. (E) Transmission electron micrograph from a patient with RARS demonstrating electron densities (black) within degenerating mitochondria (pale vacuoles, indicated by an arrow) ringing around erythroblast nuclei (photograph courtesy of Marcel Seiler, Boston Veterans Affairs Medical Center [VAMC], Boston, MA). (A-D) Equally scaled; original magnification ×1000. (E) Original magnification ∼×8000.
Morphological features of SA. (A) May-Gruenwald-Giemsa (MGG)-stained peripheral blood smear of a man with mild XLSA demonstrating hypochromia, anisocytosis, and microcytosis. (B) Iron-stained peripheral blood smear from the same patient highlighting a siderocyte (arrow). (C) MGG-stained peripheral blood smear from the patient’s mother demonstrating the dimorphic red blood cell population, including hypochromic microcytes containing Pappenheimer bodies (arrow). (D) Iron-stained bone marrow aspirate smear from a man with XLSA demonstrating iron granules (blue) ringing around late erythroblast nuclei. (E) Transmission electron micrograph from a patient with RARS demonstrating electron densities (black) within degenerating mitochondria (pale vacuoles, indicated by an arrow) ringing around erythroblast nuclei (photograph courtesy of Marcel Seiler, Boston Veterans Affairs Medical Center [VAMC], Boston, MA). (A-D) Equally scaled; original magnification ×1000. (E) Original magnification ∼×8000.
Genetic and phenotypic characteristics of SA
| Inheritance | Syndromic | Gene | Mutations | Frequency* | Age at presentation | Anemia severity | MCV | Associated abnormalities | |
|---|---|---|---|---|---|---|---|---|---|
| CSA | |||||||||
| Heme synthesis defects | |||||||||
| XLSA | X | No | ALAS2 | MS/R♂ | 100s | Infancy to adulthood | Mild to severe | ↓♂ N/↑♀ | Iron overload in the absence of transfusions |
| MS/NS/FS♀ | |||||||||
| SLC25A38 deficiency | AR | No | SLC25A38 | NS/FS/SPL/MS | ∼40 | Infancy | Severe | ↓ | Transfusional iron overload |
| EPP | AR/PSD | No | FECH | SPL/MS/NS/FS | 100s | Childhood | Mild | ↓ | Acute photosensitivity |
| Fe-S biogenesis defects | |||||||||
| GLRX5 deficiency | AR | No | GLRX5 | MS | 2 | Adulthood | Mild to severe | ↓ | Iron overload |
| HSPA9 deficiency | AR/PSD | No | HSPA9 | MS/FS/NS/SPL | 12 | Childhood | Mild to severe | N/↓ | Retinitis pigmentosa |
| HSCB deficiency | AR | No | HSCB | FS/R | 1 | Childhood | Moderate | N | None |
| XLSA/A | X | Yes | ABCB7 | MS | 5 | Childhood | Mild to moderate | ↓ | Cerebellar ataxia and hypoplasia, delayed motor development |
| Mitochondrial protein synthesis defects | |||||||||
| PMPS | SP/M | Yes | mtDNA | DEL (heteroplasmic) | 100s | Early childhood | Severe | ↑ | Lactic acidosis, exocrine pancreatic insufficiency, failure to thrive, hepatic/renal failure |
| MLASA1 | AR | Yes | PUS1 | MS, NS | ∼10 | Childhood | Mild to severe | N/↑ | Myopathy, lactic acidosis, facial dysmorphism |
| MLASA2 | AR | Yes | YARS2 | MS, FS, NS, DEL, SPL | ∼40 | Childhood | Mild to severe | N/↑ | Myopathy, lactic acidosis, cardiomyopathy |
| LARS2 deficiency | AR | Yes | LARS2 | MS | 1 | Infancy | Severe | ↑ | Lactic acidosis, cardiomyopathy, hepatopathy, seizures |
| SIFD | AR | Yes | TRNT1 | MS/FS/NS/ SPL | ∼30 | Infancy | Severe | ↓ | Immunodeficiency (B>T), aseptic febrile episodes, developmental delay, seizures, cardiomyopathy, retinitis pigmentosa, other |
| Mitochondrial respiratory protein mutations | |||||||||
| MT-ATP6-SA | SP/M | Yes | MT-ATP6 | p.Ser148Asn | 4 | Infancy to early childhood | Moderate to severe | N/↑ | Lactic acidosis, myopathy, neurological abnormalities |
| NDUFB11-SA | X | Yes | NDUFB11 | p.Phe90del | 5 | Early childhood | Moderate | N | Lactic acidosis, myopathy |
| Multifactorial | |||||||||
| TRMA | AR | Yes | SLC19A2 | NS/FS/SPL/MS | ∼50 | Early childhood | Mild to severe | ↑ | Sensorineural deafness, non-type I diabetes mellitus, optic atrophy, stroke-like episodes |
| Acquired clonal SA | |||||||||
| MDS-RS-SLD/MDS-RS-MLD | SOM | N/A | SF3B1 | Recurrent HEAT domain MS | 1000s | Adulthood | Mild to moderate | ↑/N | Iron overload, other cytopenias (MDS-RS-MLD) |
| MDS/MPN-RS-T | SOM | N/A | SF3B1 + JAK2, MPL, CALR | Recurrent HEAT domain MS + recurrent TK-activating MS | 100s | Adulthood | Mild | ↑/N | Thrombocytosis |
| Inheritance | Syndromic | Gene | Mutations | Frequency* | Age at presentation | Anemia severity | MCV | Associated abnormalities | |
|---|---|---|---|---|---|---|---|---|---|
| CSA | |||||||||
| Heme synthesis defects | |||||||||
| XLSA | X | No | ALAS2 | MS/R♂ | 100s | Infancy to adulthood | Mild to severe | ↓♂ N/↑♀ | Iron overload in the absence of transfusions |
| MS/NS/FS♀ | |||||||||
| SLC25A38 deficiency | AR | No | SLC25A38 | NS/FS/SPL/MS | ∼40 | Infancy | Severe | ↓ | Transfusional iron overload |
| EPP | AR/PSD | No | FECH | SPL/MS/NS/FS | 100s | Childhood | Mild | ↓ | Acute photosensitivity |
| Fe-S biogenesis defects | |||||||||
| GLRX5 deficiency | AR | No | GLRX5 | MS | 2 | Adulthood | Mild to severe | ↓ | Iron overload |
| HSPA9 deficiency | AR/PSD | No | HSPA9 | MS/FS/NS/SPL | 12 | Childhood | Mild to severe | N/↓ | Retinitis pigmentosa |
| HSCB deficiency | AR | No | HSCB | FS/R | 1 | Childhood | Moderate | N | None |
| XLSA/A | X | Yes | ABCB7 | MS | 5 | Childhood | Mild to moderate | ↓ | Cerebellar ataxia and hypoplasia, delayed motor development |
| Mitochondrial protein synthesis defects | |||||||||
| PMPS | SP/M | Yes | mtDNA | DEL (heteroplasmic) | 100s | Early childhood | Severe | ↑ | Lactic acidosis, exocrine pancreatic insufficiency, failure to thrive, hepatic/renal failure |
| MLASA1 | AR | Yes | PUS1 | MS, NS | ∼10 | Childhood | Mild to severe | N/↑ | Myopathy, lactic acidosis, facial dysmorphism |
| MLASA2 | AR | Yes | YARS2 | MS, FS, NS, DEL, SPL | ∼40 | Childhood | Mild to severe | N/↑ | Myopathy, lactic acidosis, cardiomyopathy |
| LARS2 deficiency | AR | Yes | LARS2 | MS | 1 | Infancy | Severe | ↑ | Lactic acidosis, cardiomyopathy, hepatopathy, seizures |
| SIFD | AR | Yes | TRNT1 | MS/FS/NS/ SPL | ∼30 | Infancy | Severe | ↓ | Immunodeficiency (B>T), aseptic febrile episodes, developmental delay, seizures, cardiomyopathy, retinitis pigmentosa, other |
| Mitochondrial respiratory protein mutations | |||||||||
| MT-ATP6-SA | SP/M | Yes | MT-ATP6 | p.Ser148Asn | 4 | Infancy to early childhood | Moderate to severe | N/↑ | Lactic acidosis, myopathy, neurological abnormalities |
| NDUFB11-SA | X | Yes | NDUFB11 | p.Phe90del | 5 | Early childhood | Moderate | N | Lactic acidosis, myopathy |
| Multifactorial | |||||||||
| TRMA | AR | Yes | SLC19A2 | NS/FS/SPL/MS | ∼50 | Early childhood | Mild to severe | ↑ | Sensorineural deafness, non-type I diabetes mellitus, optic atrophy, stroke-like episodes |
| Acquired clonal SA | |||||||||
| MDS-RS-SLD/MDS-RS-MLD | SOM | N/A | SF3B1 | Recurrent HEAT domain MS | 1000s | Adulthood | Mild to moderate | ↑/N | Iron overload, other cytopenias (MDS-RS-MLD) |
| MDS/MPN-RS-T | SOM | N/A | SF3B1 + JAK2, MPL, CALR | Recurrent HEAT domain MS + recurrent TK-activating MS | 100s | Adulthood | Mild | ↑/N | Thrombocytosis |
↓, decreased; ↑, increased; AR, autosomal recessive; DEL, deletion; EPP, erythropoietic protoporphyria; FS, frameshift; M, maternal; MCV, mean red blood cell volume; MDS/MPN, myelodysplastic syndrome/myeloproliferative neoplasm; MDS/MPN-RS-T, MDS/MPN with ring sideroblasts and thrombocytosis; MDS-RS-MLD, MDS with ring sideroblasts and multilineage dysplasia; MDS-RS-SLD, MDS with ring sideroblasts and single-lineage dysplasia; MS, missense; N, normal; N/A, not applicable; NS, nonsense; PMPS, Pearson marrow-pancreas syndrome; PSD, pseudodominant; R, regulatory; SIFD, SA, immunodeficiency, fevers, and developmental delay; SOM, somatic; SP, sporadic; SPL, splicing; TK, tyrosine kinase signaling pathway; TRMA, thiamine-responsive megaloblastic anemia; X, X-linked; XLSA, X-linked SA; XLSA/A, X-linked CSA associated with cerebellar ataxia.
Number of reported families with CSA or patients with clonal SA.
CSAs: a complex interplay of mitochondrial pathways
The CSAs are inherited diseases of mitochondrial dysfunction due to defects in heme biosynthesis, iron-sulfur cluster (ISC) biogenesis, generalized mitochondrial protein synthesis, or the synthesis of specific mitochondrial proteins involved in oxidative phosphorylation (Figure 2).7 This is not surprising given the essential nature of mitochondria in iron and heme metabolism and in energy production. More than two-thirds of the iron in the human body is in hemoglobin in erythroid cells, making the erythron uniquely susceptible to interruptions in the heme biosynthetic pathway. Like heme biosynthesis, ISC assembly is an evolutionarily conserved mitochondrial pathway that involves multiple enzymes that sequentially catalyze the reduction of organic sulfur to sulfide, complex it with iron, and deliver it to recipient apoproteins both in the mitochondria and in the cytosol.8 Although most proteins that localize to mitochondria are encoded by nuclear genes, synthesized on cytosolic ribosomes, and imported into mitochondria, several respiratory complex (RC) proteins and all the mitochondrial transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) required for mitochondrial protein synthesis are encoded by a separate mitochondrial genome.
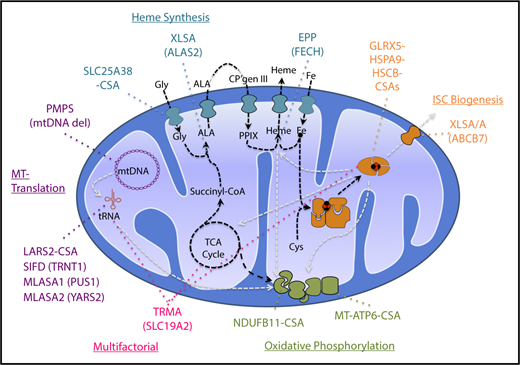
Pathways and genes implicated in CSA. Mitochondrial pathways relevant to the pathogenesis of CSA are diagrammed. Synthetic pathways are indicated by black dashed lines, whereas pathways whose products are required for other pathways implicated in CSA are shown in gray lines. Heme biosynthetic, iron-sulfur cluster biogenesis, mitochondrial (MT) translation, and oxidative phosphorylation/mitochondrial respiration pathways and the genes mutated in CSA in those pathways are highlighted in distinct colors. Thiamine-responsive megaloblastic anemia (TRMA) affects multiple pathways dependent upon thiamine. CP, coproporphyrin; Gly, glycine; MLASA, mitochondrial myopathy with lactic acidosis and SA; mtDNA, mitocondrial DNA; PMPS, Pearson marrow-pancreas syndrome; PPIX, protoporphyrin IX; TCA, tricarboxylic acid. Modified from Fleming7 with permission.
Pathways and genes implicated in CSA. Mitochondrial pathways relevant to the pathogenesis of CSA are diagrammed. Synthetic pathways are indicated by black dashed lines, whereas pathways whose products are required for other pathways implicated in CSA are shown in gray lines. Heme biosynthetic, iron-sulfur cluster biogenesis, mitochondrial (MT) translation, and oxidative phosphorylation/mitochondrial respiration pathways and the genes mutated in CSA in those pathways are highlighted in distinct colors. Thiamine-responsive megaloblastic anemia (TRMA) affects multiple pathways dependent upon thiamine. CP, coproporphyrin; Gly, glycine; MLASA, mitochondrial myopathy with lactic acidosis and SA; mtDNA, mitocondrial DNA; PMPS, Pearson marrow-pancreas syndrome; PPIX, protoporphyrin IX; TCA, tricarboxylic acid. Modified from Fleming7 with permission.
Heme synthesis and CSA
The most common and best-characterized CSA is X-linked SA (XLSA), which is typically due to missense mutations in the erythroid-specific isoform of aminolevulinic acid (ALA) synthase, ALAS2, the first step in heme biosynthesis. ALAS2 condenses glycine and succinyl-coenzyme A (CoA) to form ALA, using pyridoxal phosphate as a cofactor. Accordingly, many XLSA patients are responsive to supplementation with pyridoxine.3 Nearly 100 distinct mutations have been reported, including many that are recurrent in multiple families owing to the hypermutability of CpG dinucleotides, which also contributes to the relative frequency of de novo mutations in this disease.2 The majority of disease-associated variants occur in exons 5 and 9, the latter of which contains the pyridoxal-binding amino acid, lysine 391.9
Based on the crystal structures of prokaryotic (Rhodobacter capsulatus) and eukaryotic (Saccharomyces cerevisiae) ALA synthases,10,11 as well as functional assays in bacterially expressed human and mouse proteins, ALAS2 mutations appear to have wide-ranging effects on enzyme function, including substrate and cofactor binding, catalysis, and enzyme stability.9 Fewer than 5% of families with XLSA have mutations in a GATA1 transcription factor–binding site, responsible for high-level erythroid expression, and located in intron 1 of the gene, that result in decreased levels of messenger RNA (mRNA) encoding a structurally normal protein.12 The pathogenicity of a reported promoter variant13 has recently been called into question; it appears to be an allele present in control populations at low (0.26% in gnomAD; http://gnomad.broadinstitute.org/), but not unusually rare, frequency.
As the name would suggest, XLSA occurs preferentially in men, where it is the prototypical nonsyndromic form of hypochromic, microcytic CSA (Figure 1A-B). Clinically unaffected carrier women often have erythrocyte dimorphism (Figure 1C). Women, however, also constitute approximately one-quarter of clinically affected XLSA probands, and have a distinct clinical presentation and mutation spectrum.3,14 Unlike men who most often, though not exclusively, present within the first 2 decades of life, women with XLSA more often come to attention in mid to late adulthood with a normocytic or macrocytic, normochromic anemia that is nearly invariably associated with skewed X-chromosome inactivation of peripheral blood leukocytes.15,16 The specific cause of the skewing, indeed, whether it is clonal or not, is unknown. That affected women younger than 30 years old have been identified who are unlikely to have acquired somatic mutations associated with clonal hematopoiesis,17 and that there are several families with multiple affected women, indicating a familial predisposition to acquired X-chromosome skewing, suggests that the evolution of anemia is not clonal.18,19 Whatever the mechanism, affected women more often have more severe loss of function, and occasionally even null alleles, compared with their male counterparts. As ALAS2 is an essential gene,20 in these families, there may be a history of late-gestation demise of hydropic male fetuses.21
ALAS2 is subject to multiple negative regulatory mechanisms: (1) the mRNA is subject to translational downregulation in response to iron deficiency through an iron-responsive element (IRE) located in the 5′ untranslated region (UTR), (2) enzymatic activity is negatively regulated by a C-terminal inhibitory domain, and (3) protein levels are regulated by the CLPX/CLPP mitochondrial quality control protease.22-24 Indeed, C-terminal truncations of ALAS2 in humans and deletion of the IRE-binding protein IRP2 in mice lead to excessive protoporphyrin IX production (erythropoietic protoporphyria [EPP]) by upregulating ALAS2 activity and protein, respectively.22,25-27 This “opposite phenotype” (ie, EPP) resulting from loss of ALAS2 regulatory factors highlights the therapeutic potential for interfering with these mechanisms in XLSA to increase mutated ALAS2 expression, activity, or stability as a therapeutic strategy. Equally attractive, particularly in an era where genome editing is feasible, is the possibility of activating the housekeeping form of the enzyme, ALAS1, in erythroid cells by placing it under the control of an erythroid promoter, or by relieving heme-dependent repression mediated by the heme regulatory motif in the ALAS1 protein.28
ALAS2 has an extraordinarily low affinity, ∼1 mM, for glycine, requiring a high concentration in erythroid mitochondria for efficient heme synthesis. Although glycine is an abundant, nonessential amino acid and is readily generated from serine in both the cytosol and mitochondria, the demand for glycine in erythroid mitochondria has necessitated a professional importer, SLC25A38,29 a member of the mitochondrial carrier family of proteins, that is expressed highly and selectively in erythroblasts. SLC25A38 is an autosomal gene and recessive loss-of-function mutations cause a severe hypochromic microcytic CSA, morphologically similar to XLSA, that is unresponsive to pyridoxine.30 Also unlike XLSA, patients with SLC25A38-SA are typically transfusion-dependent. Nonsense, frameshift, and splicing mutations are the most common disease-associated alleles; approximately one-third of mutations, however, are missense variants that nearly exclusively occur at conserved amino acids at substrate contact points and within transmembrane domains (M.D.F., unpublished observation). Approximately 40 families with SLC25A38-SA have been described, and consistent with the increased frequency of rare recessive diseases such as this in consanguineous pedigrees, approximately two-thirds of patients have homozygous mutations. Some mutations, such as the most common disease variants, c.324_330del (p.Tyr109X) and c.349C>T (p.Arg117X), are recurrent in many populations, owing to their locations at a CT dinucleotide repeat and a CpG dinucleotide, respectively, and appear in many compound heterozygous patients (M.D.F., unpublished observation).
Like the other heme synthesis CSA, XLSA, the SLC25A38-SA would appear to be therapeutically targetable by pharmacological supplementation, in this case with glycine. Whereas glycine complements heme synthesis in yeast and zebrafish deficient in genes orthologous to SLC25A38,30,31 a trial of oral high-dose glycine therapy in 3 patients with SLC25A38 deficiency did not have a salutary effect.32 Although aspects of the trial design or patient compliance may have contributed to this negative result, the lack of an apparent effect might equally be due to secondary intra- or extramitochondrial toxicity of glycine therapy, including on serine-glycine interconversion and its effect on folate metabolism. Consequently, exploring these metabolic pathways in more detail in patient samples or cell or animal models may be worthwhile.
Patients with EPP due to mutations in ferrochelatase (FECH) often have a mild microcytic anemia and may, under very close inspection, have occasional ring sideroblasts in the bone marrow.33
ISC biogenesis and CSA
ISCs are catalytic cofactors and structural components of many mitochondrial and extramitochondrial proteins, including mitochondrial RCs, FECH, the cytosolic iron-regulatory protein IRP1, xanthine oxidase, and several RNA- and DNA-modifying enzymes, among many others. Mitochondrial ISC assembly can be divided conceptually into 4 stages: (1) assembly of a 2Fe-2S cluster by the core assembly complex, (2) transfer of the 2Fe-2S cluster to target proteins and transporters, and (3) assembly of a 4Fe-4S cluster, and (4) distribution of the 4Fe-4S cluster to target proteins.34 Mutations in the core ISC complex tend to yield complex neurologic and metabolic phenotypes such as Friedreich ataxia and mitochondrial myopathy. Mutations in 4Fe-4S ISC assembly often result in multiple mitochondrial deficiency syndromes (MMDSs) associated with severe neurologic symptoms, leukodystrophy, or encephalopathy. CSAs are associated with defects in the transfer stage of ISC biogenesis. HSPA9 and HSCB are a mitochondrial heat shock protein 70 homolog and cochaperone pair that transfer the nascent 2Fe-2S ISC to mitochondrial glutaredoxin 5 (GLRX5) for further distribution. Mutations in each of these components lead to nonsyndromic microcytic to normocytic SA.35-38
Of these genes, HSPA9 mutations are most common, having been described in a dozen families.37 The phenotype is due to recessive incomplete loss-of-function variants, but often appears to segregate in a pseudodominant pattern. Patients with the most severe anemia often have a null allele, such as a frameshift, in trans of a mild loss-of-function variant, as demonstrated in yeast complementation assays. Families with pseudodominant inheritance commonly have a severe allele in trans of a common (∼30% minor allele frequency) noncoding single-nucleotide polymorphism (SNP) (rs10117T) predicted to be located in an exonic splice enhancer of exon 7 of the gene that results in decreased mRNA expression.37 Although there are compelling data that this variant is causative, a small minority of patients have the more prevalent, ancestral rs10117C allele in trans of a null or severe missense variant, suggesting the possibility that the rs10117T variant is in linkage disequilibrium with the “true” causative variant. A single patient has been described with a frameshift mutation and a variant in a functionally important ETS transcription factor binding in the HSCB promoter. HSCB cooperates with HSPA9 in ISC transfer.38 Not surprisingly, as both genes are nonredundant and essential in yeast, patients with homozygous null alleles have not been described.
Two patients with missense mutations in the mitochondrial glutaredoxin, GLRX5, thought to be the immediate donor of the 2Fe-2S ISC to FECH and the 4Fe-4S cluster synthesis machinery, have been described with a moderately severe microcytic CSA.36,39,40 A missense variant in the zebrafish homolog of GLRX5 results in the anemia mutation shiraz.39 Importantly, expression of a zebrafish alas transgene lacking the IRE in the 5′UTR ameliorates the shiraz anemia, suggesting that at least a component of the pathogenesis is IRP1-dependent suppression of heme biosynthesis mediated by ALAS downregulation. Other work indicates that loss of ISC assembly factors reversibly inhibits FECH activity, which would also impair heme synthesis.41
Because the proteins of the initial stages of ISC biogenesis uniquely localize to mitochondria, and IRP1 and a number of other ISC proteins are located in the cytosol or nucleus, a component required for extramitochondrial ISC assembly must be transported out of the mitochondria to fulfill this requirement. That protein is ABCB7, an ATP-binding cassette family transporter encoded on the X-chromosome that localizes to the inner mitochondrial membrane; hemizygous missense, partial loss-of-function mutations in ABCB7 cause a rare syndromic form of microcytic X-linked CSA associated with cerebellar ataxia (XLSA/A).42-47 In XLSA/A, the anemia tends to be less clinically significant than the cerebellar dysfunction, and, indeed, 1 family with an X-linked ataxia without CSA has been shown to carry a missense allele in ABCB7.48 The substrate transported by ABCB7 is unclear, but may be an ISC complexed with glutathione.49 That mutations in ABCB7 result in CSA further suggests that the ontogeny of the ring sideroblast itself in ISC defects might not be consequent to the direct effect on mitochondria, but rather a result of a complex interplay between cytosolic ISC-dependent proteins, such as IRP1, and other mitochondrial pathways, including ALAS2 expression and heme synthesis.50
Mitochondrial RC proteins and mitochondrial protein synthesis and CSA
Generalized defects in mitochondrial protein translation or mutations in a specific mitochondrial RC protein are associated with syndromic forms of CSA. Among these phenotypes are Pearson marrow-pancreas syndrome (PMPS); mitochondrial myopathy with lactic acidosis and SA (MLASA); and SA, immunodeficiency, fevers, and developmental delay (SIFD).51,52 In each case, a broad defect in mitochondrial protein synthesis leads to CSA associated with neuromuscular disease and lactic acidosis consequent to impaired mitochondrial energy metabolism. In addition, there are rare cases of CSA with similar phenotypes that are a consequence of specific mutations in mitochondrially encoded and X-linked genes encoding components of RCs I and V.
PMPS, a protean syndrome classically associated with macrocytic CSA, lactic acidosis, exocrine pancreas insufficiency, and failure to thrive, is a disorder of early childhood that results from heteroplasmic deletions in the mitochondrial genome.53,54 Some patients present with hematological abnormalities alone, and may even exhibit “pure red cell aplasia,” being mistaken for Diamond-Blackfan anemia.55 Many often have other cytopenias. Approximately half of patients carry a 4977-bp mitochondrial DNA deletion that is common to other mitochondrial cytopathies (eg, Kearns-Sayre syndrome).56 Although the range of deletions is diverse and incompletely overlapping, at least 1 of the 22 mitochondrial tRNAs essential for mitochondrial protein translation is invariably included in the deletion. This suggests that PMPS is not the result of a deficiency of a single protein, but rather a consequence of globally suppressed mitochondrial protein synthesis, specifically certain components of RCs I, IV, and V encoded by the mitochondrial genome,
MLASA1 and MLASA2, classically characterized by the triad of muscle weakness, lactic acidosis, and normo- to macrocytic CSA, are due to mutations in pseudouridine synthase 1, PUS1,57 and the mitochondrial tyrosyl-tRNA synthetase, YARS2,58 respectively. A single patient with biallelic LARS2 mutations, the mitochondrial leucyl-tRNA synthetase, has been described to have a related, lethal mitochondrial phenotype that included CSA.59 Why deficiencies in the other mitochondrial aminoacyl-tRNA synthetases (ie, ARS2 proteins) do not result in CSA is unclear, however, many do result in other syndromic mitochondrial phenotypes. For example, other mutations in LARS2 and mutations in HARS2, among other proteins, result in Perrault syndrome: sensorineural deafness and female infertility.60 Furthermore, all patients with biallelic YARS2 mutations do not exhibit CSA, lactic acidosis, and myopathy; patients with only myopathy or CSA plus or minus lactic acidosis are relatively common.61-63 Not only is expressivity of the phenotype highly variable, so is the penetrance of the disease, with genotypically identical siblings having widely divergent hematological phenotypes, including intermittently transfusion-dependent patients with completely phenotypically normal, genetically at-risk siblings.63 Equally, the anemia in a single patient may wax and wane over time. Outside of a YARS2 mutation common in Lebanese Christians, c.156C>G (p.Phe92Leu), which appears to consistently cause moderately severe anemia, lactic acidosis, and mitochondrial myopathy, there is no clear genotype-phenotype relationship.
PUS1 is the only mitochondrial member of a family of enzymes that convert uridine on structural RNAs, such as tRNAs, to pseudouridine.64 The modified uridine residue has the capacity to form 3, rather than 2, hydrogen bonds with adenine, and in this manner it is thought to stabilize RNA structures, enhancing their activity and longevity. Fewer than 10 families, most of whom are consanguineous, have been described with MLASA1 due to PUS1 mutations. A founder allele is present in the Persian Jewish population.65 Most PUS1 mutations are missense alleles, but 1 homozygous null sibling pair has been described,66 indicating that PUS1 is not essential in mammals: a point supported by the mild phenotype and viability of Pus1−/− mice, which, like nearly all animal models of SA, do not develop bone marrow ring sideroblasts.67
SIFD is a recently recognized entity caused by incomplete loss-of-function mutations in the template-independent RNA polymerase TRNT1, also known as the CCA-adding enzyme, which catalyzes the sequential addition of cytosine, cytosine, and adenine ribonucleotides onto the 3′ terminus of all cytosolic and mitochondrial tRNAs.51,52 The tRNA 3′ adenine resulting from TRNT1 activity is the substrate for aminoacyl-tRNAs synthetases such as YARS2, and tRNAs so charged are substrates for protein synthesis. Most, but by no means all, patients with this disorder present in infancy; in utero and later childhood presentations do occur. The markedly microcytic (MCV 50-70 fL) anemia typical of this disease is in sharp contrast to other CSAs due to mitochondrial translation defects. This is likely a consequence of the fact that unlike the other disorders, TRNT1 deficiency affects cytosolic as well as mitochondrial translation, with globin proteins themselves likely being the primary targets causing microcytosis. Many patients have recurrent/periodic aseptic febrile episodes associated with diarrhea and metabolic instability, including lactic acidosis. Some patients have developmental delay, which may be severe and associated with profound cognitive deficits. By contrast, other individuals may have signs and symptoms only of isolated anemia or immunodeficiency.68-70 Still other families have just erythrocyte microcytosis and late-onset retinitis pigmentosa.71 Thus, as is true of many “new” disorders, because of genetic heterogeneity, the spectrum of TRNT1-associated disease continues to expand. In all cases, however, because the protein is essential for all protein synthesis (both mitochondrial and cytosolic), no patient has biallelic null mutations; many are homozygous missense alleles with decreased function, or compound heterozygotes with diverse missense, nonsense, frameshift, and splicing variants. One pathogenic variant in particular, c.668T>C (p.Ile223Thr), is relatively common in control populations (∼0.004% allele frequency in gnomAD) and accounts for >50% of mutant alleles in nonconsanguineous patients (M.D.F., unpublished observation).
Inflammatory episodes are a major source of morbidity and mortality in SIFD, with many patients succumbing to multiorgan failure in the context of these events. As a consequence, several patients have been treated with tumor necrosis factor-α (TNF-α) inhibitors, with some evidence of efficacy in reducing the frequency of inflammatory complications.72 This intervention is not without some rational basis, as there is some evidence that accumulation of incompletely processed tRNAs may incite a type I interferon response in a manner similar to single-stranded RNA viruses, a response that may be quelled by TNF-α inhibitors.72
In comparison with generalized mitochondrial protein synthesis defects, mutations in specific mitochondrial RC proteins are comparatively rare in patients with CSA, there being <5 families each with mutations in the X-linked NADH:ubiquinone oxidoreductase subunit B11 (NDUFB11), which encodes a component of RC-I,73 or MT-ATP6, a mitochondrially encoded subunit of ATP synthase (RC-V).74,75 In both cases, the causative variant is a unique, recurrent mutation: a deletion of a single tyrosine in a poly-tyrosine sequence in the transmembrane domain of NDUFB11 (p.Phe93del) or a serine to asparagine mutation at codon 148 in MT-ATP6 (p.Ser148Asn). The NDUFB11 anemia is a moderate to severe normocytic anemia that may be associated with mild myopathy. ATP6-SA, on the other hand, is highly variable, owing to varying degrees of heteroplasmy in the bone marrow and other tissues. Associated comorbidities include mild lactic acidosis and learning disabilities in the mildest cases; in the most severe cases, patients experience failure to thrive, mitochondrial encephalopathy, myopathy, cardiomyopathy, and severe metabolic compromise.74,75
CSA due to potential defects in multiple mitochondrial pathways
Mutations in the high-affinity thiamine transporter SLC19A2 result in thiamine-responsive megaloblastic anemia (TRMA)76-79 Ring sideroblasts are an inconstant feature of this disease, which can be treated with pharmacological doses of thiamine. There are 4 mitochondrial thiamine-dependent enzymes: transketolase, α-ketoglutarate dehydrogenase (KGDH), pyruvate dehydrogenase (PDH), and branched chain α-keto acid dehydrogenase (BCKDC), all of which are involved in carbohydrate metabolism. Both thiamine-dependent generation of succinyl CoA (involved in heme biosynthesis) and ribose sugar synthesis (related to mitochondrial RNA metabolism) may be relevant to the ring sideroblast abnormality.80 In addition, KGDH, PDH, and BCKDC all require lipoic acid as a cofactor, the synthesis of which is dependent on a mitochondrial ISC-dependent enzyme, lipoic acid synthetase (LIAS),81 further connecting thiamine metabolism to known pathogenetic mechanisms in CSA.
Acquired clonal SA: unexpected pathogenesis and therapeutic opportunities
Acquired clonal SAs, classified within the broad rubric of the myelodysplastic syndromes (MDSs) and myeloproliferative neoplasms (MPNs), are far more common than CSAs, and, until 2011, their molecular pathogenesis was essentially completely obscure. The 2016 Revised World Health Organization (WHO) Classification distinguishes 3 categories: MDS with ring sideroblasts and single-lineage dysplasia (MDS-RS-SLD), MDS with ring sideroblasts and multilineage dysplasia (MDS-RS-MLD), and MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) (collectively abbreviated as MDS-RS).82 As their names suggest, the classification is predicated on the recognition of ring sideroblasts accompanied by morphological dysplasia in 1 (characteristically the erythroid) or multiple hematopoietic lineages, or anemia and thrombocytosis with certain bone marrow features of a myeloproliferative neoplasm (most typically megakaryocytic abnormalities as seen in essential thrombocytosis), often with an erythroid hyperplasia. In 2011, NGS of germline and neoplastic DNA from patients with MDS revealed an extraordinarily strong association between somatic mutations in genes implicated in mRNA splicing (ie, spliceosome components) and the presence of ringed sideroblasts in MDSs.83-85
Even prior to the seminal NGS studies identifying SF3B1 mutations, the clonal nature of MDS-RS was supported by glucose-6-phosphate dehydrogenase isoenzyme analysis of hematopoietic cells in affected women, as well as cytogenetic studies.86,87 Chromosomal abnormalities are found in approximately one-half of cases of MDS-RS, with deletions in chromosomes 5, 11, or 20; loss of the Y chromosome; or trisomy 8 occurring most frequently.88,89 Although these abnormalities are not uncommon in MDS as a whole, Xq13 breakpoints are particularly associated with MDS-RS, and a unique variant, an isodicentric chromosome Xq13.1 (idicXq13.1), is seen only in women with MDS-RS.90-92
The strongest molecular correlate of MDS-RS is with specific somatic mutations in SF3B1, a core component of the U2 snRP spliceosome subunit that recognizes the 3′ splice acceptor site in newly transcribed mRNAs. Heterozygous, acquired clonal SF3B1 mutations are present in between 70% and 90% of MDS-RS,83-85,93,94 and, in many, it is the sole detectable clonal MDS-associated variant. Thus, mutant SF3B1 not only appears to be a driver of the MDS phenotype, but also appears to be responsible for RS formation. Befitting the clinical and pathological MDS/MPN overlap, cases of MDS/MPN-RS-T have somatic SF3B1 mutations as well as gain-of-function hematopoietic receptor tyrosine kinase–signaling mutations, most commonly JAK2 p.Val617Phe (∼60%), and less commonly other JAK2, or MPL, or CALR variants.95,96 In all cases, the SF3B1 mutations are missense variants in the C-terminal HEAT domains (amino acids 622-781), with the most common, p.Val700Glu, accounting for ∼60% of alleles; null, splicing, and other missense variants are not associated with MDS-RS. SF3B1 mutations are neomorphic alleles that result in selection of an aberrant 3′ splice site and abnormally spliced mRNAs, many of which are degraded by nonsense-mediated decay, likely leading to decreased levels of the encoded proteins.97 Mutations in several other 3′ spliceosome components, including SRSF2, ZRSR2, and PRPF8, are also seen in MDS-RS lacking SF3B1 mutations,85,98 but it is unclear whether these variants are enriched in MDS-RS beyond the MDS population as a whole.
Considerable effort has been expended to try to unify the genetics and pathogenesis of the RS in congenital and acquired clonal SA prior to and subsequent to the discovery of mutations in the spliceosome machinery in acquired SA. For example, a number of causative CSA genes were evaluated as candidate genes for MDS-RS,99 and diverse mitochondrial DNA mutations were assessed for a potential role in RS ontogeny in MDSs,100,101 with largely negative results. Given that, at present, the genes and primary pathways mutated in CSA and acquired SA appear to be mutually exclusive, and that aberrant splicing has the potential to misregulate a diverse set of genes, it is highly probable that the pathogenesis of MDS-RS is multifactorial and/or requires the epistatic interaction between multiple genes and pathways dysregulated by a splicing abnormality. Most obviously, the repertoire of dysregulated genes must simultaneously promote clonal outgrowth of the transformed hematopoietic progenitor and the development of the RS; the latter of these is considered here.
Several gene-expression studies have shown that a number of genes implicated in CSA and mitochondrial iron metabolism, including, ALAS2, HSPA9, GLRX5, and SLC25A37 (mitoferrin 1), and SLC25A38, are variably, depending upon the study, upregulated in CD34+ cells or erythroblasts from patients with MDS-RS,96,102-104 whereas the transcript encoding ABCB7 is reduced.96,102,105 This is hypothesized to result in ABCB7 deficiency and XLSA/A-like erythroblast physiology.106 The observation that the ABCB7 gene is located within the deleted region of the X chromosome in the idicXq13.1 seen in MDS-RS, and that the active X in these cases is the isodicentric X chromosome lacking ABCB7, further supports the notion that ABCB7 deficiency could underlie the MDS-RS phenotype.107,108 Reexpression of ABCB7 in MDS-RS cells mitigates growth phenotypes in primary patient cells cultured in vitro, but complementation of the sideroblastic phenotype itself has not been demonstrated.106,109 Another gene downregulated in MDS-RS is TMEM14C, which is a mitochondrial inner membrane protein that is essential for heme synthesis and erythropoiesis.104,109 Nevertheless, because we do not yet know all of the genes responsible for CSA or indeed mitochondrial iron metabolism, it cannot be said that all potential candidates and all potential mechanisms have been exhausted. For example, inappropriate upregulation of SLC25A37, responsible for erythroid mitochondrial iron uptake, or mitochondrial ferritin,110 which could potentially store iron in mitochondria, might equally be independently responsible for RS formation or contribute to an ABCB7-dependent phenotype.
Although understanding the pathogenesis of the RS in MDS-RS has merit from the perspective of mitochondrial iron biology, from a therapeutic perspective it does little to address the fundamental occupation of the bone marrow by the neoplastic clone. The results of clinical trials of spliceosome inhibitors in MDSs, which directly confront this issue, are eagerly awaited.111-114
Summary
As crystalline as the genetics of the SAs are becoming, lending themselves to rational therapies, the ontogeny of ring sideroblasts is as cloudy as it was 70 years ago when they were first described. Our limited understanding of how they arise aside, it is even unclear whether they are fundamentally detrimental to the erythroblast, causing the erythroid dysfunction. Are they a cause or consequence of disordered metabolism? Perhaps they are a physiologically programmed protective mechanism to sequester otherwise oxidatively dangerous iron. Alternatively, maybe they are simply a morphological curiosity, a riddle with no answer, never intended to be solved. Nevertheless, the knowledge of the molecular genetics of these disorders has provided a substantial foundation that will enable further therapeutic advances.
Acknowledgments
The authors thank Marian Harris for reviewing the manuscript.
This work was supported by National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases grants R24 DK099808 and R01 DK087992, and a Bridge Grant from the American Society of Hematology.
Authorship
Contribution: S.D. and M.D.F. wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mark D. Fleming, Department of Pathology, Boston Children’s Hospital, Bader 139.1, 300 Longwood Ave, Boston, MA 02115; e-mail: mark.fleming@childrens.harvard.edu.
