

Abstract
Iron deficiency anemia affects >1.2 billions individuals worldwide, and iron deficiency in the absence of anemia is even more frequent. Total-body (absolute) iron deficiency is caused by physiologically increased iron requirements in children, adolescents, young and pregnant women, by reduced iron intake, or by pathological defective absorption or chronic blood loss. Adaptation to iron deficiency at the tissue level is controlled by iron regulatory proteins to increase iron uptake and retention; at the systemic level, suppression of the iron hormone hepcidin increases iron release to plasma by absorptive enterocytes and recycling macrophages. The diagnosis of absolute iron deficiency is easy unless the condition is masked by inflammatory conditions. All cases of iron deficiency should be assessed for treatment and underlying cause. Special attention is needed in areas endemic for malaria and other infections to avoid worsening of infection by iron treatment. Ongoing efforts aim at optimizing iron salts–based therapy by protocols of administration based on the physiology of hepcidin control and reducing the common adverse effects of oral iron. IV iron, especially last-generation compounds administered at high doses in single infusions, is becoming an effective alternative in an increasing number of conditions because of a more rapid and persistent hematological response and acceptable safety profile. Risks/benefits of the different treatments should be weighed in a personalized therapeutic approach to iron deficiency.
Introduction
Iron balance is essential for all cell life. Iron homeostatic mechanisms evolved to avoid iron excess and the generation of harmful reactive oxygen species by reutilizing body iron and limiting its uptake from the environment. The inevitable other side of the coin is the easy development of iron deficiency.
Iron deficiency is the depletion of total-body iron, especially of macrophage and hepatocyte iron stores. Because the largest amount of iron is consumed for hemoglobin (Hb) synthesis to produce 200 billion erythrocytes daily, anemia is the more evident sign of iron deficiency, and iron deficiency anemia is often considered synonymous with iron deficiency. However, iron deficiency is a broader condition that often precedes the onset of anemia or indicates deficiency in organs/tissues other than those involved in erythropoiesis, such as skeletal muscles and the heart, the latter highly iron dependent for myoglobin and energy production to sustain mechanical contraction.
This article reviews the mechanisms of adaptation to iron deficiency and related anemia, examines how improved knowledge is influencing treatment, and discusses areas that remain uncertain from the biological and clinical perspectives.
Epidemiology
According to the Global Burden of Disease Study 2016, iron deficiency anemia is 1 of the 5 leading causes of years lived with disability burden and is the first cause in women.1 Adopting the World Health Organization–recommended cutoff for anemia (Hb <13 g/dL in males, <12 g/dL in females, <11g/dL during pregnancy), a worldwide survey showed that in 2010, anemia still affected one third of the population, with approximately half of the cases resulting from iron deficiency. The estimate is that ∼1.24 billion individuals experience iron deficiency anemia, although with huge variations from low- to high-income countries.2 The global prevalence of iron deficiency without anemia remains elusive, although the suggested figure is at least double that of iron deficiency anemia. The problem becomes even more relevant if we take into account functional iron deficiency, which occurs when iron is hardly mobilized from stores, as in chronic inflammations/infections or when the vigorous erythropoietic expansion by exogenous or endogenous erythropoietin (EPO) causes an acute disproportion between iron demand and supply.
Globally, iron deficiency anemia has relevant medical and social impacts, accounting for impairment of cognitive performance in young children,3 adverse outcomes of pregnancy for both mothers and newborns,4 decreased physical and working capacities in adults, and cognitive decline in the elderly.5,6 From available data, the relative contribution of iron deficiency to these negative outcomes is difficult to disassociate from that of anemia.
Pathophysiology of iron deficiency
Iron deficiency deeply affects iron homeostasis, inducing adaptive mechanisms on the hepcidin-ferroportin (FPN) axis, the iron regulatory protein (IRP)/iron responsive element (IRE) machinery, and other regulators. The aim is to optimize iron usage by erythropoiesis and to counteract the physiological inhibition of iron absorption.
Mechanisms of adaptation
Systemic regulation
Liver hepcidin is the master hormone that physiologically limits iron entry into plasma. Binding to its receptor FPN, hepcidin blocks iron export both by occluding the exporter central cavity7 and by inducing its degradation.8 Because of the high FPN expression on professional iron exporter cells, such as enterocytes and macrophages, hepcidin suppression in iron deficiency enhances both iron absorption and its release from macrophages to plasma. Multiple factors downregulate hepcidin transcription (Figure 1). The BMP-SMAD signaling pathway is repressed, because in iron deficiency, expression of BMP6 ligand is low,9 the BMP coreceptor HJV is cleaved by TMPRSS6,10 and TFR2 is removed from the cell surface.11 In addition, the histone deacetylase HDAC3 erases activation markers from the hepcidin locus,12 providing an epigenetic contribution to hormone suppression. The function of ERFE, released by erythroid cells stimulated by erythropoietin,13 is less relevant in iron deficiency without anemia, because hepcidin is downregulated when iron deficiency is induced in Erfe−/− mice.12 However, ERFE plays a role in the presence of anemia and hypoxia.13
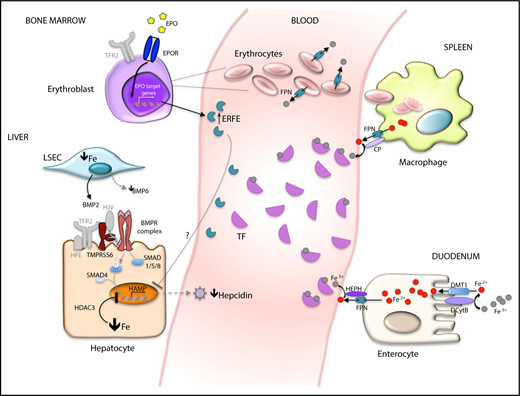
Mechanisms of hepcidin inhibition in iron deficiency anemia. Main cells/organs involved in hepcidin (HAMP) inhibition in iron deficiency are illustrated. In the hepatocytes, bone morphogenic protein (BMP)-SMAD signaling, the main activator of hepcidin, is low because low levels of BMP6 are produced by liver sinusoidal endothelial cells (L-SEC), the BMP coreceptor hemojuvelin (HJV) is cleaved from the hepatocyte surface by the transmembrane serine protease 6 (TMPRSS6), and the second transferrin receptor (TFR2) is not stabilized on the cell surface in the absence of the ligand diferric transferrin (TF). Low hepcidin levels increase iron absorption by enterocytes and recycling by macrophages through increased activity of the iron exporter FPN. In mild iron deficiency in the absence of hypoxia, increased EPO sensitivity is due to the loss of TFR2 on erythroblast surfaces. Histone deacetylase 3 (HIDAC3) participates in hepcidin suppression by erasing markers of activation at the hepcidin locus. In iron deficiency anemia, hypoxia increases EPO. Increased ERFE fully blocks the hepcidin pathway, although the molecular mechanism of hepcidin inhibition by ERFE remains unknown (?). BMPR, BMP receptor; CP, ceruloplasmin; DCYTB, duodenal cytochrome B; DMT1, divalent metal transporter 1; EPOR, EPO receptor; HEPH, hephestin.
Mechanisms of hepcidin inhibition in iron deficiency anemia. Main cells/organs involved in hepcidin (HAMP) inhibition in iron deficiency are illustrated. In the hepatocytes, bone morphogenic protein (BMP)-SMAD signaling, the main activator of hepcidin, is low because low levels of BMP6 are produced by liver sinusoidal endothelial cells (L-SEC), the BMP coreceptor hemojuvelin (HJV) is cleaved from the hepatocyte surface by the transmembrane serine protease 6 (TMPRSS6), and the second transferrin receptor (TFR2) is not stabilized on the cell surface in the absence of the ligand diferric transferrin (TF). Low hepcidin levels increase iron absorption by enterocytes and recycling by macrophages through increased activity of the iron exporter FPN. In mild iron deficiency in the absence of hypoxia, increased EPO sensitivity is due to the loss of TFR2 on erythroblast surfaces. Histone deacetylase 3 (HIDAC3) participates in hepcidin suppression by erasing markers of activation at the hepcidin locus. In iron deficiency anemia, hypoxia increases EPO. Increased ERFE fully blocks the hepcidin pathway, although the molecular mechanism of hepcidin inhibition by ERFE remains unknown (?). BMPR, BMP receptor; CP, ceruloplasmin; DCYTB, duodenal cytochrome B; DMT1, divalent metal transporter 1; EPOR, EPO receptor; HEPH, hephestin.
Local mechanisms increase intestinal iron absorption. Hypoxia-inducible factor 2α (HIF2α) upregulates the expression of both the brush border machinery (DMT1 and DCYTB) that uptakes iron from the lumen and the iron exporter FPN at the basolateral membrane by binding hypoxia-responsive elements of these gene promoters.14
Macrophages rapidly recycle iron derived from the phagocytosis of senescent red cells (Figure 1). However, the absolute amount of iron recycled from hypochromic erythrocytes by heme-oxygenase 1 decreases in parallel with the severity of iron deficiency, because Hb content per cell (mean corpuscular Hb [MCH]) is reduced. A novel mechanism related to erythrocyte FPN, which is highly expressed in iron deficiency, may contribute to maintaining circulating iron levels.15 Low serum hepcidin levels ultimately determine the amount of iron entering the circulation (Figure 1).
Cellular regulation
Cellular iron content is controlled by IRPs that in iron deficiency bind stem-loop sequences (IREs) in the untranslated regions (UTRs) of iron genes to posttranscriptionally coordinate proteins of iron absorption, export, use, and storage.16 Binding to 3′UTR IREs, IRPs stabilize the messenger RNA of TFRC and DMT1; binding to 5′UTR IREs, they repress translation of ferritin, FPN, 5′-aminolevulinate synthase 2 (ALAS2), and HIF2α. To avoid deleterious iron retention in iron-deficient enterocytes and maturing erythroblasts, an alternative isoform of FPN lacking 5′UTR IRE escapes IRP control15 while remaining sensitive to the hepcidin effect.
Other IRP-independent mechanisms optimize iron use in low-iron states. mTOR inhibition activates tristetraprolin, which reduces both TFR1 and FPN expression to save iron for tissue metabolic needs.17 Cells may recover their own iron stored in ferritin. In iron deficiency, ferritin is delivered to autophagosomes for degradation (ferritinophagy) by nuclear receptor coactivator 4, which in contrast is proteasome degraded in iron-replete cells.18 Reduced ferritinophagy makes NcoA4 knockout mice susceptible to hypoferremia and iron deficiency.19 Ferritinophagy has been shown to provide iron for erythroid differentiation in vitro20 and in zebrafish.21 It has not been assessed whether ferritin is reduced in plasma when it undergoes ferritinophagy. Although serum ferritin is the best biomarker of iron deficiency, the mechanisms of its release as well as its function in the circulation remain mysterious.22
Iron-restricted erythropoiesis
Iron restriction limits the expansion of early erythropoiesis and optimizes iron use by terminal erythropoiesis. In vitro iron deprivation blunts the EPO responsiveness of early progenitors through inactivation of iron-dependent aconitase, which suppresses isocitrate production.23 Accordingly, iron or isocitrate treatment restores erythroid lineage differentiation.24 EPO is not elevated in mice with iron deficiency without anemia.25,26 However, in the same condition, terminal erythropoiesis is modified, with decreased apoptosis and increased number of late erythroblasts. The same phenotype, expression of increased EPO sensitivity, is recapitulated by the genetic loss of the EPOR partner TFR2 in mice; this condition mimics iron deficiency,27 because TFR2 is lost from the membrane when diferric TF is reduced.11,28
With the development of anemia and hypoxia, EPO levels increase exponentially, and multiple mediators, such as erythroferrone,13 GDF15,29 and PDGF-BB,30 suppress hepcidin to enhance iron supply. In this process, a role of soluble TFR (sTFR), an accepted biomarker of iron deficiency,31 although reasonable, remains unproven.
Because of the increased number of erythroblasts and limited iron supply, heme content per cell is reduced. Globin translation is also impaired by low heme; the stress sensor heme-regulated inhibitor (HRI) phosphorylates the elongation initiation factor 2a (eIF2A) to block translation, concomitantly increasing ATF4, which inhibits the translation regulator mTOR.32 The heme/globin coordination improves erythropoiesis, producing microcytic (low mean corpuscular volume)/hypochromic (low MCH) erythrocytes. The optimization of erythropoiesis might preserve iron for vital functions within a global body economy. However, the mechanism is not fully effective, because even in the absence of anemia, other organs may become iron deficient.
Etiology of iron deficiency
Individuals at risk
Reflecting high iron requirements, infants, preschool children (age <5 years), young menstruating women, and women in the second/third trimester of pregnancy and postpartum are the most affected groups.33,34 Adolescents also are susceptible to iron deficiency because of rapid growth.
In Western countries, other healthy individuals may be at risk. These include vegetarians, especially vegans, because of diet restriction and blood donors.35 The RISE study, which evaluated the iron status of >2000 frequent blood donors in the United States, showed that two thirds of women and half of men were iron deficient.36 Elite endurance athletes are at risk because of inflammation-induced increased hepcidin and blood losses. Females are more affected in all the groups listed here.
Iron deficiency with or without anemia may be isolated or secondary to a causative disorder or occur in the context of multiple pathological conditions (eg, in the elderly). Iron deficiency is usually acquired and exceptionally inherited.
Acquired iron deficiency
In developing countries, iron deficiency anemia is nutritional, resulting from reduced intake of bioavailable iron (Table 1), and often associated with infections causing hemorrhages, such as hookworm infestation or schistosomiasis. In Western societies, other than in individuals at risk, iron depletion results from chronic bleeding and/or reduced iron absorption, disorders that may be more relevant than anemia itself (Table 1). For this reason, considering age, sex, clinical history, and symptoms, identification of the underlying cause is an essential part of the patient’s workup.33,34
Main causes of absolute iron deficiency/iron deficiency anemia
| Type of cause | Condition | Pathophysiologic mechanism |
|---|---|---|
| Increased iron requirements | Infants, preschool children, adolescents | Rapid growth |
| Pregnant women: second and third trimesters | Expansion of maternal and fetal erythroid mass | |
| ESA treatment | Acute expansion of erythroid mass | |
| Low iron intake | Malnutrition* | Insufficient dietary iron: low heme iron or scarcely bioavailable iron (eg, chelated by phytates) |
| Vegetarians, vegans | ||
| Decreased intestinal iron absorption | Gastrectomy, duodenal bypass, bariatric surgery | Decreased absorptive surface |
| Gluten-induced enteropathy | ||
| Autoimmune atrophic gastritis | Increased pH | |
| Helicobacter pylori infection | Increased pH and blood loss | |
| Drugs: proton pump inhibitors, H2 blockers | Blocking of gastric acid secretion | |
| Genetic IRIDA† | High serum hepcidin levels | |
| Chronic blood loss | Hookworm infestation* | Bleeding from gastrointestinal tract |
| Gastrointestinal benign and malignant lesions | ||
| Salicylates, corticosteroids, nonsteroidal anti-inflammatory drugs | ||
| Heavy menses, hematuria | Bleeding from genitourinary system | |
| Intravascular hemolysis (PNH, march hemoglobinuria) | Urinary loss of hemoglobin (iron) | |
| Drugs: anticoagulants, antiplatelet compounds | Systemic bleeding | |
| Defects of hemostasis (hereditary hemorrhagic telangectasia, von Willebrand disease) | ||
| Frequent blood donors | Repeated blood letting | |
| Multiple causes (absolute iron deficiency associated with inflammation) | Chronic infections in malnutrition* | Reduced intake, increased proinflammatory cytokines |
| Chronic kidney disease | Decreased iron absorption, increased blood loss, reduced hepcidin excretion and increased production, drugs, ESAs | |
| Chronic systolic heart failure | Decreased iron absorption, increased inflammation, blood loss | |
| Inflammatory bowel diseases | Decreased iron absorption, increased blood loss, high hepcidin | |
| Postoperative anemia of major surgery | Blood loss, increased proinflammatory cytokines |
| Type of cause | Condition | Pathophysiologic mechanism |
|---|---|---|
| Increased iron requirements | Infants, preschool children, adolescents | Rapid growth |
| Pregnant women: second and third trimesters | Expansion of maternal and fetal erythroid mass | |
| ESA treatment | Acute expansion of erythroid mass | |
| Low iron intake | Malnutrition* | Insufficient dietary iron: low heme iron or scarcely bioavailable iron (eg, chelated by phytates) |
| Vegetarians, vegans | ||
| Decreased intestinal iron absorption | Gastrectomy, duodenal bypass, bariatric surgery | Decreased absorptive surface |
| Gluten-induced enteropathy | ||
| Autoimmune atrophic gastritis | Increased pH | |
| Helicobacter pylori infection | Increased pH and blood loss | |
| Drugs: proton pump inhibitors, H2 blockers | Blocking of gastric acid secretion | |
| Genetic IRIDA† | High serum hepcidin levels | |
| Chronic blood loss | Hookworm infestation* | Bleeding from gastrointestinal tract |
| Gastrointestinal benign and malignant lesions | ||
| Salicylates, corticosteroids, nonsteroidal anti-inflammatory drugs | ||
| Heavy menses, hematuria | Bleeding from genitourinary system | |
| Intravascular hemolysis (PNH, march hemoglobinuria) | Urinary loss of hemoglobin (iron) | |
| Drugs: anticoagulants, antiplatelet compounds | Systemic bleeding | |
| Defects of hemostasis (hereditary hemorrhagic telangectasia, von Willebrand disease) | ||
| Frequent blood donors | Repeated blood letting | |
| Multiple causes (absolute iron deficiency associated with inflammation) | Chronic infections in malnutrition* | Reduced intake, increased proinflammatory cytokines |
| Chronic kidney disease | Decreased iron absorption, increased blood loss, reduced hepcidin excretion and increased production, drugs, ESAs | |
| Chronic systolic heart failure | Decreased iron absorption, increased inflammation, blood loss | |
| Inflammatory bowel diseases | Decreased iron absorption, increased blood loss, high hepcidin | |
| Postoperative anemia of major surgery | Blood loss, increased proinflammatory cytokines |
ESA, erythropoiesis-stimulating agent; H2 antagonists, histamine receptor blockers; IRIDA, iron-refractory iron deficiency anemia; PNH, paroxysmal nocturnal hemoglobinuria.
More common in developing countries.
Rarely resulting from gene mutations other than TMPRSS6.100
Absolute iron deficiency may be masked by comorbidities (eg, in the elderly, and in the setting of renal failure). Anemia in the elderly has multiple causes.37 Iron deficiency accounts for ∼30% of cases, resulting from low intake, reduced absorption (atrophic gastritis, use of proton pump inhibitors), gastrointestinal blood losses (antithrombotic drugs, angiodysplasia, peptic ulcer, hemorrhoids, and even colorectal cancer). Unfortunately, being obscured by comorbidities, it often remains undiagnosed,38 while even mild anemia worsens the outcome of associated disorders and influences mortality.39 Patients with chronic kidney disease (CKD) are prone to absolute iron deficiency because of reduced absorption40 and blood loss at dialysis, at an estimated rate of up to 2 to 3 g per year.41 However, high hepcidin levels and inflammation, which reduce iron mobilization from stores, may mask absolute deficiency. A recognized cause of dysregulation of iron metabolism is obesity, which may lead to iron deficiency, especially after bariatric surgery because of global absorption impairment (Table 1).42 In Western countries, as a result of increased life expectancy, these types of iron deficiency are expected to increase in coming years.
Considering the need for balancing iron demand and supply, specific clinical settings are characterized by acute restriction of iron for erythropoiesis. The best-known example is treatment with erythropoiesis-stimulating agents. Another example is postoperative anemia that follows major surgery. Recovery from anemia may be limited or delayed because of preexisting unrecognized iron deficiency that becomes evident after surgery and/or cytokine-induced defective iron mobilization.
Genetics of iron deficiency
IRIDA43 is a rare recessive condition resulting from mutations of TMPRSS6,44,45 leading to an inability to cleave the BMP coreceptor HJV and inhibit hepcidin.10 High hepcidin in IRIDA patients impairs iron absorption, counteracting an essential compensatory mechanism to sustain erythropoiesis. IRIDA patients are refractory to oral iron supplementation.46,47 IV iron is indicated when anemia is severe, but it may be only partially effective. In adults, especially men, anemia may be less evident than in children, while iron deficiency and microcytosis persist.48
Populations studies suggest that susceptibility to iron deficiency is in part influenced by genetics. Studies of blood donors have strengthened the hypothesis that genetic variants of iron genes, especially TMPRSS6 and HFE, reported to influence iron parameters49,50 and hepcidin,51 may predispose to or protect individuals from iron deficiency.52
In a novel murine model, genetic iron deficiency anemia was caused by loss of the enzyme of the sulfur assimilation pathway bisphosphate-3′-nucleotidase (Bpnt1). Iron deficiency anemia characterizes both germinal and intestinal conditional Bpnt1 knockout mice, establishing a novel link between sulfur and iron homeostasis.53
Diagnosing iron deficiency
Clinical signs and symptoms of iron deficiency anemia are limited and often neglected. The most important, fatigue, is unspecific. Alterations of epithelial cells such as dry mouth, cheilitis, atrophic glossitis, Plummer-Vinson pharyngeal webs, and hair loss are observed in longstanding deficiency. Restless leg syndrome reveals iron deficiency in a proportion of cases.54,55 In the elderly, iron deficiency anemia may cause heart failure or angina. For a detailed discussion of symptoms in iron deficiency anemia, readers are referred elsewhere.34,56
A correct diagnosis requires laboratory tests. Low serum ferritin levels are the hallmark of absolute iron deficiency, reflecting exhausted stores. Levels <30 mg/L are the accepted threshold that identifies mild cases; in the presence of anemia, ferritin levels are usually lower (<10-12 mg/L). In the absence of inflammations/infections, serum ferritin shows the best correlation with bone marrow stainable iron, once the gold standard in assessing depletion of iron stores.33,34
Measuring TF saturation (<16%) is unnecessary for diagnosis, although it has diagnostic value in functional deficiency when serum ferritin is unreliable. Hepcidin levels, which are low/undetectable in absolute iron deficiency, are also unnecessary. Exceptions are the rare IRIDA patients who show low TF saturation and normal/high hepcidin and serum ferritin levels, reflecting increased macrophage iron. Measuring serum hepcidin may be diagnostic of this atypical iron deficiency, provided that inflammation is excluded.57
sTFR and its relationship to ferritin (sTFR/logFt index) are good indicators of iron-deficient erythropoiesis,58,59 but tests to measure these indicators are scarcely available in clinics. Reduction of MCH and mean corpuscular volume and increased (>6% in CKD) hypochromic red cells (with MCH <28 pg) occur relatively late because of the erythrocyte lifespan. Reticulocyte Hb content may reveal rapid changes in erythropoietic activity. Early reduction (<26 pg) may occur after erythropoiesis-stimulating agent treatment and early increase after iron supplementation.60 When heme is low, zinc is incorporated into protoporphyrin-IX, levels of which become elevated and measurable in mature erythrocytes.60
All tissues are assumed to be iron deficient when ferritin is low. No specific test assesses tissue (eg, cardiac or muscle) iron deficiency when ferritin is unreliable, such as in inflammation. Perception of this deficiency by patients is highly variable. Clinical diagnosis relies on deterioration of the specific organ (eg, heart) function or on unspecific symptoms, the most popular being fatigue. Alternatively, the diagnosis is based on a positive outcome after iron supplementation, such as in heart failure.61
To correctly diagnose iron deficiency in the context of multiple comorbidities, such as in inflammation, ferritin threshold <100 mg/L or even higher values are suggested, in combination with low (<20%) TF saturation.62 Although these arbitrary cutoffs likely overestimate iron deficiency, they are largely used for therapeutic decisions. The diagnosis of absolute iron deficiency is also challenging in the elderly; proposed cutoffs between >30 and <100 mg/L are based on small studies.37,38 This supports the need for well-designed prospective clinical trials and development of biomarkers for tissue iron deficiency.
Advances and controversies in the treatment of iron deficiency
The etiological cause of iron deficiency should be addressed in all cases and, whenever possible, eliminated. Iron treatment should be started immediately, even in the absence of anemia, especially in symptomatic patients.63,64 A systematic review of the efficacy of iron supplementation in iron-deficient nonanemic individuals concluded that treatment (any type) increased Hb and ferritin and reduced self-reported fatigue but did not improve physical performance or maximal oxygen consumption.65
The choice of iron compound and the route of administration are largely dependent on the presence and degree of anemia, reversibility of the underlying cause, clinical status (age, sex, longstanding vs recent onset), and in some instances patient preference.
Oral iron supplementation
Iron salts such as iron sulfate, fumarate, and gluconate remain a mainstay of therapy in absolute iron deficiency. Mounting evidence indicates that low doses are more effective and better tolerated than the traditionally recommended 100 to 200 mg of elementary iron per day. Because absorption of nonheme iron is modest (5% to 28% at the fastest),66 high doses may result in ROS-mediated toxicity of nonabsorbed iron on intestinal mucosa. Common adverse effects, such as nausea, vomiting, constipation, or diarrhea, may lead to noncompliance with therapy in 30% to 70% of cases67 and jeopardize the prolonged (several months) treatment planned. Importantly, even a mild increase in serum iron activates hepcidin to limit iron absorption. This physiological response was exploited to design the most appropriate dose and schedule of oral iron administration in iron-deficient nonanemic women. In short-term studies that used stable iron isotopes, supplementation with iron sulfate (60-240 mg) induced hepcidin increase for up to 48 hours, limiting the absorption of the subsequent doses.68 In another trial in which participants were randomly assigned to receive 60 mg of iron per day for 14 days or on alternate day for 28 days, fractional iron absorption was significantly greater in the latter group (21.8% vs 16.3%). In a study comparing 2 groups of women who were receiving 120 mg of iron sulfate per day either as a single or 2 divided doses, the first group showed smaller serum hepcidin increases.69 Altogether these elegant studies indicate that changing the administration from daily to alternate-day schedules and from divided to single doses increases the efficacy of treatment in iron-deficient nonanemic individuals and has the potential to improve tolerability. An ongoing study in women with iron deficiency anemia70 is assessing whether the alternate-day protocol should also be recommended in the presence of anemia,71 when hypoxia further increases intestinal iron absorption and fully suppresses hepcidin.14
Other adverse effects of unabsorbed iron include alterations in the composition of the gut microbiome, with reduction of beneficial Lactobacillus and Bifidobacterium bacteria, enhancement of potential pathogens (Enterobacteriaceae), and increased inflammation and diarrhea, as shown in African children.72,73
The minimal dose used for iron supplementation is 60 mg per day. Lower doses (37.5 mg per day) of oral iron have proven useful in blood donors to limit deferrals from donations.36
A prophylactic treatment with iron sulfate (60 mg in adults and 30 mg in children) has been recommended in world areas characterized by high prevalence of iron deficiency anemia.74 However, the validity of universal supplementation in countries with high prevalence of malaria and/or other infections is controversial. Epidemiological75 and in vitro studies have shown that iron deficiency is an adaptation process protecting from Plasmodium virulence and that its correction may increase infection severity.76,77 Recent evidence shows that FPN expressed in erythrocytes is functional and reduced by the high hepcidin levels induced by iron supplementation. This would increase erythrocyte iron content, favoring the parasite growth.15 In these cases, iron supplementation should occur in association with antimalarial treatment.78 Another problem is related supplemented iron causing gut dysbiosis and diarrhea. To avoid the latter effects, a future solution is the development of iron compounds bioavailable only to humans and not to pathogens.
There is great interest in the development of compounds better tolerated than iron salts; numerous compounds have been proposed (eg, sucrosomial iron, heme iron polypeptide, iron containing nanoparticles), but studies are limited.79 Sucrosomial iron has been tested in patients with CKD,80 but the mechanism of absorption and the real benefits are uncertain. In the same condition, the phosphate binder iron ferric citrate simultaneously corrects both hyperphosphatemia and iron deficiency; its double effect is being tested in a clinical trial in CKD.81 A phase 3 trial of ferric maltol provided positive results on iron deficiency anemia in inflammatory bowel diseases.82 Rigorously designed clinical trials are needed to confirm the efficacy of these iron preparations.
The natural compound extracted from the bark of the Taiwanese tree hinokitiol restores iron transport in cells lacking transporters, such as DMT1 or FPN.83 Exploiting the iron gradient that, in the absence of the transporter, is formed across membranes, hinokitiol restores transport direction both in vitro and in zebrafish, but no data are available on its chronic use in mice.
IV iron
The alternative for patients intolerant or unresponsive to oral compounds is IV iron.47 Once limited by the risk of severe hypersensitivity reactions, this route of administration is currently more widely used as a result of the improved safety profile of last-generation compounds. Established indications to IV iron are reduced absorption capacity in the presence of gastrointestinal disorders or bariatric surgery, severe anemia (Hb <7-8 g/dL), high hepcidin resulting from concomitant inflammation, and rarely IRIDA and when a fast recovery is desirable (Table 2). Advantages are the more rapid effect and the negligible gastrointestinal toxicity.67 IV iron is more effective than oral iron in CKD patients treated with erythropoiesis-stimulating agents41,84 and avoids oxidative damage to the intestinal mucosa in active inflammatory bowel diseases.85 In the latter disorders, IV iron preserves the normal microbiome, which would be disrupted by oral iron.40 The European Crohn’s and Colitis Organization recommends IV iron as a first-line therapy for patients with active disease and Hb <10 g/dL and oral iron in inactive disease/mild anemia patients,86 the latter being more likely to have absolute iron deficiency.
Indication for IV iron therapy
| Condition | Reason |
|---|---|
| Oral iron intolerance | Persistent gastrointestinal adverse effects |
| Oral iron refractoriness | Defective absorption: gastrectomy, duodenal bypass, bariatric surgery |
| Intestinal disorders (selected cases): IBD, atrophic gastritis, Helicobacter pylori infection, gluten enteropathy | |
| Genetic forms (IRIDA) | |
| No Hb improvement after 4 wk of oral therapy | |
| Severe anemia (Hb <7-8 g/dL) | Need for rapid Hb improvement |
| Second and third trimesters of pregnancy | Need for rapid Hb increase; often intolerance to oral preparations |
| ESA treatment | More effective than oral iron in CKD |
| Chronic blood loss difficult to manage with oral iron | Heavy uterine bleeding |
| Hereditary disorders of hemostasis | |
| Other | Postoperative anemia of major surgery |
| Chronic systolic heart failure |
| Condition | Reason |
|---|---|
| Oral iron intolerance | Persistent gastrointestinal adverse effects |
| Oral iron refractoriness | Defective absorption: gastrectomy, duodenal bypass, bariatric surgery |
| Intestinal disorders (selected cases): IBD, atrophic gastritis, Helicobacter pylori infection, gluten enteropathy | |
| Genetic forms (IRIDA) | |
| No Hb improvement after 4 wk of oral therapy | |
| Severe anemia (Hb <7-8 g/dL) | Need for rapid Hb improvement |
| Second and third trimesters of pregnancy | Need for rapid Hb increase; often intolerance to oral preparations |
| ESA treatment | More effective than oral iron in CKD |
| Chronic blood loss difficult to manage with oral iron | Heavy uterine bleeding |
| Hereditary disorders of hemostasis | |
| Other | Postoperative anemia of major surgery |
| Chronic systolic heart failure |
IBD, inflammatory bowel disease.
IV iron is available in different forms; iron gluconate and iron sucrose require repeated infusions, whereas ferric carboxymaltose, ferumoxytol, low molecular weight iron dextran, and iron isomaltoside may be administered in high doses to rapidly replace the total iron deficit (usually 1-1.5 g) in 1 or 2 infusions.56 The stable carbohydrate shell of the latter compounds prevents free iron release, a feature that increases their safety.56 The high-dose schedule avoids repeated hospital visits (eg, for patients with reduced mobility, such as the elderly, for whom oral therapy can be particularly disturbing)38 and is convenient when a fast recovery is needed (such as in the second and third trimesters of pregnancy or in postpartum anemia87 and the prevention of repeated cycles of therapy [eg, in heavy uterine bleeding]).88 In addition to the prompt Hb increase, this protocol rapidly reconstitutes stores,89 making the advantages (single access, accelerated recovery, limited need for blood tests) outweigh the disadvantages (cost, invasiveness, risk of reactions). However, this decision should be carefully made on an individual basis.
High-dose IV iron may increase Hb or iron stores before surgery predicted to induce heavy bleeding. This is a kind of prevention of acute postoperative anemia and an alternative to blood transfusions, which are associated with several postoperative complications, including infections. Patient blood management programs that limit blood transfusions by perioperative iron use reduce morbidity and negative prognoses in high-risk interventions.90 A randomized trial of IV iron administration at postoperative day 1 vs standard care showed less anemia and reduction of transfusions and infections in the iron arm in major orthopedic and abdominal surgeries.91 The iron infusion approach might be especially valuable in surgery candidates prone to iron deficiency, such as young women or patients with colorectal cancer.92
An important issue concerning IV iron is safety. Because iron is a growth factor for several pathogens, iron therapy is contraindicated in infections. The risk of infection after IV iron is still a matter of controversy. Increased risk was found in a meta-analysis evaluating trials of IV iron to spare transfusions,93 and caution was suggested in dialysis patients.94 Another meta-analysis of >10 000 patients receiving different IV compounds or oral iron or placebo did not find different risks of infection.95 Long-term studies are needed in patients with different disorders.
Hypophosphatemia after ferricarboxymaltose is usually transient and reversible, although rarely, severe cases have been reported after repeated infusions.96 Minor/moderate infusion reactions (nausea, pruritus, urticaria, flushing, back or thoracic pain), often self-limited, may be observed in 1:200 infusions and more serious reactions (hypotension, dyspnea) in 1:200 000.95 Although globally IV treatment seems safe, the number of reported patients is usually too limited to detect extremely rare anaphylactic reactions, such as those once caused by high molecular weight iron dextran. Their unclear pathogenesis is ascribed to the release of iron particles in the circulation and has been interpreted as a “complement activation-related pseudo-allergy.”97(p5029) Personnel who administer IV iron must be prepared to manage any type of reaction, including exceptionally severe ones.98
The superior efficacy of IV vs oral iron is undisputable and expected; the long-term adverse effects of ROS generation in cases of therapy-induced positive iron balance have been scarcely explored, although overtreatment might occur in functional rather than in absolute iron deficiency. A recent analysis in CKD concluded that patients seemed to tolerate positive iron balance, because iron that was not used was safely stored in reticule-endothelial cells.99 However, in the absence of data and iron toxicity tests, it is advisable to regularly assess iron status when high doses are repeatedly administered.
Future perspectives
Although advances in understanding iron metabolism and regulation are systematically providing novel insights, additional studies are needed before iron therapy becomes a personalized approach in all cases. These studies should aim at discovering markers of tissue iron deficiency, investigate novel schedules of iron administration based on iron physiology, provide clearer indications to high-dose IV iron, and contribute long-term evaluations of treatment outcomes.
Acknowledgments
The author thanks Domenico Girelli for his valuable advice and criticism and Alessia Pagani for help with the figure.
Authorship
Contribution: C.C. conceived, wrote, and reviewed the paper.
Conflict-of-interest disclosure: C.C. is an advisor for Vifor Iron Core and has received honoraria from Vifor Pharma.
Correspondence: Clara Camaschella, Division of Genetics and Cell Biology, San Raffaele Scientific Institute, Via Olgettina, 58, 20132 Milan, Italy; e-mail: camaschella.clara@hsr.it.
