

Key Points
Immune responses to FVIII sequence variants encoded by ns-SNPs do not contribute appreciably to inhibitor development in African Americans.
African American HA subjects with an intron-22 inversion had a 2- to 3-times-higher inhibitor incidence than whites with the same mutation.
Abstract
African American hemophilia A (HA) patients experience a higher incidence of neutralizing anti-factor VIII (FVIII) antibodies (“inhibitors”) vis-à-vis white patients. Nonsynonymous single-nucleotide polymorphisms (ns-SNPs) in the F8 gene encoding FVIII-H484, FVIII-E1241, and FVIII-V2238 are more prevalent in African Americans. This study tested the hypothesis that immune responses to these sites provoke inhibitors. Blood samples were obtained from 174 African American and 198 white HA subjects and their F8 gene sequences determined. Major histocompatibility complex class II binding and T-cell recognition of polymorphic sequences were evaluated using quantitative binding assays and HLA-DRB1 tetramers. Peptides corresponding to 4 common ns-SNPs showed limited binding to 11 HLA-DRB1 proteins. CD4 T cells from 22 subjects treated with FVIII products having sequences at residues FVIII-484, 1241, and 2238 differing from those of putative proteins encoded by their F8 genes did not show high-avidity tetramer binding, whereas positive-control staining of tetanus-specific CD4 T cells was routinely successful. African Americans with an intron-22 inversion mutation showed a 2-3 times-higher inhibitor incidence than whites with the same mutation (odds ratio = 2.3 [1.1-5.0, P = .04]), but this did not correlate with any of the ns-SNPs. We conclude that immune responses to “sequence-mismatched” FVIII products are unlikely to contribute appreciably to the inhibitor incidence in African Americans.
Introduction
Hemophilia A (HA) is an X-linked disorder caused by mutations in the F8 gene resulting in lack of circulating or defective factor VIII (FVIII). Approximately 25% of severe HA patients develop neutralizing antibodies (“inhibitors”) following FVIII infusions, which can cause severe bleeding that is difficult and expensive to manage.1 The development of inhibitory antibodies requires T-cell help. For T-cell stimulation to occur, 1 or more peptides must contain epitopes that bind effectively to major histocompatibility complex (MHC) class II (eg, HLA-DRB1) on antigen-presenting cells. If this class II-peptide complex is then recognized by a T-cell receptor, class II-peptide–T-cell receptor interactions lead to proliferation and cytokine secretion promoting anti-FVIII antibody production, class switching, and affinity maturation. Infusions of therapeutic FVIII expose patients’ immune systems to amino acid sequences absent from their endogenous hemophilic FVIII (if any), and recognition of these sequences by T-effector cells can lead to inhibitor development.
Mild and moderately severe HA is generally caused by minor FVIII sequence variations.2 The incidence of inhibitors in this population is low because they typically are not exposed to as much therapeutic FVIII as severely affected patients and the region(s) of infused FVIII that differ from their endogenous FVIII sequences are usually not recognized as epitopes. However, some of these patients do develop clinically significant inhibitors, especially if they receive intensive FVIII treatment in a setting of trauma or inflammation.3-5 Several studies have demonstrated that mild HA inhibitor subjects showed HLA-restricted T-cell responses to epitopes that included the wild-type FVIII sequence at the hemophilic missense site.6-8 Thus, even slight sequence variations in therapeutic FVIII could contribute to inhibitor risk.
African American HA patients have been reported to experience an increased inhibitor incidence.9-11 Three nonsynonymous single-nucleotide polymorphisms (ns-SNPs) in the F8 gene encode non-hemophilia-causing amino acid sequence variations (FVIII-H484, FVIII-E1241, and FVIII-V2238) found in a significant fraction of individuals with black African ancestry.12-14 Earlier studies13,14 identified F8 haplotypes H1 (encoding FVIII-R484, D1241, M2238) and H2 (encoding FVIII-R484, E1241, M2238) (Figure 1) in 93% and 7% of 86 white and 35% and 37% of 67 African American HA subjects studied, respectively, whereas F8 genes encoding FVIII-H484 (haplotype H4) and FVIII-V2238 (haplotypes H3 + H5) were found in 4% and 23% of the African American subjects, respectively, but not in whites. Additional ns-SNPs are being identified by large-scale DNA sequencing efforts, leading to a growing appreciation of the possibility that some patients’ MHC class II may present “sequence-mismatched” regions of infused FVIII and stimulate T-effector cells, leading to inhibitor development.15-18 Recombinant FVIII proteins with R484, either D1241 or E1241, and M2238 are currently used therapeutically. Many HA patients are thus exposed to FVIII products having amino acid sequences mismatched with the putative protein encoded by their hemophilic F8 gene.

FVIII sequence variants encoded by F8 ns-SNPs and haplotypes. (A) Schematic illustration of the FVIII protein, which consists of domains A1, A2, B, A3, C1, and C2. The F8 regions a1, a2, and ap are short stretches of acidic residues. The locations of 3 ns-SNPs encoding amino acid variations found in African and African American populations are indicated. (B) FVIII variants corresponding to 6 human haplotypes found in whites (primarily H1 and H2) and African Americans (primarily H1-H5). Haplotype H6 was found in an Asian subject of an earlier study.13 Shaded boxes indicate amino acid variants (single-letter code) that are not present in currently available recombinant FVIII products).
FVIII sequence variants encoded by F8 ns-SNPs and haplotypes. (A) Schematic illustration of the FVIII protein, which consists of domains A1, A2, B, A3, C1, and C2. The F8 regions a1, a2, and ap are short stretches of acidic residues. The locations of 3 ns-SNPs encoding amino acid variations found in African and African American populations are indicated. (B) FVIII variants corresponding to 6 human haplotypes found in whites (primarily H1 and H2) and African Americans (primarily H1-H5). Haplotype H6 was found in an Asian subject of an earlier study.13 Shaded boxes indicate amino acid variants (single-letter code) that are not present in currently available recombinant FVIII products).
A 2009 study of 76 African American HA subjects14 reported a higher prevalence of inhibitors associated with F8 haplotypes H3 + H4 compared with H1 + H2. The present study used 3 approaches to investigate relationships between F8 haplotypes, race, and inhibitor risk. First, the correlations between F8 haplotypes and inhibitor history were evaluated for 174 African American and 198 white HA subjects. Second, binding of FVIII peptides with sequences encoded by 4 ns-SNPs to 11 common MHC(HLA-DRB1) proteins was measured, as MHC binding is a prerequisite for T-cell stimulation. Third, CD4 T cells from 22 subjects who had been treated with “sequence-mismatched” FVIII products and 5 controls were analyzed using peptide-loaded MHC tetramers in an attempt to identify T-cell responses to sequences encoded by ns-SNPs in the F8 gene.
Methods
Subjects’ clinical status and polymorphism analysis
Four hundred seventeen African American and white HA subjects, 41 non-HA, 2 HA inhibitor subjects of other races, and 3 autoimmune subjects were enrolled in the Personalized Approaches to Therapies for Hemophilia (PATH) study, 365 with severe HA and 7 with an intron-22 inversion mutation and clotting activity ≥1% normal. These 372 subjects are referred to as the study cohort. All provided written informed consent in accordance with the principles of Helsinki. Questionnaires filled out at their clinics summarized their clinical histories. Blood samples were obtained and genomic DNA isolated using standard protocols. Peripheral blood mononuclear cells (PBMCs) were isolated from heparin-anticoagulated blood by Ficoll-Paque density gradient centrifugation and cryopreserved.
DNA sequences at ns-SNPs encoding FVIII-R484H, FVIII-D1241E, and FVIII-M2238V were determined by polymerase chain reaction (PCR), a DNA digestion assay, high-resolution melting reactions, and Sanger sequencing. To the extent possible, the FVIII-484, FVIII-1241, and FVIII-2238 sequence loci were cross-checked by comparing reverse transcription PCR (RT-PCR), melting curves, DNA digests, and sequencing data. In several cases, the sequence chromatograms were checked visually to make the final sequence call. HLA-DRB1 typing was performed by the UCLA Immunogenetics Laboratory (Los Angeles, CA) and additional F8 sequence data were provided by the Texas Biomedical Research Institute (San Antonio, TX). Pairwise correlations between inhibitor status and race, haplotypes, individual ns-SNPs, HLA-DRB1, and age were evaluated using the Fisher exact 2-way test, followed by logistic regression analysis adjusting for age, F8 haplotype, family relationships, and potential exposure to “sequence-mismatched” FVIII.
Peptide-MHC binding assays and predictions
Sixteen 20-mer FVIII peptides (Table 1), plus non-FVIII reference peptides with high-affinity binding to specific recombinant HLA-DRB1 molecules and/or known to elicit HLA-DRB1-restricted T-cell responses (supplemental Table 1, available on the Blood Web site), were used for competition binding assays to determine 50% inhibitory concentration (IC50) values. The peptides corresponded to both versions of sequences encoded by the 4 ns-SNPs identified previously,13 and they were designed to potentially allow binding to recombinant MHC(HLA-DRB1) proteins in multiple registers within the peptide-binding groove.19 The ability of 11 HLA-DRB1 recombinant proteins, whose corresponding alleles are found in ∼74% and ∼40% of European and African American populations, respectively,20 to present peptides containing these sequence variants was characterized by quantitative peptide-MHC binding assays measuring displacement of biotinylated reference peptides specific for each HLA-DRB1.8 Binding affinity predictions were calculated using ProPred21 (www.imtech.res.in/raghava/propred) and NetMHCIIPan222 (www.cbs.dtu.dk/services/NetMHCIIpan/) algorithms. ProPred predicts HLA-DRB1 binding regions using a matrix-based algorithm.21,23 Lower percentages indicate stronger binding. NetMHCIIpan2 utilizes artificial neural networks to predict affinities as IC50 values.
Sequences of peptides corresponding to regions encoded by ns-SNPs
| Peptide pool | Peptides | FVIII residues | Sequence* |
|---|---|---|---|
| F8 ns-SNP pool 1 | F8-M2238-1 | 2224-2243 | NNPKEWLQVDFQKTMKVTGV |
| F8-M2238V-1 | 2224-2243 | NNPKEWLQVDFQKTVKVTGV | |
| F8-M2238-2 | 2232-2251 | VDFQKTMKVTGVTTQGVKSL | |
| F8-M2238V-2 | 2232-2251 | VDFQKTVKVTGVTTQGVKSL | |
| F8 ns-SNP pool 2 | F8-R484-1 | 470-489 | SRPYNIYPHGITDVRPLYSR |
| F8-R484H-1 | 470-489 | SRPYNIYPHGITDVHPLYSR | |
| F8-R484-2 | 478-497 | HGITDVRPLYSRRLPKGVKH | |
| F8-R484H-2 | 478-497 | HGITDVHPLYSRRLPKGVKH | |
| F8 ns-SNP pool 3 | F8-R776-1 | 762-781 | PENDIEKTDPWFAHRTPMPK |
| F8-R776G-1 | 762-781 | PENDIEKTDPWFAHGTPMPK | |
| F8-R776-2 | 770-789 | DPWFAHRTPMPKIQNVSSSD | |
| F8-R776G-2 | 770-789 | DPWFAHGTPMPKIQNVSSSD | |
| F8 ns-SNP pool 4 | F8-D1241-1 | 1227-1246 | LFLLSTRQNVEGSYDGAYAP |
| F8-D1241E-1 | 1227-1246 | LFLLSTRQNVEGSYEGAYAP | |
| F8-D1241-2 | 1235-1254 | NVEGSYDGAYAPVLQDFRSL | |
| F8-D1241E-2 | 1235-1254 | NVEGSYEGAYAPVLQDFRSL |
| Peptide pool | Peptides | FVIII residues | Sequence* |
|---|---|---|---|
| F8 ns-SNP pool 1 | F8-M2238-1 | 2224-2243 | NNPKEWLQVDFQKTMKVTGV |
| F8-M2238V-1 | 2224-2243 | NNPKEWLQVDFQKTVKVTGV | |
| F8-M2238-2 | 2232-2251 | VDFQKTMKVTGVTTQGVKSL | |
| F8-M2238V-2 | 2232-2251 | VDFQKTVKVTGVTTQGVKSL | |
| F8 ns-SNP pool 2 | F8-R484-1 | 470-489 | SRPYNIYPHGITDVRPLYSR |
| F8-R484H-1 | 470-489 | SRPYNIYPHGITDVHPLYSR | |
| F8-R484-2 | 478-497 | HGITDVRPLYSRRLPKGVKH | |
| F8-R484H-2 | 478-497 | HGITDVHPLYSRRLPKGVKH | |
| F8 ns-SNP pool 3 | F8-R776-1 | 762-781 | PENDIEKTDPWFAHRTPMPK |
| F8-R776G-1 | 762-781 | PENDIEKTDPWFAHGTPMPK | |
| F8-R776-2 | 770-789 | DPWFAHRTPMPKIQNVSSSD | |
| F8-R776G-2 | 770-789 | DPWFAHGTPMPKIQNVSSSD | |
| F8 ns-SNP pool 4 | F8-D1241-1 | 1227-1246 | LFLLSTRQNVEGSYDGAYAP |
| F8-D1241E-1 | 1227-1246 | LFLLSTRQNVEGSYEGAYAP | |
| F8-D1241-2 | 1235-1254 | NVEGSYDGAYAPVLQDFRSL | |
| F8-D1241E-2 | 1235-1254 | NVEGSYEGAYAPVLQDFRSL |
The polymorphic sequences at FVIII residues 484, 776, 1241, and 2238 are bolded.
Tetramer-guided epitope mapping
MHC class II (HLA-DRB1) tetramer staining experiments24 were carried out using CD4 T cells from 27 HA subjects with the relevant F8 gene polymorphisms whose HLA-DRB1 alleles matched the available tetramer reagents: (1) 15 African American and 1 white HA subject (15 severe and 1 mild HA, 7 with a historic peak inhibitor >1 Bethesda unit [BU]/mL) whose F8 gene encoded V2238 and therefore had been treated with a therapeutic FVIII product mismatched at this site with respect to the endogenous sequence encoded by their hemophilic F8 gene; (2) 3 African American, 1 white, and 1 southeast (SE) Asian severe HA subject (3 with and 2 without a historic peak inhibitor >1 BU/mL) whose F8 gene encoded M2238 as controls (the SE Asian subject was enrolled as an inhibitor-positive control); (3) 1 African American severe HA subject with a historic peak inhibitor titer of 11 BU/mL whose F8 gene encoded H484; and (4) 5 severe HA subjects whose endogenous F8 gene encoded D1241, who had at least 1 HLA-DRB1 shown to bind FVIII-E1241 peptides with moderate to strong affinity, and whose clinical records indicated they had been treated with FVIII-1241E.
Peptides containing FVIII-M2238 and FVIII-V2238, or FVIII-R484 and FVIII-H484, or FVIII-D1241 and FVIII-E1241, were used to stimulate CD4 T cells and were loaded onto tetramers, first in pools of 4 peptides and then as single peptides. Positive control experiments carried out using aliquots of PBMCs used peptides corresponding to tetanus epitopes.25 Tetramer-positive cells (loaded with FVIII or tetanus peptides) were single-cell sorted and also sorted at 250 cells per well using a FACSAria II (BD Biosciences) into 96-well round-bottom plates containing 75 μL of 20% human serum T-cell medium, in an attempt to generate T-cell clones and polyclonal lines, respectively. These cells were expanded by stimulating with 2 μg/mL phytohemagglutinin (PHA) and 100 000 irradiated PBMCs from subjects with mismatched HLA-DRB1 alleles in 100 μL of 15% human serum T-cell medium. After 24 hours, 25 μL of natural human interleukin-2 (IL-2) (Hemagen) diluted 2.5-fold in 15% human serum T-cell media was added to each well. As cultures expanded, filling the bottom of round-bottom wells (∼2 weeks), they were transferred to flat-bottom wells and an equal volume of fresh 15% human serum T-cell medium containing a 1:10 dilution of natural human IL-2 was added every 48 hours. After 3 weeks, the cells were restimulated with PHA and irradiated HLA-mismatched PBMCs. Cells were given an equal volume of fresh 15% human serum T-cell medium containing a 1:10 dilution of natural human IL-2 24 to 48 hours after restimulation and every 2 to 3 days thereafter for 14 days. The expanded cells were then stained with the same peptide-loaded tetramers used for sorting to determine whether they corresponded to T-cell clones or polyclonal lines that specifically bound the tetramer.
Results
Subjects’ clinical status and polymorphism analysis
Clinical status and correlations between F8 polymorphisms/haplotypes, HA-causing mutations, and inhibitor histories are summarized in Tables 2-6 and supplemental Tables 2-3. HA-causing mutations were determined for 274 of 372 and F8 haplotypes for 362 of 372 subjects. Polymorphisms encoding variants FVIII-D1241E and FVIII-M2238V showed no significant correlation with inhibitor development, nor did combinations of ns-SNPs comprising F8 haplotypes 1-5. Similar results were obtained when 323 severe unrelated HA probands were analyzed (supplemental Table 2), and when only high-responder inhibitors (peak titer ≥5 BU/mL) were classified as “inhibitor-positive” (supplemental Table 3). The most noteworthy difference among the various subgroups was that African American subjects with an intron-22 inversion mutation were 2-3 times as likely to experience an inhibitor as white subjects with the same mutation (adjusted odds ratio [OR] = 2.3 [1.1-5.0, 95% confidence interval (CI)], P = .04, Table 6). This remained significant when only high-responder inhibitors (peak titer ≥5 BU/mL) were classified as “inhibitor-positive” (supplemental Table 3). Subgroups with non-intron-22-inversion mutations were too small to calculate meaningful correlations between other risk factors and inhibitor incidence. Clinical records indicated that up to 91%, 71%, and 100% of subjects with haplotypes H1, H2, and (H3-H5), respectively, had been infused or transfused with “sequence-mismatched” FVIII. This subgroup, which included all subjects who had ever been treated with plasma-derived FVIII or other blood-derived products, including blood transfusions or FVIII inhibitor bypass agent, had a significantly higher inhibitor risk (Table 6).
Clinical characteristics of total study cohort and of the proband subset
| Entire cohort | Probands* | |||||
|---|---|---|---|---|---|---|
| Total | INH(−)† | INH(+) | Total | INH(−) | INH(+) | |
| Total subjects | 372 | 259 | 113 | 330 | 230 | 100 |
| African American | 174 | 113 | 61 | 145 | 94 | 51 |
| White | 198 | 146 | 52 | 185 | 136 | 49 |
| Severe HA | 365 (98%) | 323 (98%) | ||||
| Haplotype | ||||||
| H1 | 213 | 153 | 60 | 191 | 137 | 54 |
| H2 | 117 | 78 | 39 | 104 | 71 | 33 |
| H3 | 28 | 20 | 8 | 22 | 15 | 7 |
| H4 | 1 | 0 | 1 | 1 | 0 | 1 |
| H5 | 3 | 1 | 2 | 2 | 0 | 2 |
| Intron-22 inversion | 151 | 100 | 51 | 138 | 93 | 45 |
| Intron-1 inversion | 12 | 7 | 5 | 10 | 6 | 4 |
| Missense mutation | 53 | 43 | 10 | 47 | 39 | 8 |
| Mutation unknown | 106 | 73 | 33 | 89 | 58 | 31 |
| Age at enrollment, y | ||||||
| 2-3, African American | 8 | 6 | 2 | 7 | 5 | 2 |
| 2-3, White | 7 | 3 | 4 | 6 | 2 | 4 |
| 4-17, African American | 67 | 48 | 19 | 57 | 40 | 17 |
| 4-17, White | 72 | 49 | 23 | 65 | 43 | 22 |
| 18+, African American | 99 | 59 | 40 | 81 | 49 | 32 |
| 18+, White | 119 | 94 | 25 | 114 | 91 | 23 |
| High-responder inhibitors, ≥5 BU/mL | ||||||
| African American | 41/174 (24%) | 35/145 (24%) | ||||
| White | 33/198 (17%) | 31/185 (17%) | ||||
| Entire cohort | Probands* | |||||
|---|---|---|---|---|---|---|
| Total | INH(−)† | INH(+) | Total | INH(−) | INH(+) | |
| Total subjects | 372 | 259 | 113 | 330 | 230 | 100 |
| African American | 174 | 113 | 61 | 145 | 94 | 51 |
| White | 198 | 146 | 52 | 185 | 136 | 49 |
| Severe HA | 365 (98%) | 323 (98%) | ||||
| Haplotype | ||||||
| H1 | 213 | 153 | 60 | 191 | 137 | 54 |
| H2 | 117 | 78 | 39 | 104 | 71 | 33 |
| H3 | 28 | 20 | 8 | 22 | 15 | 7 |
| H4 | 1 | 0 | 1 | 1 | 0 | 1 |
| H5 | 3 | 1 | 2 | 2 | 0 | 2 |
| Intron-22 inversion | 151 | 100 | 51 | 138 | 93 | 45 |
| Intron-1 inversion | 12 | 7 | 5 | 10 | 6 | 4 |
| Missense mutation | 53 | 43 | 10 | 47 | 39 | 8 |
| Mutation unknown | 106 | 73 | 33 | 89 | 58 | 31 |
| Age at enrollment, y | ||||||
| 2-3, African American | 8 | 6 | 2 | 7 | 5 | 2 |
| 2-3, White | 7 | 3 | 4 | 6 | 2 | 4 |
| 4-17, African American | 67 | 48 | 19 | 57 | 40 | 17 |
| 4-17, White | 72 | 49 | 23 | 65 | 43 | 22 |
| 18+, African American | 99 | 59 | 40 | 81 | 49 | 32 |
| 18+, White | 119 | 94 | 25 | 114 | 91 | 23 |
| High-responder inhibitors, ≥5 BU/mL | ||||||
| African American | 41/174 (24%) | 35/145 (24%) | ||||
| White | 33/198 (17%) | 31/185 (17%) | ||||
Probands were defined as the first member of each family group enrolled.
INH, inhibitor. The INH(+) subjects included 13 (entire cohort) and 11 (severe HA probands) with chart notes indicating they had a previous inhibitor, but with no numeric peak titer recorded.
F8 haplotype subgroup characteristics
| F8 haplotype | Race and inhibitor history (%) | |||||
|---|---|---|---|---|---|---|
| African American | White | No inhibitor | Inhibitor | Peak titer ≥5BU/mL | Total | |
| H1 | 64 (30) | 149 (70) | 153 (72) | 60 (28) | 36 (17) | 213 |
| H2 | 78 (67) | 39 (33) | 78 (67) | 39 (33) | 26 (22) | 117 |
| H3 | 27 (96) | 1 (4) | 20 (71) | 8 (29) | 6 (21) | 28 |
| H4 | 1 | 0 | 0 | 1 | 1 | 1 |
| H5 | 3 | 0 | 1 | 2 | 2 | 3 |
| F8 haplotype | Race and inhibitor history (%) | |||||
|---|---|---|---|---|---|---|
| African American | White | No inhibitor | Inhibitor | Peak titer ≥5BU/mL | Total | |
| H1 | 64 (30) | 149 (70) | 153 (72) | 60 (28) | 36 (17) | 213 |
| H2 | 78 (67) | 39 (33) | 78 (67) | 39 (33) | 26 (22) | 117 |
| H3 | 27 (96) | 1 (4) | 20 (71) | 8 (29) | 6 (21) | 28 |
| H4 | 1 | 0 | 0 | 1 | 1 | 1 |
| H5 | 3 | 0 | 1 | 2 | 2 | 3 |
HA-causing mutations: F8 haplotype distributions
| F8 mutation | H1, n = 213 | H2, n = 117 | H3, n = 28 | H4, n = 1 | H5, n = 3 |
|---|---|---|---|---|---|
| Intron-22 inversion (+) | 90 | 50 | 7 | 0 | 3 |
| Intron-22 inversion (−) | 96 | 56 | 17 | 1 | 0 |
| Intron-22 inversion status unknown | 27 | 11 | 4 | 0 | 1 |
| Intron-1 inversion (+) | 7 | 2 | 3 | 0 | 0 |
| Intron-1 inversion (−) | 166 | 89 | 12 | 1 | 2 |
| Intron-1 inversion status not tested | 40 | 26 | 13 | 0 | 1 |
| Missense mutation | 22 | 26 | 4 | 0 | 0 |
| Stop, insertion, deletion, frameshift | 32 | 17 | 2 | 0 | 0 |
| Mutation unknown | 63 | 22 | 12 | 1 | 1 |
| F8 mutation | H1, n = 213 | H2, n = 117 | H3, n = 28 | H4, n = 1 | H5, n = 3 |
|---|---|---|---|---|---|
| Intron-22 inversion (+) | 90 | 50 | 7 | 0 | 3 |
| Intron-22 inversion (−) | 96 | 56 | 17 | 1 | 0 |
| Intron-22 inversion status unknown | 27 | 11 | 4 | 0 | 1 |
| Intron-1 inversion (+) | 7 | 2 | 3 | 0 | 0 |
| Intron-1 inversion (−) | 166 | 89 | 12 | 1 | 2 |
| Intron-1 inversion status not tested | 40 | 26 | 13 | 0 | 1 |
| Missense mutation | 22 | 26 | 4 | 0 | 0 |
| Stop, insertion, deletion, frameshift | 32 | 17 | 2 | 0 | 0 |
| Mutation unknown | 63 | 22 | 12 | 1 | 1 |
Pairwise analyses of inhibitor risk
| Race* | INH(−) | INH(+)† | Odds | OR‡ | 95% CI | P | |
|---|---|---|---|---|---|---|---|
| Viel et al,14 76 AA subjects, HA all severities | |||||||
| H3 + H4 | AA | 10 | 9 | 0.9 | 3.4 | 1.1-10.2 | .03 |
| H1 + H2 | AA | 45 | 12 | 0.3 | |||
| 173 haplotyped AA subjects | |||||||
| H3 + H4 + H5 | AA | 20 | 11 | 0.6 | 1.0 | 0.5-2.4 | 1.0 |
| H1 + H2 | AA | 93 | 49 | 0.5 | |||
| 362 haplotyped AA + W subjects§ | |||||||
| H3 + H4 + H5 | AA + W | 21 | 11 | 0.5 | 1.2 | 0.6-2.6 | .7 |
| H1 + H2 | AA + W | 231 | 99 | 0.4 | |||
| All AA subjects | 113 | 60 | 0.5 | 1.5 | 0.9-2.3 | .1 | |
| All W subjects | 139 | 50 | 0.4 | ||||
| 1241 E | AA + W | 98 | 48 | 0.5 | 1.2 | 0.8-1.9 | .4 |
| 1241 D | AA + W | 154 | 62 | 0.4 | |||
| 2238 V | AA + W | 21 | 10 | 0.5 | 1.1 | 0.5-2.4 | .8 |
| 2238 M | AA + W | 231 | 100 | 0.4 | |||
| Intron-22 inversion (+) | AA + W | 99 | 50 | 0.5 | 1.4 | 0.9-2.3 | .2 |
| Intron-22 inversion (−) | AA + W | 125 | 45 | 0.4 | |||
| Intron-22 inversion (+) | AA | 31 | 26 | 0.8 | 2.4 | 1.2-4.8 | .02 |
| Intron-22 inversion (+) | W | 68 | 24 | 0.4 | |||
| Intron-22 inversion (−) | AA | 59 | 26 | 0.4 | 1.5 | 0.8-3.0 | .3 |
| Intron-22 inversion (−) | W | 66 | 19 | 0.3 | |||
| Intron-22 inversion (+) | AA | 31 | 26 | 0.8 | 1.9 | 0.9-3.8 | .08 |
| Intron-22 inversion (−) | AA | 59 | 26 | 0.4 | |||
| Intron-22 inversion (+) | W | 68 | 24 | 0.4 | 1.2 | 0.6-2.4 | .6 |
| Intron-22 inversion (−) | W | 66 | 19 | 0.3 | |||
| Intron-22 inversion (+), 1241 E | AA | 20 | 18 | 0.9 | 1.2 | 0.4-3.8 | .8 |
| Intron-22 inversion (+), 1241 D | AA | 11 | 8 | 0.7 | |||
| Intron-22 inversion (+), 2238 V | AA | 4 | 5 | 1.3 | 1.6 | 0.4-6.7 | .7 |
| Intron-22 inversion (+), 2238 M | AA | 27 | 21 | 0.8 | |||
| Intron-22 inversion (+), age 18+ y | AA | 16 | 19 | 1.2 | 2.5 | 0.8-7.7 | .1 |
| Intron-22 inversion (+), age <18 y | AA | 15 | 7 | 0.5 | |||
| Age 18+ y | AA | 59 | 39 | 0.7 | 1.7 | 0.9-3.2 | .1 |
| Age <18 y | AA | 54 | 21 | 0.4 | |||
| HLA-DR15 (H2) | AA + W | 10 | 13 | 1.3 | 1.9 | 0.7-5.0 | .3 |
| HLA-DR15 (H1) | AA + W | 30 | 21 | 0.7 | |||
| HLA-DR15 | AA | 22 | 18 | 0.8 | 1.0 | 0.4-2.3 | 1.0 |
| HLA-DR15 | W | 21 | 18 | 0.9 |
| Race* | INH(−) | INH(+)† | Odds | OR‡ | 95% CI | P | |
|---|---|---|---|---|---|---|---|
| Viel et al,14 76 AA subjects, HA all severities | |||||||
| H3 + H4 | AA | 10 | 9 | 0.9 | 3.4 | 1.1-10.2 | .03 |
| H1 + H2 | AA | 45 | 12 | 0.3 | |||
| 173 haplotyped AA subjects | |||||||
| H3 + H4 + H5 | AA | 20 | 11 | 0.6 | 1.0 | 0.5-2.4 | 1.0 |
| H1 + H2 | AA | 93 | 49 | 0.5 | |||
| 362 haplotyped AA + W subjects§ | |||||||
| H3 + H4 + H5 | AA + W | 21 | 11 | 0.5 | 1.2 | 0.6-2.6 | .7 |
| H1 + H2 | AA + W | 231 | 99 | 0.4 | |||
| All AA subjects | 113 | 60 | 0.5 | 1.5 | 0.9-2.3 | .1 | |
| All W subjects | 139 | 50 | 0.4 | ||||
| 1241 E | AA + W | 98 | 48 | 0.5 | 1.2 | 0.8-1.9 | .4 |
| 1241 D | AA + W | 154 | 62 | 0.4 | |||
| 2238 V | AA + W | 21 | 10 | 0.5 | 1.1 | 0.5-2.4 | .8 |
| 2238 M | AA + W | 231 | 100 | 0.4 | |||
| Intron-22 inversion (+) | AA + W | 99 | 50 | 0.5 | 1.4 | 0.9-2.3 | .2 |
| Intron-22 inversion (−) | AA + W | 125 | 45 | 0.4 | |||
| Intron-22 inversion (+) | AA | 31 | 26 | 0.8 | 2.4 | 1.2-4.8 | .02 |
| Intron-22 inversion (+) | W | 68 | 24 | 0.4 | |||
| Intron-22 inversion (−) | AA | 59 | 26 | 0.4 | 1.5 | 0.8-3.0 | .3 |
| Intron-22 inversion (−) | W | 66 | 19 | 0.3 | |||
| Intron-22 inversion (+) | AA | 31 | 26 | 0.8 | 1.9 | 0.9-3.8 | .08 |
| Intron-22 inversion (−) | AA | 59 | 26 | 0.4 | |||
| Intron-22 inversion (+) | W | 68 | 24 | 0.4 | 1.2 | 0.6-2.4 | .6 |
| Intron-22 inversion (−) | W | 66 | 19 | 0.3 | |||
| Intron-22 inversion (+), 1241 E | AA | 20 | 18 | 0.9 | 1.2 | 0.4-3.8 | .8 |
| Intron-22 inversion (+), 1241 D | AA | 11 | 8 | 0.7 | |||
| Intron-22 inversion (+), 2238 V | AA | 4 | 5 | 1.3 | 1.6 | 0.4-6.7 | .7 |
| Intron-22 inversion (+), 2238 M | AA | 27 | 21 | 0.8 | |||
| Intron-22 inversion (+), age 18+ y | AA | 16 | 19 | 1.2 | 2.5 | 0.8-7.7 | .1 |
| Intron-22 inversion (+), age <18 y | AA | 15 | 7 | 0.5 | |||
| Age 18+ y | AA | 59 | 39 | 0.7 | 1.7 | 0.9-3.2 | .1 |
| Age <18 y | AA | 54 | 21 | 0.4 | |||
| HLA-DR15 (H2) | AA + W | 10 | 13 | 1.3 | 1.9 | 0.7-5.0 | .3 |
| HLA-DR15 (H1) | AA + W | 30 | 21 | 0.7 | |||
| HLA-DR15 | AA | 22 | 18 | 0.8 | 1.0 | 0.4-2.3 | 1.0 |
| HLA-DR15 | W | 21 | 18 | 0.9 |
The study cohort consisted of all severe HA subjects plus 7 intron-22 inversion subjects with reported mild or moderate severity HA. These 7 subjects were included because, given the fact that intact FVIII protein cannot be translated from mRNA of inversion mutation patients, their clotting activity was presumably due to other coagulation factors besides FVIII. Therefore, it would not be expected to influence anti-FVIII immune responses. All of the pairwise comparisons of individual F8 haplotypes, and all possible subgroupings of these haplotypes, were evaluated for African American (AA), white (W), and AA + W racial groups (not shown). None of these comparisons showed a statistically significant association of F8 haplotype with inhibitor risk.
Relative risks of developing an inhibitor were evaluated for the indicated subgroups of AA and W HA subjects.
Positive inhibitor status (INH(+)) indicates the clinical record for this subject showed he had at least 1 measured inhibitor titer of ≥0.6 BU/mL.
ORs and probabilities were determined using the calculator at http://vassarstats.net/odds2x2.html to determine Fisher exact 2-way probabilities. P values <.05 were considered significant.
Haplotypes could not be assigned to 10 of the 372 subjects due to lack of DNA or poorly resolved sequence data.
Logistic regression analyses of inhibitor risk
| Race* | INH(−) | INH(+)† | Adjusted OR‡ | 95% CI | P | |
|---|---|---|---|---|---|---|
| Model 1: 347 AA + W subjects,§age 4+ y | ||||||
| H3 + H4 + H5 | AA + W | 19 | 11 | 1.2 | 0.5-2.8 | .7 |
| H1 + H2 | AA + W | 224 | 93 | |||
| Potentially exposed to “sequence-mismatched” FVIII¶ | AA + W | 198 | 92 | 2.3 | 1.1-4.8 | .03 |
| Not exposed to “sequence-mismatched” FVIII | AA + W | 45 | 12 | |||
| Intron-22 inversion (+) | AA | 30 | 25 | 2.3 | 1.1-5.0 | .04 |
| Intron-22 inversion (+) | W | 67 | 24 | |||
| Intron-22 inversion (−) | AA | 57 | 25 | 1.4 | 0.7-3.0 | .3 |
| Intron-22 inversion (−) | W | 65 | 18 | |||
| Intron-22 inversion (unknown) | AA | 20 | 8 | 0.3 | 0.07-1.7 | .2 |
| Intron-22 inversion (unknown) | W | 4 | 4 | |||
| Model 2: 165 AA subjects, age 4+ y | ||||||
| H3 + H4 + H5 | AA | 18 | 11 | 1.3 | 0.5-3.1 | .6 |
| H1 + H2 | AA | 89 | 47 | |||
| Potentially exposed to “sequence-mismatched” FVIII | AA | 81 | 51 | 2.9 | 1.1-7.5 | .026 |
| Not exposed to “sequence-mismatched” FVIII | AA | 26 | 7 | |||
| Intron-22 inversion (+) | AA | 30 | 25 | 2.5 | 1.0-7.0 | .7 |
| Intron-22 inversion (−) | AA | 57 | 25 | 1.2 | 0.4-3.0 | .7 |
| Intron-22 inversion (unknown) | AA | 20 | 8 |
| Race* | INH(−) | INH(+)† | Adjusted OR‡ | 95% CI | P | |
|---|---|---|---|---|---|---|
| Model 1: 347 AA + W subjects,§age 4+ y | ||||||
| H3 + H4 + H5 | AA + W | 19 | 11 | 1.2 | 0.5-2.8 | .7 |
| H1 + H2 | AA + W | 224 | 93 | |||
| Potentially exposed to “sequence-mismatched” FVIII¶ | AA + W | 198 | 92 | 2.3 | 1.1-4.8 | .03 |
| Not exposed to “sequence-mismatched” FVIII | AA + W | 45 | 12 | |||
| Intron-22 inversion (+) | AA | 30 | 25 | 2.3 | 1.1-5.0 | .04 |
| Intron-22 inversion (+) | W | 67 | 24 | |||
| Intron-22 inversion (−) | AA | 57 | 25 | 1.4 | 0.7-3.0 | .3 |
| Intron-22 inversion (−) | W | 65 | 18 | |||
| Intron-22 inversion (unknown) | AA | 20 | 8 | 0.3 | 0.07-1.7 | .2 |
| Intron-22 inversion (unknown) | W | 4 | 4 | |||
| Model 2: 165 AA subjects, age 4+ y | ||||||
| H3 + H4 + H5 | AA | 18 | 11 | 1.3 | 0.5-3.1 | .6 |
| H1 + H2 | AA | 89 | 47 | |||
| Potentially exposed to “sequence-mismatched” FVIII | AA | 81 | 51 | 2.9 | 1.1-7.5 | .026 |
| Not exposed to “sequence-mismatched” FVIII | AA | 26 | 7 | |||
| Intron-22 inversion (+) | AA | 30 | 25 | 2.5 | 1.0-7.0 | .7 |
| Intron-22 inversion (−) | AA | 57 | 25 | 1.2 | 0.4-3.0 | .7 |
| Intron-22 inversion (unknown) | AA | 20 | 8 |
The study cohort consisted of all F8-haplotyped severe HA subjects plus 7 intron-22 inversion subjects with reported mild or moderate severity HA.
Relative risks of developing an inhibitor were evaluated for the indicated subgroups of African American (AA) and white (W) HA subjects.
Positive inhibitor status (INH(+)) indicates the clinical record showed at least 1 measured inhibitor titer of ≥0.6 BU/mL.
The multivariate logistic analyses adjusted for age (under or over age 18), F8 haplotype, family relationships, and potential exposure to “sequence-mismatched” FVIII. P values <.05 were considered significant.
Subjects age 4 y and over were considered to be multiply infused with FVIII.
Subjects potentially exposed to “sequence-mismatched” FVIII included all who had been exposed to blood products, for example, blood transfusions or Factor Eight Inhibitor Bypass Agent to control bleeding. The risk factors calculated for this subgroup should be interpreted with caution due to possible confounding from clinical conditions requiring treatment with blood-derived products. Comparisons of individual (eg, H2 vs H1) and alternatively grouped (eg, H1 vs H3 + H4 + H5) F8 haplotypes (not shown) showed no statistically significant differences in inhibitor risk.
Peptide-MHC binding assays and predictions
The affinities of 16 20-mer FVIII peptides, corresponding to both sequences encoded by 4 common ns-SNPs, and with flanking sequences of different lengths to allow potential binding to HLA-DRB1 in ≥2 binding registers, are shown in Table 7 and summarized briefly below. Representative competition binding curves are shown in Figure 2.
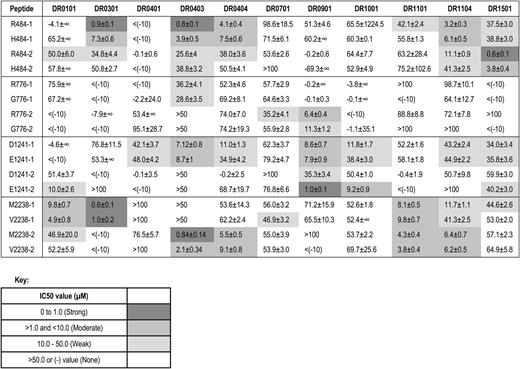
Binding affinities (IC50 values in µM) of FVIII peptides encoded by ns-SNPs to HLA-DRB1 proteins

Binding of FVIII and positive control peptides to recombinant HLA-DR0301. Representative competition binding curves for FVIII peptides binding to recombinant HLA-DR proteins. Serially diluted F8-SNP pool 2238 peptides and nonbiotinylated Myo-137-148 reference peptide (which binds HLA-DR0301 with IC50 = 3µM, see supplemental Table 3) as a positive control were incubated with biotinylated Myo-137-148 in a 96-well enzyme-linked immunosorbent assay (ELISA) plate coated with the recombinant extracellular domain of HLA-DR0301. Europium-labeled streptavidin was added as described above and plates were read on a Victor 2D time-resolved fluorometer. Fluorescence counts were analyzed, sigmoidal binding curves simulated, and IC50 values calculated for each peptide using Sigmaplot software. M2238-1 and V2238-1 peptides competed with the reference peptide whereas M2238-2 and V2238-2 showed no binding, indicating the former 2 peptides contain the minimal sequence required to fit into the peptide-binding groove for HLA-DR0301.
Binding of FVIII and positive control peptides to recombinant HLA-DR0301. Representative competition binding curves for FVIII peptides binding to recombinant HLA-DR proteins. Serially diluted F8-SNP pool 2238 peptides and nonbiotinylated Myo-137-148 reference peptide (which binds HLA-DR0301 with IC50 = 3µM, see supplemental Table 3) as a positive control were incubated with biotinylated Myo-137-148 in a 96-well enzyme-linked immunosorbent assay (ELISA) plate coated with the recombinant extracellular domain of HLA-DR0301. Europium-labeled streptavidin was added as described above and plates were read on a Victor 2D time-resolved fluorometer. Fluorescence counts were analyzed, sigmoidal binding curves simulated, and IC50 values calculated for each peptide using Sigmaplot software. M2238-1 and V2238-1 peptides competed with the reference peptide whereas M2238-2 and V2238-2 showed no binding, indicating the former 2 peptides contain the minimal sequence required to fit into the peptide-binding groove for HLA-DR0301.
R484H.
R484 peptides had strong affinity for DR0301, DR0403, and DR1501; H484 peptides had moderate affinity for DR0301, DR0403, and DR1501; both R484 and H484 peptides had moderate affinity for DR1104 and DR0404.
R776G.
R776 peptides had weak-to-moderate affinity for DR0901.
D1241E.
D1241 and E1241 peptides had moderate affinity for DR0403. E1241 peptides had moderate-to-strong affinity for DR0901 and moderate affinity for DR1001 and DR0101. D1241 peptides had weak-to-moderate affinity for DR0901.
M2238V.
Both M2238 and V2238 peptides had strong affinity for DR0301, strong-to-moderate affinity for DR0403 and moderate affinity for DR0101, DR1104, DR1101, and DR0404.
To summarize, effective peptide-MHC binding is required for T-cell engagement by antigen-presenting cells. Therefore, peptides with weak or no binding affinity to particular MHC proteins would be highly unlikely to provoke CD4 T-cell responses restricted to the corresponding MHC alleles. Several FVIII peptides differing by just 1 ns-SNP–encoded amino acid bound to particular HLA-DRB1 proteins with different affinities. The most striking difference between these pairs was for peptide E1241-2, which bound DR0901 with higher affinity than peptide D1241-2, suggesting that, if a similar peptide was naturally processed and presented on antigen-presenting cells of a patient who expressed a hemophilic FVIII protein with FVIII-D1241, this might break preexisting tolerance to FVIII-D1241.
The computer algorithms ProPred and NetMHCIIpan2 correctly identified many of the FVIII peptides that bound to a given HLA-DRB1 protein, for example, the FVIII-M2238 variant to DR0301 and DR1104 and the E1241 variant to DR0101, but they also incorrectly predicted many binding interactions that were not confirmed experimentally (supplemental Tables 4-5), as seen in epitope-mapping studies of other antigens.26 Specifically, of 128 HLA-DRB1 binding interactions that were both measured experimentally and evaluated using the ProPred program, 35 of 39 predicted nonbinders did not bind to the corresponding HLA-DRB1 proteins and 49 of 89 FVIII peptides predicted to bind with weak-to-strong affinity bound to the HLA-DRB1 proteins with IC50 values ≤50 μM. Of 176 HLA-DRB1 binding interactions that were both measured experimentally and evaluated using the NetMHCIIpan2 program, 60 of 85 predicted nonbinders did not bind to the corresponding HLA-DRB1 proteins and 39 of 91 FVIII peptides predicted to bind with weak-to-strong affinity bound to the HLA-DRB1 proteins with IC50 values ≤50 μM. Clearly, the predictions of nonbinding peptides were confirmed more often than the predictions of effective peptide-MHC binding.
Tetramer-guided epitope mapping
Twenty-two subjects whose T-cell responses were evaluated using tetramers had clinical records indicating that they had been infused with “sequence-mismatched” FVIII products, and 5 additional control subjects whose F8 gene encoded M2238 were also tested (Table 8). None of the experiments using tetramers loaded with FVIII peptides produced unambiguous, tetramer-high staining. In contrast, 24 of 32 experiments carried out using tetramers loaded with tetanus peptides produced promising tetramer staining of the CD4high cells, with over half of these showing strong, unambiguous staining of a tetramer-high population (Figure 3, supplemental Figure 1). Some of the cultures stimulated with F8 SNP-Pool 2238 peptides showed low-level, possibly nonspecific, staining by tetramers loaded with these peptides. One of these was from a SE Asian control subject who had been partially tolerized and whose F8 gene encoded FVIII-M2238 (Figure 3A). His immune response was queried further, as he had a current inhibitor and so was expected to have FVIII-specific T cells. His cells were incubated for 2 more days following the initial tetramer-staining experiment and then cultured with tetramers loaded with the individual FVIII peptides. Despite weak tetramer positivity (Figure 3B), the small number of tetramer-positive CD4 T cells were single-cell sorted and expanded in culture in an unsuccessful attempt to generate FVIII-specific T-cell clones and lines (Figure 3C). Attempts were also made to isolate FVIII-specific T-cell clones and polyclonal lines recognizing polymorphic sequences at FVIII-2238 from subjects GIS-002-033 and GIS-007-029 (both tolerized to FVIII) and GIS-007-002 (no inhibitor history) (supplemental Table 6) by sorting the few tetramer-positive cells and culturing them under expansion conditions. No FVIII-specific clones were obtained, indicating the background-level tetramer staining was nonspecific or that the cells had low avidity for tetramers or were anergic (eg, in successfully tolerized subjects). Similar negative results were obtained using cells from 5 subjects infused with FVIII products mismatched at residue FVIII-1241 (Table 8, supplemental Table 6, and supplemental Figure 1). In contrast, 22 of 32 positive control experiments carried out in parallel using CD4 T cells from 25 of the 27 HA subjects produced strong staining by tetramers loaded with tetanus-derived peptides, and borderline tetanus-positive staining was seen for cells from 2 additional subjects25 (Figure 3D, supplemental Figure S1, and supplemental Table 6).
Clinical and genetic characteristics of 27 HA subjects whose T-cell responses were analyzed
| HA subject | Age (y) | Race* | HA severity | Recent titer, peak titer† | ITI? | FVIII mutation, HGVS‡ | FVIII mutation, legacy§ | DRB1 alleles | FVIII variant analyzed |
|---|---|---|---|---|---|---|---|---|---|
| 001-043 | 18+ | AA | Mild | Never | No | ND | ND | 0301, 0701|| | V2238 |
| 002-033 | 4-17 | AA | Severe | 0 (1 y ago), 17 (11 y ago) | Yes | ND | ND | 1503, 1503 | V2238 |
| 009-035 | 18+ | AA | Severe | Never | No | Int22 | Int22 | 0804, 1101g¶ | V2238 |
| 016-005 | 18+ | AA | Severe | 0 (2 y ago), 14 (16 y ago) | No | ND | ND | 0101, 1301 | V2238 |
| 004-022 | 4-17 | AA | Severe | Never | No | Int22 | Int22 | 1201g, 1302 | V2238 |
| 009-034 | 18+ | AA | Severe | 0 (2 y ago), 9 (17 y ago) | Yes | Int22 | Int22 | 0804, 1302 | V2238 |
| 011-052 | 2-3 | AA | Severe | Never | No | ND | ND | 0901, 1102 | V2238 |
| 015-006 | 4-17 | AA | Severe | 0 (2 y ago), 1.3 (6 y ago) | No | Int22 | Int22 | 1202, 1302 | V2238 |
| 002-017 | 4-17 | AA | Severe | Never | No | c.791T>C, p.Leu264Pro | CTG→CCG, L245P | 1101, 1201g | V2238 |
| 004-020 | 4-17 | AA | Severe | 0 (2 y ago), 32 (6 y ago) | Yes | Int1 | Int1 | 0301, 0804 | V2238 |
| 016-027 | 4-17 | AA | Severe | 0 (2 y ago), 6 (13 y ago) | No | ND | ND | 0301, 1401 | V2238 |
| 004-005 | 18+ | W | Severe | Never | No | ND | ND | 0701, 0804 | V2238 |
| 004-013 | 4-17 | AA | Severe | Never | No | Int1 | Int1 | 1201g, 1501 | V2238 |
| 007-002 | 18+ | AA | Severe | Never | No | ND | ND | 0101, 0804 | V2238 |
| 011-001 | 4-17 | AA | Severe | 15 (2 wk ago), 309 (no date) | Yes | Int22 | Int22 | 1101, 0701 | V2238 |
| 002-025 | 2-3 | AA | Severe | 0 (same day), <0.5 (1 y ago) | Yes | c.3637del, Ile1213Phefs*5 | ΔA, E1191-I1194 | 0301, 0804 | V2238 |
| 005-019 | 18+ | SEA | Severe | 1.0 (4 mo ago), 132 (no date) | Yes | Int22 | Int22 | 1101, 0301 | M2238 |
| 013-011 | 18+ | W | Severe | Never | No | c.3637dup, p.Ile1213Asnfs*28 | +A, E1191-I1194 | 1101, 0103 | M2238 |
| 013-013 | 4-17 | AA | Severe | Never | No | c.1171C>T, p.Arg3191Cys | CGC→TGC, R372C | 0301, 1401g | M2238 |
| 014-016 | 4-17 | AA | Severe | 0.3 (5 mo ago), 34 (no date) | Yes | c.5961dup, p.Glu1988Argfs*4 | GAG→AGAG, E1969 | 0301, 0301 | M2238 |
| 008-006 | 2-3 | AA | Severe | 13 (same day), 87 (5 wk later) | No | c.6967C>T, p.Arg2323Cys | CGC→TGC, R2304C | 0301, 1301 | M2238 |
| 011-016 | 18+ | AA | Severe | 0 (2 y ago), 11 (20 y ago) | Yes | ND | ND | 0701, 1302 | H484 |
| 013-024 | 4-17 | AA | Severe | 0 (1 y ago), 1126 (∼10 y ago) | Yes | Int22 | Int22 | 0701, 1001 | D1241 |
| 009-003 | 2-3 | W | Severe | 0 | No | ND | ND | 0404, 0901 | D1241 |
| 011-020 | 18+ | W | Severe | 0 | No | ND | ND | 0701, 0901 | D1241 |
| 005-015 | 18+ | W | Mild | 2-7 (4 y ago), 250 (7 y ago) | No | A2201P (from chart) | 0101, 1503 | D1241 | |
| 007-029 | 18+ | W | Severe | 0 (2 y ago), 280 (21 y ago) | Yes | ND | ND | 0101, 0701 | D1241 |
| HA subject | Age (y) | Race* | HA severity | Recent titer, peak titer† | ITI? | FVIII mutation, HGVS‡ | FVIII mutation, legacy§ | DRB1 alleles | FVIII variant analyzed |
|---|---|---|---|---|---|---|---|---|---|
| 001-043 | 18+ | AA | Mild | Never | No | ND | ND | 0301, 0701|| | V2238 |
| 002-033 | 4-17 | AA | Severe | 0 (1 y ago), 17 (11 y ago) | Yes | ND | ND | 1503, 1503 | V2238 |
| 009-035 | 18+ | AA | Severe | Never | No | Int22 | Int22 | 0804, 1101g¶ | V2238 |
| 016-005 | 18+ | AA | Severe | 0 (2 y ago), 14 (16 y ago) | No | ND | ND | 0101, 1301 | V2238 |
| 004-022 | 4-17 | AA | Severe | Never | No | Int22 | Int22 | 1201g, 1302 | V2238 |
| 009-034 | 18+ | AA | Severe | 0 (2 y ago), 9 (17 y ago) | Yes | Int22 | Int22 | 0804, 1302 | V2238 |
| 011-052 | 2-3 | AA | Severe | Never | No | ND | ND | 0901, 1102 | V2238 |
| 015-006 | 4-17 | AA | Severe | 0 (2 y ago), 1.3 (6 y ago) | No | Int22 | Int22 | 1202, 1302 | V2238 |
| 002-017 | 4-17 | AA | Severe | Never | No | c.791T>C, p.Leu264Pro | CTG→CCG, L245P | 1101, 1201g | V2238 |
| 004-020 | 4-17 | AA | Severe | 0 (2 y ago), 32 (6 y ago) | Yes | Int1 | Int1 | 0301, 0804 | V2238 |
| 016-027 | 4-17 | AA | Severe | 0 (2 y ago), 6 (13 y ago) | No | ND | ND | 0301, 1401 | V2238 |
| 004-005 | 18+ | W | Severe | Never | No | ND | ND | 0701, 0804 | V2238 |
| 004-013 | 4-17 | AA | Severe | Never | No | Int1 | Int1 | 1201g, 1501 | V2238 |
| 007-002 | 18+ | AA | Severe | Never | No | ND | ND | 0101, 0804 | V2238 |
| 011-001 | 4-17 | AA | Severe | 15 (2 wk ago), 309 (no date) | Yes | Int22 | Int22 | 1101, 0701 | V2238 |
| 002-025 | 2-3 | AA | Severe | 0 (same day), <0.5 (1 y ago) | Yes | c.3637del, Ile1213Phefs*5 | ΔA, E1191-I1194 | 0301, 0804 | V2238 |
| 005-019 | 18+ | SEA | Severe | 1.0 (4 mo ago), 132 (no date) | Yes | Int22 | Int22 | 1101, 0301 | M2238 |
| 013-011 | 18+ | W | Severe | Never | No | c.3637dup, p.Ile1213Asnfs*28 | +A, E1191-I1194 | 1101, 0103 | M2238 |
| 013-013 | 4-17 | AA | Severe | Never | No | c.1171C>T, p.Arg3191Cys | CGC→TGC, R372C | 0301, 1401g | M2238 |
| 014-016 | 4-17 | AA | Severe | 0.3 (5 mo ago), 34 (no date) | Yes | c.5961dup, p.Glu1988Argfs*4 | GAG→AGAG, E1969 | 0301, 0301 | M2238 |
| 008-006 | 2-3 | AA | Severe | 13 (same day), 87 (5 wk later) | No | c.6967C>T, p.Arg2323Cys | CGC→TGC, R2304C | 0301, 1301 | M2238 |
| 011-016 | 18+ | AA | Severe | 0 (2 y ago), 11 (20 y ago) | Yes | ND | ND | 0701, 1302 | H484 |
| 013-024 | 4-17 | AA | Severe | 0 (1 y ago), 1126 (∼10 y ago) | Yes | Int22 | Int22 | 0701, 1001 | D1241 |
| 009-003 | 2-3 | W | Severe | 0 | No | ND | ND | 0404, 0901 | D1241 |
| 011-020 | 18+ | W | Severe | 0 | No | ND | ND | 0701, 0901 | D1241 |
| 005-015 | 18+ | W | Mild | 2-7 (4 y ago), 250 (7 y ago) | No | A2201P (from chart) | 0101, 1503 | D1241 | |
| 007-029 | 18+ | W | Severe | 0 (2 y ago), 280 (21 y ago) | Yes | ND | ND | 0101, 0701 | D1241 |
AA, African American; SEA, SE Asian; W, white.
Inhibitor titers in BU/mL were not available for all subjects on the day of sample collection, therefore the most recently determined titers are reported.
HGVS: Human Genome Variation Society nomenclature, in which residue 1 is the first residue of the signal peptide. Int1, intron-1 inversion; Int22, intron-22 inversion; ND, not determined.
Legacy nomenclature, in which residue 1 is the first residue of the FVIII protein after signal peptide removal.
HLA-DRB1 identifiers in bold indicate that the corresponding HLA-DRB1 tetramers were used to analyze CD4 T-cell responses for these subjects. Cells from mild HA subject 001-043 were analyzed because his F8 gene encoded FVIII-V2238. Cells from mild HA subject 005-015 were analyzed because he had a recently resolved inhibitor. Five of 15 subjects with an inhibitor history did not receive ITI. One of these (008-006) had his inhibitor diagnosed at enrollment and 1 (005-015) had received FVIII only to support surgery.
Alleles with the suffix “g” are the most likely of multiple alternative genotypes encoding identical protein sequences in the HLA-DRB1 antigen recognition site.

Representative tetramer-guided epitope mapping results. (A) Analysis of an initial possible tetramer-positive response to F8-SNP-Pool 2238 peptides. CD4 cells from severe HA subject GIS-005-019, whose F8 gene encodes the M2238 sequence, were stimulated with irrelevant peptides (FVIII-A2 peptide pool 6), tetanus peptides (TT506-525, a known HLA-DRB1*0301–restricted epitope or TT946-965, a known HLA-DRB1*11:01–restricted epitope), or with F8 ns-SNP peptide pool 2238 peptides for 2 weeks in culture and then stained with phycoerythrin (PE)-labeled HLA-DR0301 and DR1101 tetramers loaded with the same peptides used for stimulation. Cultures stimulated with tetanus peptide showed the expected strong T-cell staining by tetramers loaded with these known T-cell epitopes. Cultures stimulated with F8 SNP-Pool 2238 peptides showed low-level, possibly nonspecific, staining by DR0301 and DR1101 tetramers loaded with these peptides. (B) Decoding the pooled-peptide results using the individual peptides comprising F8 SNP-Pool 2238. CD4 cells were cultured for 2 more days following the initial tetramer staining experiment (A) and then stained using HLA-DR1101 and DR0301 tetramers loaded with the 4 individual peptides comprising the original peptide pool. Again, low-level tetramer staining was seen using tetramers loaded with several of these individual 2238 peptides. (C) Attempts to isolate T-cell clones recognizing sequences encoded by ns-SNPs in F8. Similarly, tetramer-positive T cells from the cultures stimulated with F8 SNP-Pool 2238 (shown in panel A) were single-cell sorted and expanded with PHA for 6 weeks in an attempt to isolate FVIII-specific T-cell clones. They were then incubated with fluorescent HLA-DR0301 and DR1101 tetramers loaded with the F8 SNP-Pool 2238 peptides. Representative results are shown for 2 of these expanded cultures. The PHA expansion did not produce any T-cell clones recognizing the pooled peptides, indicating the earlier “borderline” tetramer-positive staining was likely nonspecific. These negative staining results are in distinct contrast to our earlier studies, in which T-cell responses to “sequence-mismatched” FVIII products in mild HA subjects were identified initially by tetramer staining of CD4 T cells and then unambiguously verified by isolation of T-cell clones and lines with HLA-restricted responses to the mismatched FVIII sequence.7,8,40,41 (D) Isolation of tetanus-specific T-cell clones as positive controls. T cells showing positive staining using tetramers loaded with tetanus peptides (A) were single-cell sorted onto 96-well plates and expanded with PHA for 6 weeks to isolate tetanus-specific T-cell clones, as positive controls. Representative staining results are shown for 2 of 12 and 2 of 32 T-cell clones isolated from these cultures, stained with DR0301 and DR1101 tetramers loaded with tetanus peptides TT 506-525 and TT946-965, respectively.
Representative tetramer-guided epitope mapping results. (A) Analysis of an initial possible tetramer-positive response to F8-SNP-Pool 2238 peptides. CD4 cells from severe HA subject GIS-005-019, whose F8 gene encodes the M2238 sequence, were stimulated with irrelevant peptides (FVIII-A2 peptide pool 6), tetanus peptides (TT506-525, a known HLA-DRB1*0301–restricted epitope or TT946-965, a known HLA-DRB1*11:01–restricted epitope), or with F8 ns-SNP peptide pool 2238 peptides for 2 weeks in culture and then stained with phycoerythrin (PE)-labeled HLA-DR0301 and DR1101 tetramers loaded with the same peptides used for stimulation. Cultures stimulated with tetanus peptide showed the expected strong T-cell staining by tetramers loaded with these known T-cell epitopes. Cultures stimulated with F8 SNP-Pool 2238 peptides showed low-level, possibly nonspecific, staining by DR0301 and DR1101 tetramers loaded with these peptides. (B) Decoding the pooled-peptide results using the individual peptides comprising F8 SNP-Pool 2238. CD4 cells were cultured for 2 more days following the initial tetramer staining experiment (A) and then stained using HLA-DR1101 and DR0301 tetramers loaded with the 4 individual peptides comprising the original peptide pool. Again, low-level tetramer staining was seen using tetramers loaded with several of these individual 2238 peptides. (C) Attempts to isolate T-cell clones recognizing sequences encoded by ns-SNPs in F8. Similarly, tetramer-positive T cells from the cultures stimulated with F8 SNP-Pool 2238 (shown in panel A) were single-cell sorted and expanded with PHA for 6 weeks in an attempt to isolate FVIII-specific T-cell clones. They were then incubated with fluorescent HLA-DR0301 and DR1101 tetramers loaded with the F8 SNP-Pool 2238 peptides. Representative results are shown for 2 of these expanded cultures. The PHA expansion did not produce any T-cell clones recognizing the pooled peptides, indicating the earlier “borderline” tetramer-positive staining was likely nonspecific. These negative staining results are in distinct contrast to our earlier studies, in which T-cell responses to “sequence-mismatched” FVIII products in mild HA subjects were identified initially by tetramer staining of CD4 T cells and then unambiguously verified by isolation of T-cell clones and lines with HLA-restricted responses to the mismatched FVIII sequence.7,8,40,41 (D) Isolation of tetanus-specific T-cell clones as positive controls. T cells showing positive staining using tetramers loaded with tetanus peptides (A) were single-cell sorted onto 96-well plates and expanded with PHA for 6 weeks to isolate tetanus-specific T-cell clones, as positive controls. Representative staining results are shown for 2 of 12 and 2 of 32 T-cell clones isolated from these cultures, stained with DR0301 and DR1101 tetramers loaded with tetanus peptides TT 506-525 and TT946-965, respectively.
In conjunction, these results indicate that deleterious immune responses to FVIII regions encoded by nonhemophilic ns-SNPs occur much less frequently than might have been anticipated based on peptide-MHC binding data and/or T-cell epitope prediction algorithms. This was true even for HA patients whose MHC class II (HLA-DRB1) were capable of binding FVIII peptides that were mismatched to the sequence encoded by their hemophilic F8 gene (Table 7).
Discussion
African American HA patients have been reported to have an increased incidence of FVIII inhibitors vis-à-vis white patients.9-11 An earlier study of 76 black HA subjects suggested that infusions with “sequence-mismatched” FVIII may be a risk factor for inhibitor development.14 Subsequent studies have not shown a statistical link between F8 polymorphisms and inhibitor incidence.27-29 The present study focuses primarily on inhibitor incidence in patients with severe HA. Seven subjects who had an intron-22 inversion mutation but baseline clotting activity ≥1% normal were also included, as they are not expected to circulate FVIII. We thus avoided possible confounding due to variable inhibitor risks associated with mutations causing mild/moderately severe HA.
No significant correlations were seen between inhibitor history and either F8 haplotypes or the individual ns-SNPs encoding FVIII-D1241E and FVIII-M2238V variants. Only 1 severe HA subject had the polymorphism encoding FVIII-H484. A logistic regression analysis adjusting for age range, F8 haplotype, and potential exposure to “sequence-mismatched” FVIII was carried out in which all subjects who had received plasma-derived FVIII and/or any other blood product were assumed to have been exposed to “sequence-mismatched” FVIII. These subjects showed an increased inhibitor incidence but there was no correlation with F8 haplotypes (Table 6). No firm conclusions can be drawn from this apparent association because the sequences of the infused FVIII could be confirmed only for subjects treated with recombinant full-length FVIII products. Furthermore, other product-related, genetic, or environmental differences affecting immunogenicity, for example, between subjects requiring or not requiring blood transfusions, or treated with single vs multiple FVIII products, could not be quantified or ruled out.
African Americans with an intron-22 inversion mutation had a 2- to 3-times-higher inhibitor risk than white subjects with the same mutation, consistent with a recent report of inhibitor incidence in a South African cohort.28 Multiple-exon deletions have been shown to carry the largest inhibitor risk for patients with severe HA, whereas missense mutations and small insertions/deletions carry lower risks.2 The race-associated inhibitor risks associated with these types of mutations could not be determined because the subgroups in this study were too small. Previous studies investigating possible associations between HLA and inhibitor incidence have identified weak associations with HLA alleles including HLA-DR15,30,31 however, the diversity of HLA repertoires in inhibitor patients is consistent with the multifactorial origin of inhibitor development.32-35 A possibly increased inhibitor risk was seen for subjects with HLA-DR15 and the H2 haplotype (Table 5), but this was not seen for those with peak titers ≥5 BU/mL (supplemental Table 3). Future studies will evaluate additional genetic and environmental factors34,36-38 that influence race-associated differences in inhibitor risk.
Peptide-MHC binding assays and predictions, as well as tetramer analyses of T cells, were carried out to assess the potential risk of immune responses to sequences encoded by polymorphic sites in the F8 gene. Competition binding assays identified several HLA-DRB1 with moderate- to high-affinity binding to FVIII peptides with polymorphic sequences at residues 484, 1241, and 2238 (Table 7). A fourth polymorphic site encoding FVIII-R776G (identified in a SE Asian subject)13 was also investigated. The 2 computer algorithms tended to overpredict productive peptide-MHC binding (supplemental Tables 4-5). Such predictions will nevertheless be useful in future studies to determine the MHC-binding registers within peptides shown experimentally to bind specific HLA-DRB1 proteins.
MHC class II binding is required for peptide presentation on antigen-presenting cells. Individuals whose MHC effectively bind these peptides could be at higher risk for inhibitor development, but only if similar FVIII peptides are actually processed and presented by their antigen-presenting cells and their T-cell repertoire includes T-effector cells capable of recognizing these peptide-MHC complexes. It is important to emphasize that MHC binding of synthetic FVIII peptides does not in itself indicate a higher inhibitor risk. However, a lack of effective peptide-MHC binding suggests strongly that this peptide would not provoke a T-cell response restricted to the corresponding MHC allele. Approximately 85% of the peptide binding assays carried out for peptides encoded by ns-SNPs at FVIII positions 484, 776, 1241, and 2238 indicated weak or no binding of these sequences to 11 HLA-DRB1 that together are found in ∼40% of the African American population.20 The polymorphism-encoded FVIII sequences did not bind the following HLA-DRB1 proteins: DR0401, 0701, 0901, and 1001 (R484H); DR0301, 1101, and 0701 (D1241E); DR0401, 0901, and 1001 (M2238V); and all but 2 of the HLA-DRB1 proteins tested (R776G), indicating HLA-DRB1–restricted CD4 T-cell responses restricted to these peptide/allele combinations would be highly unlikely. Eight of the 16 subjects whose F8 genes encoded FVIII-V2238 never developed a clinically significant inhibitor even though 2 had HLA-DRB1 (HLA-DR0101 and HLA-DR1101) that bound mismatched FVIII-M2238 peptides with moderate affinity. An additional 2 subjects expressed HLA-DR0301, which bound with high affinity to a peptide having the mismatched M2238 sequence (Table 7).
All African American subjects with endogenous F8 sequences encoding FVIII-H484 or FVIII-V2238 (and 1 subject with FVIII-V2238 who self-identified as white) had been infused with “sequence-mismatched” FVIII. Peptides containing R484H or M2238V bound with strong-to-moderate affinity to HLA-DRB1 proteins DR1501, DR0301, DR1104, DR0403, and DR0404 (R484H) and to DR0101, DR0301, DR0403, DR0404, DR1101, and DR1104 (M2238V), respectively. Together, the corresponding HLA-DRB1 alleles are found in ∼11% and ∼20% of the African American population.20,39 Peptides containing D1241E bound with strong-to-moderate affinity to HLA-DRB1 proteins DR0403 and DR0901. Interestingly, 2 subjects with an HLA-DRB1*09:01 allele, who had been treated with FVIII-E1241, had no inhibitor history and no T-cell response to FVIII-E1241 peptides (Table 8, supplemental Figure 1).
We and others have used tetramer-based protocols to query CD4 T-cell responses in mild HA subjects, unambiguously identifying T-cell epitopes corresponding to wild-type FVIII sequences at hemophilic mutation sites by isolation of FVIII-specific T-cell clones and lines.6-8,40,41 The relative incidence of inhibitors in mild HA patients varies with mutation, with several recurrent mutations such as R593C and R2150H having a stronger association with inhibitor development.6,42-45 The present study did not identify T-cell epitopes corresponding to the most common F8 polymorphisms in the African American population, D1241E and M2238V, whereas control experiments carried out using aliquots of the same blood samples routinely produced distinct tetramer-positive populations from which tetanus-specific T-cell clones and polyclonal lines could be expanded.
The immunogenicity of FVIII sequences encoded by non-HA-causing ns-SNPs has not yet been evaluated using blood samples from HA subjects receiving their initial FVIII infusions, and some patients may develop effector T-cell responses to these sequences. Epitope maturation or drift following initial inhibitor development may also have occurred in the subjects of the present study. T-cell responses restricted to other HLA loci such as HLA-DQ or HLA-DRB3/4/5 have also not yet been evaluated. Only 1 African American subject had the polymorphism encoding FVIII-H484. The FVIII-R484 peptide bound with moderate-to-high affinity to 5 HLA-DRB1 proteins tested (Table 7), suggesting this polymorphism could confer risk of an immune response to FVIII products mismatched at this site in some patients. However, the low prevalence of this F8 variant indicates that immune responses to epitopes containing FVIII-R484 could not account for an appreciable fraction of inhibitors in African Americans.
The present study evaluated samples and data from 372 African American and white HA subjects. Both statistical analysis and T-cell stimulation assays strongly indicated that currently available FVIII products have not provoked T-cell responses to potential neoepitopes encoded by ns-SNPs in an appreciable fraction of African American HA patients. Although T-cell stimulation by such neoepitopes may well occur in some patients, their frequency is unlikely to exceed the inhibitor incidence associated with most F8 missense mutations.2,45-47 In order to better define subsets of patients that might be at risk for immune responses to FVIII sequences encoded by these ns-SNPs, additional studies of patients with potentially higher-risk HLA alleles (that bind these sequences with moderate-to-high affinity), and of patients receiving initial FVIII infusions, are warranted. The present study confirmed that African American patients with severe HA experience a higher inhibitor incidence than white patients, and African American subjects with the most common HA-causing mutation, an intron-22 inversion, were found to have a two- to threefold-higher inhibitor risk. Future studies of the complex etiology of inhibitor development will address this significant health disparity.
The online version of this article contains a data supplement.
There is an on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all PATH Study Investigators, oversight committees, and their staff for their essential contributions to this study, and are grateful to all subjects and their families for generous blood donations and support.
This work was supported by National Institutes of Health National Heart, Lung, and Blood Institute grant 1RC2-HL101851, and unrestricted hemophilia research grants from CSL Behring and Bayer (K.P.P.).
Authorship
Contribution: D.G., R.A.E., S.N.F., E.A.J., M.S.E., M.L., R.J.H., and K.P.P. designed and performed experiments, analyzed data, and wrote the paper; and J.C.B., J.W., and D.C.M. enrolled patients, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: K.P.P. has received unrestricted hemophilia research funding from CSL Behring, Pfizer, and Bayer. The remaining authors declare no competing financial interests.
A complete list of the members of the Personalized Approaches to Therapies for Hemophilia (PATH) Study Investigators appears in the Acknowledgments section of the online data supplement.
The opinions or assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense or the Uniformed Services University of the Health Sciences.
Correspondence: Kathleen P. Pratt, Department of Medicine (MED) A3075, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Rd, Bethesda, MD, 20814; e-mail: kathleen.pratt@usuhs.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal