

Abstract
β-Thalassemia (BT) is an inherited genetic disorder that is characterized by ineffective erythropoiesis (IE), leading to anemia and abnormal iron metabolism. IE is an abnormal expansion of the number of erythroid progenitor cells with unproductive synthesis of enucleated erythrocytes, leading to anemia and hypoxia. Anemic patients affected by BT suffer from iron overload, even in the absence of chronic blood transfusion, suggesting the presence of ≥1 erythroid factor with the ability to modulate iron metabolism and dietary iron absorption. Recent studies suggest that decreased erythroid cell differentiation and survival also contribute to IE, aggravating the anemia in BT. Furthermore, hypoxia can also affect and increase iron absorption. Understanding the relationship between iron metabolism and IE could provide important insights into the BT condition and help to develop novel treatments. In fact, genetic or pharmacological manipulations of iron metabolism or erythroid cell differentiation and survival have been shown to improve IE, iron overload, and anemia in animal models of BT. Based on those findings, new therapeutic approaches and drugs have been proposed; clinical trials are underway that have the potential to improve erythrocyte production, as well as to reduce the iron overload and organ toxicity in BT and in other disorders characterized by IE.
Iron metabolism
Hepcidin is the master iron regulator, which is regulated by iron demand, iron stores, erythropoiesis, hypoxia, and inflammation.1,2 It is synthesized primarily in the liver and secreted in the bloodstream.1,2 Once in circulation, hepcidin recognizes the membrane-bound protein ferroportin, which is the only iron exporter (Figure 1A).1,3 Hepcidin binding to ferroportin causes the hepcidin-ferroportin complex to be internalized and degraded by lysosomes, preventing iron delivery from the intracellular milieu to the extracellular milieu (Figure 1A).1,3 Because ferroportin is expressed in enterocytes, parenchymal hepatic cells, and macrophages, the relative abundance of hepcidin in circulation, as well as that of ferroportin on extracellular membranes, controls dietary iron absorption in the duodenum, iron storage in the liver, and iron recycling in macrophages (Figure 1A).1 Heterozygous mutations in ferroportin can lead to ferroportin disease, an autosomal dominant iron-loading disorder that is characterized by genetic hyperferritinemia.4
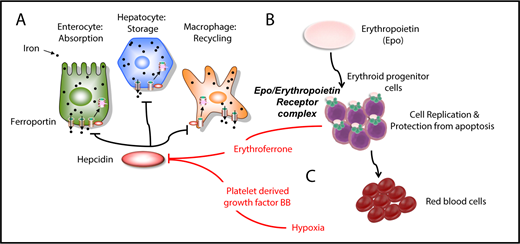
Iron metabolism and erythropoiesis. (A) Hepcidin is a master regulator of iron metabolism. It is produced in the liver in response to iron demand, iron storage, anemia, and inflammation. Once secreted, it targets and degrades the iron exporter ferroportin. (B) EPO is the main hormone controlling erythropoiesis. It works by modulating or interacting with a variety of proteins that control iron intake, cell replication, cell survival, and sensitivity to EPO itself. (C) Erythroferrone is secreted by erythroid progenitor cells and suppresses hepcidin in the liver. Another factor, PDGF-BB, suppresses hepcidin under hypoxic conditions.
Iron metabolism and erythropoiesis. (A) Hepcidin is a master regulator of iron metabolism. It is produced in the liver in response to iron demand, iron storage, anemia, and inflammation. Once secreted, it targets and degrades the iron exporter ferroportin. (B) EPO is the main hormone controlling erythropoiesis. It works by modulating or interacting with a variety of proteins that control iron intake, cell replication, cell survival, and sensitivity to EPO itself. (C) Erythroferrone is secreted by erythroid progenitor cells and suppresses hepcidin in the liver. Another factor, PDGF-BB, suppresses hepcidin under hypoxic conditions.
A series of proteins that control hepcidin expression have been identified in animal models and patients with abnormal iron metabolism.1 In the liver, the ligand bone morphogenetic protein 2 (BMP2) is required for activation of hepcidin synthesis, primarily at the basal or steady-state level, whereas the intracellular iron-dependent response is primarily triggered by BMP6, which is expressed by liver endothelial cells.5-8 BMP ligands bind a protein complex on hepatic cells that includes transforming growth factor-β (TGF-β)–like receptors indicated as type I (ALK2 and ALK3) and type II (BMPR2 and ACVR2A).6,8-10 The complex includes the coreceptors hemojuvelin and neogenin, which regulate iron homeostasis by interacting directly with BMP ligands to induce hepcidin expression.6,8,11 BMPR2 and ACVR2A recognize the BMP6/2 ligands and, upon recognition, activate the type I receptors. Those, in turn, phosphorylate the SMAD1/5/8 complex, which, together with SMAD4, translocates to the nucleus as a multiprotein complex, activating hepcidin expression.1
Another important player in hepcidin regulation is the transmembrane serine protease TMPRSS6 or matriptase-2.1 TMPRSS6 attenuates hepcidin expression, and its mutations are responsible for iron refractory iron deficiency anemia, which is characterized by inappropriately high levels of hepcidin.12 In contrast, mutations in hepcidin, hemojuvelin, BMP6, and other genes that positively control hepcidin expression (eg, HFE and TFR2, see next paragraph) lead to hemochromatosis in humans and mice.1
Iron in circulation is normally bound to transferrin, which delivers it to the organs.1 The relative amount of iron-bound transferrin molecules is “sensed” by a group of proteins known as TFR1, HFE, and TFR2.1 Increasing levels of transferrin saturation (TSat) activate the complex HFE/TFR2 by a mechanism that is incompletely understood but seems to act through the same SMAD complex activated by BMP6/2.1
Intestinal hypoxia-inducible factor 2α (HIF2α) is an important contributor to iron absorption under conditions of iron deficiency, erythropoiesis, and hepcidin deficiency.13,14 HIF2α belongs to the family of hypoxia-inducible transcription factors, which control the cellular and systemic responses to oxygen deficiency.15 Ferroportin, as well as other iron-related molecules, is a direct HIF2α target gene.13 Initial studies indicated that ferroportin is upregulated in the duodenum of animals with β-thalassemia (BT), which are anemic and hypoxic.16 Subsequent studies showed that HIF2α is a critical regulator of the expression of ferroportin and other iron-related molecules in the duodenum, because selective deletion of HIF2α in enterocytes significantly reduced iron absorption in BT mice.17 This strongly suggests that HIF2α dysfunction makes a significant contribution to the iron overload that occurs in BT.
Normal erythropoiesis
Erythropoiesis is the mechanism that produces red blood cells (RBCs). The main function of RBCs is to deliver oxygen via hemoglobin (Hb) molecules. In humans, after 12 months of age, RBCs contain mostly adult Hb that is made of α-globin and β-globin chains (the Hb tetramer α2β2).18,19 Each chain carries an individual heme molecule.18,19 The synthesis of β and α chains must be balanced; otherwise, the relative excess of the β or α chain can lead to α-thalassemia or BT, respectively.18,19
Erythropoiesis requires several steps, starting when burst-forming unit erythroid precursor cells develop into colony-forming unit erythroid cells.20 The colony-forming unit erythroid cells develop successively into proerythroblasts, basophilic cells, polychromatophilic cells, and orthochromatic erythroid cells, which are enucleated to form reticulocytes and, subsequently, mature RBCs.20 In humans at homeostasis, the proliferation, differentiation, and survival of those cells are balanced to generate roughly 2 million RBCs per second.1
Erythropoietin (EPO) is the master erythroid hormone made in the kidney in response to tissue oxygenation and is absolutely required to sustain erythropoiesis in vitro and in vivo (Figure 1B).21 By binding to the EPO receptor (EPOR) on the surface of erythroid cells, EPO allows the proliferation and survival of erythroid cells and determines the production of RBCs at homeostasis and under conditions of hypoxia (Figure 1B).22 EPOR activation induces the phosphorylation of JAK2 kinase and the transcription factor STAT5.23-25 In turn, the JAK2/STAT5 pathway activates the antiapoptotic protein Bcl-xL and the iron-intake molecule transferrin receptor-1 (TFR1).26,27 Furthermore, the EPO-stimulated phosphatidylinositol 3-kinase (PI3K)/AKT pathway has been identified as an important mediator of cell survival and proliferation.28 The PI3K/AKT pathway also mediates EPO-induced phosphorylation of GATA-1, 1 of the major erythroid transcription factors.29 GATA-1 modulates the expression of genes for globin chains and ALAS2, which controls the first step of heme biosynthesis.30
Iron-bound TFR1 also activates the PI3K/Akt and MAPK pathways.31 In particular, it has been shown that polymeric immunoglobulin (Ig)A1 (pIgA1) can enhance erythropoiesis through its ability to bind TFR1, suggesting pIgA1 as a novel erythropoiesis-stimulating agent.31 TFR2, expressed on the surface of erythroid cells, also modulates the sensitivity of erythroid cells to EPO, likely through the cargo receptor Scribble and its effect on AKT signaling.32,33
Additional factors also modulate erythropoiesis. It has been shown in vitro that stem cell factor and glucocorticoids support the self-renewal and slow down the differentiation of early erythroid progenitors.34,35 Also, TGF-β–like ligands (eg, growth differentiation factor 11 [GDF11]) may modulate erythropoiesis. GDF11, activins, GDF8/myostatin, and other members of the TGF-β superfamily signal through a combination of type I and type II receptors, all of which are transmembrane serine/threonine kinases.36-39 Two type II receptors (ACVR2A and ACVR2B) have been demonstrated to bind multiple TGF-β–like ligands, including activin A and B, inhibin A and B, Nodal, GDF11, GDF8, and a subset of BMP ligands (BMP2, 3, and 7).36-39 GDF11 binding to ACVR2A or ACVR2B results in the recruitment of ALK4, ALK5, or ALK7.40 The resulting complexes phosphorylate the SMAD2/3 complex, which then undergoes multimerization with the comediator SMAD4, translocates into the nucleus, and recruits additional transcriptional activators or repressors to regulate the transcription of target genes.36-39 It has been proposed that, in BT, reactive oxygen species (ROS) mediate the overexpression of GDF11 and the activation of SMAD2/3 signaling, which limits the differentiation of erythroid cells.41
Cell survival is also an important factor that controls erythropoiesis at homeostasis and under disease conditions. It has been postulated that negative-regulatory feedback operates in a paracrine fashion during erythropoiesis. In that model, mature erythroblasts express the Fas ligand (FasL; or CD95L), a protein belonging to the tumor necrosis factor family. Immature erythroblasts express FasR or CD95R, the corresponding receptor.42 The interaction between FasL and FasR results in caspase-8 activation and GATA-1 cleavage, which blocks erythroid differentiation and maturation.42 Normally, that mechanism contributes to normal homeostatic erythropoiesis.42 However, in patients affected by the relatively low-risk myelodysplastic syndrome (MDS) and in mouse models of BT, the FasL/FasR pathway has been reported to be expressed at pathological levels, contributing to anemia.41,43
In addition to being regulated on the molecular level, erythropoietic activity is controlled by the microenvironment. Macrophages play an important role in modulating normal and stress erythropoiesis. That process occurs within a structure called the erythroblastic island, in which a macrophage is surrounded by erythroblasts at all stages of maturation.44,45 When macrophages are depleted, the number of erythroid cells is greatly reduced in normal erythropoiesis or when there is an increased demand for erythroid cells, indicating that the macrophages at the centers of erythroblastic islands play a supporting role in erythropoiesis, which involves direct cellular interactions.46
The final maturation of erythroid cells requires the activation of proteins that control oxidative stress, inflammation, and apoptosis, such as FOXO3A and caspase-3.47-49 In erythroid cells under normal circumstances, the function of those proteins is to facilitate enucleation rather than to induce apoptosis or inflammatory pathways.50,51 Because caspase-3 can target and inactivate GATA-1, erythroid cells use inducible heat shock protein 70 (HSP70), a ubiquitous chaperone, to protect GATA-1 in the nucleus from caspase-3–mediated proteolysis during caspase activation.47-49
Stress erythropoiesis, ineffective erythropoiesis, and increased iron absorption
Erythropoiesis requires iron to sustain Hb and RBC synthesis, so it is not surprising that increased production of erythroid progenitor cells requires an increased iron supply.52,53 Iron absorption is increased when more RBCs are required (under hypoxic or anemic conditions).19,54,55
Anemic or hypoxic stress induces a physiological response to increase oxygen delivery to the tissues. Under those conditions, the rate of erythrocyte production can increase by ∼10-fold.1,56 The response to increase RBC production is defined as stress erythropoiesis and depends on the formation of stress erythroid progenitors, which respond to unique signals. Stress erythropoiesis requires stem cell factor, as well as GDF15, BMP4, and hedgehog, to promote the rapid expansion of early stress erythroid progenitors.56-58
Ineffective erythropoiesis (IE) is a particular form of anemia in which an increase in erythroid cells fails to produce a corresponding increase in RBCs. As a consequence, iron absorption is still increased in response to stress, but the iron is deposited in the organs rather than being used to generate more erythrocytes.19,54,55
Several potential mechanisms have been proposed through which erythropoiesis and hypoxia could increase iron absorption (Figure 1C). Erythropoiesis was proposed to directly suppress hepcidin expression by studies that identified erythroferrone, a hormone secreted by erythroblasts, as the main “erythroid regulator” (Figure 1C).52,53,59 Under anemic conditions in the presence of expanding erythroid progenitors, the role of erythroferrone is to increase and expedite iron delivery to the bone marrow by decreasing hepcidin levels, thereby allowing increased iron absorption and the release of iron stores into the blood plasma.60,61 Erythroferrone is synthesized as a consequence of EPOR/JAK2/STAT5 pathway activation in erythroid cells, but the mechanism by which it suppresses hepcidin in the liver has not been elucidated.59
A recent study described another mechanism of hepcidin suppression that is associated with increased RBC requirement but is independent of EPO production and erythroferrone synthesis.62,63 Under hypoxic conditions, platelet-derived growth factor-BB (PDGF-BB) was significantly increased in the circulation (Figure 1C).62-64 Furthermore, administration of PDGF-BB resulted in suppression of hepcidin expression, whereas inactivation of PDGF-BB suppressed hypoxia-induced suppression of hepcidin.62,63 The mechanism by which PDGF-BB mediates the suppression of hepcidin has not been elucidated, and it is unclear what role PDGF-BB–mediated hepcidin inhibition may play in IE.
IE in BT: causes and consequences
Several disorders involve different levels of IE, but the causes and consequences of IE have been best characterized in BT. BT develops as a consequence of mutations in the β-globin gene. Those mutations reduce or abolish the synthesis of the corresponding β-globin chain and impair RBC synthesis.19,54,55,67 Decreased β-globin synthesis and failure of RBC production can have different degrees of clinical severity.19,54,55,67 Patients affected by thalassemia major (TM), also known as transfusion-dependent thalassemia (TDT), are diagnosed early with severe anemia, hepatosplenomegaly, and failure to thrive.19,54,55 Patients with TM require chronic blood transfusions. Iron chelation therapy is also required to prevent the morbidities and mortality due to the iron overload that develops as a result of frequent blood transfusions.19,54,55 In contrast, patients affected by thalassemia intermedia, also known as non-TDT (NTDT), do not require blood transfusions early in life; however, the disease often progresses, resulting in splenomegaly and worsening anemia over time.19,54,55 Many patients with NTDT eventually develop iron overload because IE leads to increased iron absorption.19,54,55 Patients with NTDT often require iron chelation.19,54,55
In BT, the normal balance of α-globin and β-globin chains is lost, leading to a relative excess of α-globin proteins.19,54,55 IE in BT is triggered by the relative excess of α-globin chains.19,54,55 The unbound α-globin chains perturb erythropoiesis via several mechanisms. Because unbound α-globin bind free heme molecules in the absence of β-globin chains, they form toxic insoluble aggregates, called hemichromes, that precipitate and damage RBC membranes.68,69 They also trigger the formation of ROS, leading to oxidative stress.68,70 That combination leads to apoptosis of a subset of polychromatophilic erythroid cells.71 If the polychromatophilic cells succeed in differentiating into orthochromatic cells, many of the reticulocytes derived from the enucleation of those orthochromatic cells fail to generate erythrocytes. Furthermore, the few erythrocytes that are produced are abnormal in size and structure and have a reduced lifespan.72
The excess of free α-globin chains also negatively modulates GATA-1 activity. In BT, free α-globin chains bind HSP70.73 Because HSP70 is sequestered and GATA-1 is no longer protected, caspase-3 cleaves and degrades GATA-1, further impairing RBC production via end-stage maturation arrest and apoptosis of erythroid progenitors.73
It has been suggested that ROS, in combination with GDF11, a ligand of ACVR2A and ACVR2B, play an important role in erythroid maturation arrest and cell death in BT.41 GDF11 is increased in splenic erythroblasts from thalassemic mice and in sera from humans with BT.41 GDF11 is a member of the TGF-β superfamily, which includes other GDFs, TGF-βs, activins, and BMPs.74 It has been proposed that GDF11 impairs erythropoiesis in BT by activating SMAD2/3 signaling, which limits erythroid cell differentiation.41,75,76 Furthermore, GDF11 may also inhibit the apoptosis of early erythroid progenitors by suppressing the Fas-FasL pathway, further contributing to erythroid expansion and IE in BT.41
In patients with TM, frequent blood transfusions suppress endogenous (ineffective) erythropoiesis, and iron overload develops primarily from the accumulation of donor-derived RBCs.19,54,55 In contrast, iron overload develops from increased iron absorption in patients with NTDT.19,54,55 Studies in mice and clinical observations suggest a dynamic model in which IE and iron metabolism interact without reaching a stable state, worsening the pathophysiology of the disease. In that working model, hepcidin is suppressed by the increased production of erythroferrone in the absence of blood transfusion and during the early stages of the disease (Figure 2A).61 As iron accumulates over time in the organs, hepcidin synthesis increases (Figure 2B), likely as a result of increasing TSat and increasing iron concentrations in the liver; hepcidin synthesis triggered by those 2 mechanisms activates the BMP/SMAD and HFE/TFR2 complexes, respectively.1 Of note, these hepcidin levels are in the normal range, but reduced if normalized to parameters such as liver iron concentration or serum ferritin ratio.77 However, if hepcidin levels are in the normal range, we can speculate that additional mechanism(s) may contribute to the increased iron absorption observed in these individuals. As the disease advances, chronic stress erythropoiesis and unresolved anemia aggravate marrow expansion and extramedullary erythropoiesis, which occur primarily in the spleen (Figure 2C) and liver.19,54,55 Our model proposes that, as splenomegaly worsens through a process that is likely mediated by increased eryptosis, more RBCs are sequestered in the spleen, exacerbating the anemia and hypoxia (Figure 2D).78 Hypoxia stabilizes HIF2α in enterocytes, promoting the expression of molecules that increase iron absorption, such as ferroportin (Figure 2E).16,17 Therefore, even if the levels of hepcidin increase over time in individuals with BT, the net balance between hepcidin and ferroportin in the duodenum is such that iron absorption is sustained (Figure 2B,E).
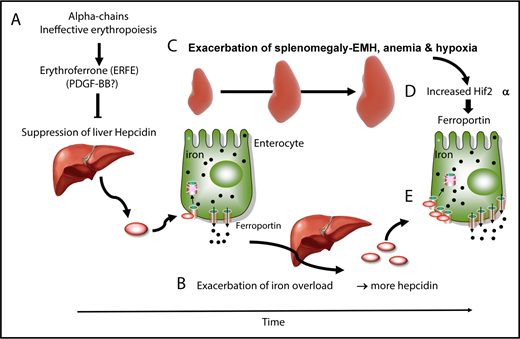
Causes and consequences of β-thalassemia. (A) During the early stages of BT, as well as in the absence of transfusion or when transfusion is inadequate, IE and erythroid expansion are responsible for hepcidin suppression, likely through erythroferrone. (B) This leads to increased iron absorption and iron overload. (C) Over time, splenomegaly occurs, exacerbating extramedullary erythropoiesis, RBC sequestration, anemia, and hypoxia. In particular, hypoxia stabilizes a transcription factor called HIF2α in enterocytes (D), which increases expression of ferroportin, among other iron-related molecules, in the duodenum (E). Therefore, the relative balance of hepcidin vs ferroportin is such that iron absorption is still increased, despite the fact that iron overload is promoting hepcidin expression.
Causes and consequences of β-thalassemia. (A) During the early stages of BT, as well as in the absence of transfusion or when transfusion is inadequate, IE and erythroid expansion are responsible for hepcidin suppression, likely through erythroferrone. (B) This leads to increased iron absorption and iron overload. (C) Over time, splenomegaly occurs, exacerbating extramedullary erythropoiesis, RBC sequestration, anemia, and hypoxia. In particular, hypoxia stabilizes a transcription factor called HIF2α in enterocytes (D), which increases expression of ferroportin, among other iron-related molecules, in the duodenum (E). Therefore, the relative balance of hepcidin vs ferroportin is such that iron absorption is still increased, despite the fact that iron overload is promoting hepcidin expression.
Thinking outside the box
Broadly, to think outside the box means to avoid proposing obvious solutions to difficult problems, thereby achieving unexpected results with unorthodox thinking and approaches. From that perspective, the mechanisms leading to IE have been revised and re-elaborated in the last few years. Erythroid expansion and maturation arrest have been introduced, together with reduced cell survival, as major contributors to the IE in BT. Furthermore, the role of iron has been expanded from that of a major contributor to organ iron toxicity to that of a modulator of IE. Based on those novel notions, 2 classes of approaches and novel drugs have been developed to improve IE in BT.
Improving anemia by iron restriction
A few studies introduced the notion that iron restriction could improve anemia in BT (Figure 3A).79-83 That is a counterintuitive argument, because iron deficiency is usually associated with iron-restrictive erythropoiesis, reduced synthesis of heme molecules (which contain iron), and anemia. In BT, heme molecules are included in normal Hb molecules, as well as in the insoluble aggregates known as hemichromes, which are 1 of the major contributors to IE.55 Because heme is a major component of hemichromes, it has been postulated that iron restriction could decrease TSat and erythroid iron intake, limiting heme synthesis (Figure 3A). That can limit the production of hemichromes, which, in turn, limits the toxicity associated with the excess of α-globin chains. The reduction in hemichrome formation can also improve the quality of the RBCs produced, increasing the lifespan and the number of RBCs in circulation (Figure 3A). Additional beneficial effects of iron restriction are a reduction in hypoxia and EPO synthesis, with an improvement in extramedullary erythropoiesis and the ratio of erythroid progenitors/RBCs (Figure 3A).
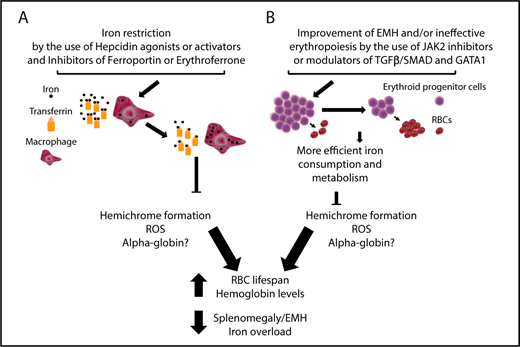
Targeting iron metabolism or IE to improve anemia and iron overload in BT. (A) Iron restriction can be achieved by using hepcidin agonists or activators or inhibitors of ferroportin, HIF2α, and erythroferrone to reduce iron intake and increase iron sequestration into macrophages, decreasing TSat. This limits hemichrome formation, ROS, and, potentially, free α-globin chain accumulation, resulting in improvements to RBC lifespan, anemia, extramedullary erythropoiesis and also iron overload. (B) Improvement of IE with drugs that inhibit JAK2 or modulate TGFβ/SMAD (eg, luspatercept) and GATA-1 activity. Use of JAK2 inhibitors will only decrease the number of erythroid progenitors and improve splenomegaly. In the event of the use of TGFβ/SMAD and GATA-1 modulators, it is expected that, as the differentiation of erythroid progenitors increases and the relative number of RBC increases, iron consumption improves and, as a consequence, hemichrome formation is reduced. In this last case, the end points are similar to those observed with the use of iron-restrictive agents, although the extent of each improvement can vary based on the activity and characteristics of each drug. Of note, the challenge in the development of GATA-1 modulators would require identifying compounds that protect GATA-1 in cells in which HSP70 is sequestered by the excess of α-globin chains, but not in cells in which GATA-1 is fully active, to prevent undesirable effects.
Targeting iron metabolism or IE to improve anemia and iron overload in BT. (A) Iron restriction can be achieved by using hepcidin agonists or activators or inhibitors of ferroportin, HIF2α, and erythroferrone to reduce iron intake and increase iron sequestration into macrophages, decreasing TSat. This limits hemichrome formation, ROS, and, potentially, free α-globin chain accumulation, resulting in improvements to RBC lifespan, anemia, extramedullary erythropoiesis and also iron overload. (B) Improvement of IE with drugs that inhibit JAK2 or modulate TGFβ/SMAD (eg, luspatercept) and GATA-1 activity. Use of JAK2 inhibitors will only decrease the number of erythroid progenitors and improve splenomegaly. In the event of the use of TGFβ/SMAD and GATA-1 modulators, it is expected that, as the differentiation of erythroid progenitors increases and the relative number of RBC increases, iron consumption improves and, as a consequence, hemichrome formation is reduced. In this last case, the end points are similar to those observed with the use of iron-restrictive agents, although the extent of each improvement can vary based on the activity and characteristics of each drug. Of note, the challenge in the development of GATA-1 modulators would require identifying compounds that protect GATA-1 in cells in which HSP70 is sequestered by the excess of α-globin chains, but not in cells in which GATA-1 is fully active, to prevent undesirable effects.
Agents that act or may act to restrict iron absorption and erythroid iron intake include apotransferrin, agonists or inducers of hepcidin, and inhibitors of ferroportin, HIF2α, or erythroferrone (Figure 3A). Genetic models and novel drugs have been tested in animals with BT, showing improvement of the phenotype, as predicted by the model.17,79,83-89 Additional information on the use of some of these drugs can be found in the recent review by Casu et al.90
Ameliorating anemia by improving erythroid differentiation
It might be possible to improve IE by limiting erythroid expansion, removing the block in maturation arrest, and/or improving cell survival (Figure 3B). Some drugs that target those mechanisms are being tested in clinical trials (as described in the next paragraphs).
Given the central role of the EPO/JAK2 axis in BT, it has been proposed that targeting JAK2 could be a way to reverse splenomegaly.19,91,92 If that approach is successful, it could be used as an alternative to splenectomy.19,91,92 Based on that hypothesis and preclinical studies, a clinical trial with ruxolitinib, a JAK1/JAK2 inhibitor, was performed.93 The trial was a single-arm open-label multicenter phase 2a study that explored the efficacy and safety of ruxolitinib in regularly transfused patients with BT and spleen enlargement.93 The major positive conclusion of the trial was that treatment with ruxolitinib led to a sustained reduction in spleen size.93 However, limited differences in the pretransfusion Hb levels and transfusion requirements were observed; for this reason, further trials were not proposed.93 The main conclusion from this study is that the use of JAK1/2 inhibitors represents a valid option for patients with BT and splenomegaly, especially if these drugs will be administered for a short time to avoid the potential side effects (ie, thrombocytopenia) that are observed in patients treated over the long-term who are affected by myeloproliferative disorders.92
Sotatercept (ACE-011) and luspatercept (ACE-536) contain the extracellular domains of ACVR2A and ACVR2B, respectively, fused to the Fc domain of human IgG1. Both drugs act as ligand traps for TGF-β–like molecules.94 Sotatercept was originally developed to treat bone loss disorders, but clinical studies unexpectedly revealed increased hematocrit and Hb levels in treated patients.95 Luspatercept was generated with a single amino acid modification to outcompete activins and bind with higher affinity to GDF11.
RAP-011 and RAP-536, the mouse counterparts of sotatercept and luspatercept, contain the extracellular domains of mouse Acvr2A and Acvr2B, respectively, fused to the Fc domain of mouse IgG.94 In mouse models of BT and MDS, RAP-536 and RAP-011 had effects similar to those observed in wild-type mice (increased RBC and Hb synthesis), but they also had some disease-specific effects. In BT animals, the drugs increased the differentiation of late erythroid cells and concomitantly decreased the survival of early erythroid progenitors, with a beneficial effect on IE and anemia (Figure 3B). Furthermore, they improved iron metabolism, because the levels of hepcidin increased, and iron overload improved.41,75 The quality and lifespan of RBCs improved, and there was a reduction in hemichromes.41,75 However, the improvement in iron metabolism was secondary to the amelioration of IE (Figure 3B). Based on those initial positive results, clinical trials were initiated. In patients affected by TDT, NTDT, and MDS, sotatercept and luspatercept showed encouraging results in phase 1 and 2 clinical trials, with a dose-dependent improvement in anemia and a satisfactory drug safety profile.96 A phase 3 clinical trial is currently ongoing.
Conclusions and future directions
The results of preclinical studies indicate that drugs that target iron intake and recycling have great potential to improve IE in BT. However, those studies do not address the question of whether iron restriction influences the synthesis of α-globin chains or the relative toxicity of α-globin chains in the absence of heme. There are additional unanswered questions regarding the role of the different forms of iron-bound transferrin molecules (apotransferrin, mono-transferrin, and holo-transferrin) on erythroid cell maturation, the modulation of EPO sensitivity and hepcidin synthesis, and whether iron can directly or indirectly modulate erythroid differentiation under conditions of IE. Future studies will need to address those questions.
Regarding the drugs that target IE, the mechanism of action of the trap ligands ACE011 and ACE536 may be more complex than what was initially proposed (ie, the inhibition of GDF11). For instance, trap ligands for GDF11 are also able to stimulate RBC synthesis in healthy humans and mice,41,95 but GDF11 expression has not been reported in healthy erythroblasts from mice or humans.41 GDF11 is expressed in the pancreas, intestine, kidney, skeletal muscle, heart, developing nervous system, olfactory system, and retina, but it is structurally similar to GDF8/myostatin.38 In fact, some GDF11 expression data have been questioned because of the inability of available antibodies to discriminate between GDF11 and GDF8.97 GDF11 trap ligands seem to be quite promiscuous in targeting additional molecules, which may also contribute to normal erythropoiesis and IE.97 Additionally, reduced activity of Gdf11 and similar ligands, like GDF8, seem to be associated with increased cardiovascular events.98 In conclusion, further studies are required to elucidate the role of GDF11 and other molecules that, through the regulation of TGF-β–like pathways, may control normal erythropoiesis and IE.
Despite several open questions, new drugs have great potential to modify the current clinical treatments for BT. Preclinical and clinical data suggest that all of the new drugs may increase Hb levels in BT by 1-2 g/dL.54,90 Based on their independent mechanisms of action, it is logical to hypothesize that different drugs that target iron metabolism and IE could be combined to produce even greater increases in RBC synthesis and Hb levels, respectively, further improving the phenotype of patients affected by BT. Furthermore, additional drugs (like agents that reactivate fetal Hb) and gene therapy approaches are being developed, which hold great promise.99,100 Altogether, these new approaches have the potential to radically modify the way in which patients affected BT will be treated or cured.
Acknowledgment
This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grants R01 DK095112 and R01 DK090554 (S.R.).
Authorship
Contribution: S.R. wrote the manuscript.
Conflict-of-interest disclosure: S.R. is a consultant for Ionis Pharmaceuticals, Disc Medicine, Meira GTx, and Protagonist Therapeutics.
Correspondence: Stefano Rivella, Division of Hematology, Department of Pediatrics, Children’s Hospital of Philadelphia, Cell and Molecular Biology Graduate Group, University of Pennsylvania, Abramson Research Center, Room 316B, 3615 Civic Center Blvd, Philadelphia, PA 19104; e-mail: rivellas@e-mail.chop.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal