Abstract
COVID-19 is a primary respiratory illness that is frequently complicated by systemic involvement of the vasculature. Vascular involvement leads to an array of complications ranging from thrombosis to pulmonary edema secondary to loss of barrier function. This review will address the vasculopathy of COVID-19 with a focus on the role of the endothelium in orchestrating the systemic response to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. The endothelial receptor systems and molecular pathways activated in the setting of COVID-19 and the consequences of these inflammatory and prothrombotic changes on endothelial cell function will be discussed. The sequelae of COVID-19 vascular involvement at the level of organ systems will also be addressed, with an emphasis on the pulmonary vasculature but with consideration of effects on other vascular beds. The dramatic changes in endothelial phenotypes associated with COVID-19 has enabled the identification of biomarkers that could help guide therapy and predict outcomes. Knowledge of vascular pathogenesis in COVID-19 has also informed therapeutic approaches that may control its systemic sequelae. Because our understanding of vascular response in COVID-19 continues to evolve, we will consider areas of controversy, such as the extent to which SARS-CoV-2 directly infects endothelium and the degree to which vascular responses to SARS-CoV-2 are unique or common to those of other viruses capable of causing severe respiratory disease. This conceptual framework describing how SARS-CoV-2 infection affects endothelial inflammation, prothrombotic transformation, and barrier dysfunction will provide a context for interpreting new information as it arises addressing the vascular complications of COVID-19.
The quiescent endothelium
The endothelium lines the inner surface of all blood vessels and provides a critical interface between the circulation and organ-specific tissues. To understand the endothelial contribution to the vasculopathy of COVID-19, one must first consider functions that the endothelial surfaces serve in healthy physiology. Endothelium originates from the hemangioblast, the embryonic precursor of immune cells, and endothelial cells serve a critical role in immune surveillance, functioning both in adaptive and innate immunity.1,2 Endothelium also provides an anticoagulant surface and maintains barrier integrity. Specific constitutive cytoprotective circuits and distinct cellular features maintain endothelial quiescence.
Cytoprotective signaling
Several signaling pathways in healthy endothelium actively maintain its resting state. The endothelial cell–enriched Tie2 receptor tyrosine kinase is constitutively activated by its oligomeric ligand, angiopoietin-1, secreted by perivascular cells (Figure 1, panel 1). Constitutive Tie2 activation is potently anti-inflammatory, inhibiting transcription factor NF-κB, thereby blocking proinflammatory effects mediated by several cytokines. Stimulation of Tie2 also prevents induction of TF and exposure of surface phosphatidylserine and is thereby antithrombotic.3 Ligation of Tie2 is critical in maintenance of barrier function by supporting cortical actin formation at the periphery of the cell and stabilizing adherens junctions.4 Highly relevant to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is cytoprotective signaling associated with the angiotensin pathway. ACE2 generates angiotensin-(1-7), a peptide that interacts with the Mas1 receptor to mediate anti-inflammatory and antithrombotic signaling (Figure 1, panel 2).5,6 Cleavage of PAR1 by APC is a second endothelial cytoprotective pathway that has similar actions as Tie2 activation (Figure 1, panel 3).7,8 S1P receptors are important for maintenance of barrier function as well.9
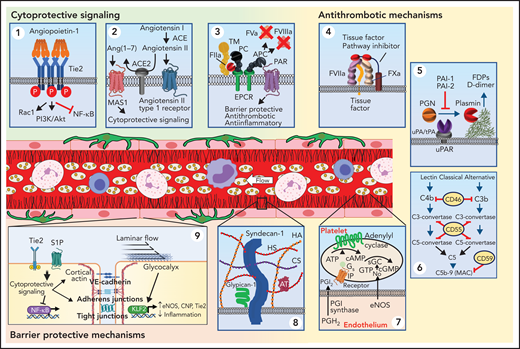
Endothelial mechanisms that maintain vascular quiescence and vessel patency. The quiescent endothelium depicted in the center of the figure features a rich glycocalyx covering its inner surface (black lines) and is surrounded by pericytes (dark green) on its out surface. Circulating cells move freely within its lumen. The endothelium has several receptor systems that mediate constitutive cytoprotective signaling (green background). (Panel 1) Activation of Tie2 (blue) by multimeric angiopoitein-1 (orange). Clustering of Tie2 by angiopoieitin-1 results in phosphorylation of the Tie2 cytoplasmic tail. Stimulation of Tie2 also promotes barrier function via activation of Rac1. (Panel 2) Although ACE produces angiotensin II, which can stimulate inflammatory signaling, the resting endothelium expresses ACE2, which cleaves angiotensin II into Ang(1-7). Ang(1-7) binds to MAS1, resulting in cytoprotective signaling. (Panel 3) The endothelium expresses thrombomodulin (TM), which binds thrombin (IIa) and occludes its fibrin binding site, modifying its substrate specificity. TM is closely associated with the endothelial protein C receptor (EPCR), which binds protein C (PC) enabling it to be cleaved by thrombin and generate activated protein C (APC). APC is a potent anticoagulant that cleaves factors Va and VIIIa. It also cleaves protease-activated receptor 1 (PAR1) at a noncanonical cleavage site, stimulating cytoprotective signaling. Antithrombotic mechanisms are indicated with yellow background. (Panel 4) The endothelium expresses tissue factor pathway inhibitor-β (TFPIβ), which associates with the membrane surface via a glycosylphosphatidylinositol (GPI) anchor. TFPIβ binds to both factor VIIa (FVIIa) and factor Xa (FXa) via its kunitz domains (red), thereby inhibiting the ability of tissue factor (TF) to activate coagulation. (Panel 5): Fibrin is cleared from the vasculature by plasmin degradation. Endothelial cells secrete both tissue plasminogen activator (tPA) and urokinase plasminogen activation (uPA), which binds to the urinokinase plasminogen activator receptor (uPAR). Plasminogen activators convert plasminogen to plasmin, which cleaves fibrin, resulting in generation of fibrin degradation products (FDPs) and d-dimer. Plasminogen activator inhibitors 1 and 2 (PAI-1 and PAI-2) inhibit tPA and uPA. (Panel 6) Several surface proteins inhibit inappropriate complement activation. These include the type 1 membrane protein CD46 that inactivates C3b and C4b, the GPI-linked membrane protein CD55 (or complement decay-accelerating factor) that prevents formation of C3-convertase and C5-convertase, and the GPI-linked membrane protein CD59 that prevents C9 polymerization, thereby blocking formation of the membrane attack complex (MAC). (Panel 7) The endothelium also elaborates nitric oxide (NO) and PGI2 to maintain platelets in a resting state. These mechanisms maintain blood flow as depicted in the schematic of a resting venule in the center of the figure: endothelium (pink rectangular cells), glycocalyx (black), platelets (beige), neutrophils (pink nucleated cells), monocytes (irregular cells with purple nucleus), and lymphocytes (blue). (Panel 8) Healthy endothelium is coated with a thick glycocalyx consisting of heparan sulfate and chondroitin sulfate attached to syndecan, hyaluronan, and glypican-1. This physical barrier buffers oncotic forces across the vessel wall and limits interaction with leukocytes and platelets. (Panel 9) Maintenance of endothelial cell barrier function (orange background) is an active process. Fluid and leukocyte extravasation is prevented by tight junctions containing junctional adhesion molecules and claudins and adherens junctions containing VE-cadherin. Activation of Tie2 by Angpt-1 or the sphingosine 1-phosphate receptor 1 (S1PR1) by sphingosine 1-phosphate (S1P) maintains cortical actin networks and promotes VE-cadherin adherens junctions at the cell surface. Cytoprotective signaling through EPCR and Tie2 inhibits activation of the inflammatory and prothrombotic transcription factor NF-κB. Laminar blood flow activates transcription factors KLF2 and KLF4, which promote maintenance of vascular tone via expression of nitric oxide synthase and C-natriuretic peptide (CNP) and reduce inflammation and thrombosis by increasing Tie2 and TM expression. Tonic bradykinin (BK) signaling through the constitutive bradykinin receptor 2 (B2R) and Angiotensin1-7 (Ang1-7) activation of the Mas1 receptor promotes vascular tone via NO synthase and suppressing inflammation. ACE, angiotensin-converting enzyme.
Endothelial mechanisms that maintain vascular quiescence and vessel patency. The quiescent endothelium depicted in the center of the figure features a rich glycocalyx covering its inner surface (black lines) and is surrounded by pericytes (dark green) on its out surface. Circulating cells move freely within its lumen. The endothelium has several receptor systems that mediate constitutive cytoprotective signaling (green background). (Panel 1) Activation of Tie2 (blue) by multimeric angiopoitein-1 (orange). Clustering of Tie2 by angiopoieitin-1 results in phosphorylation of the Tie2 cytoplasmic tail. Stimulation of Tie2 also promotes barrier function via activation of Rac1. (Panel 2) Although ACE produces angiotensin II, which can stimulate inflammatory signaling, the resting endothelium expresses ACE2, which cleaves angiotensin II into Ang(1-7). Ang(1-7) binds to MAS1, resulting in cytoprotective signaling. (Panel 3) The endothelium expresses thrombomodulin (TM), which binds thrombin (IIa) and occludes its fibrin binding site, modifying its substrate specificity. TM is closely associated with the endothelial protein C receptor (EPCR), which binds protein C (PC) enabling it to be cleaved by thrombin and generate activated protein C (APC). APC is a potent anticoagulant that cleaves factors Va and VIIIa. It also cleaves protease-activated receptor 1 (PAR1) at a noncanonical cleavage site, stimulating cytoprotective signaling. Antithrombotic mechanisms are indicated with yellow background. (Panel 4) The endothelium expresses tissue factor pathway inhibitor-β (TFPIβ), which associates with the membrane surface via a glycosylphosphatidylinositol (GPI) anchor. TFPIβ binds to both factor VIIa (FVIIa) and factor Xa (FXa) via its kunitz domains (red), thereby inhibiting the ability of tissue factor (TF) to activate coagulation. (Panel 5): Fibrin is cleared from the vasculature by plasmin degradation. Endothelial cells secrete both tissue plasminogen activator (tPA) and urokinase plasminogen activation (uPA), which binds to the urinokinase plasminogen activator receptor (uPAR). Plasminogen activators convert plasminogen to plasmin, which cleaves fibrin, resulting in generation of fibrin degradation products (FDPs) and d-dimer. Plasminogen activator inhibitors 1 and 2 (PAI-1 and PAI-2) inhibit tPA and uPA. (Panel 6) Several surface proteins inhibit inappropriate complement activation. These include the type 1 membrane protein CD46 that inactivates C3b and C4b, the GPI-linked membrane protein CD55 (or complement decay-accelerating factor) that prevents formation of C3-convertase and C5-convertase, and the GPI-linked membrane protein CD59 that prevents C9 polymerization, thereby blocking formation of the membrane attack complex (MAC). (Panel 7) The endothelium also elaborates nitric oxide (NO) and PGI2 to maintain platelets in a resting state. These mechanisms maintain blood flow as depicted in the schematic of a resting venule in the center of the figure: endothelium (pink rectangular cells), glycocalyx (black), platelets (beige), neutrophils (pink nucleated cells), monocytes (irregular cells with purple nucleus), and lymphocytes (blue). (Panel 8) Healthy endothelium is coated with a thick glycocalyx consisting of heparan sulfate and chondroitin sulfate attached to syndecan, hyaluronan, and glypican-1. This physical barrier buffers oncotic forces across the vessel wall and limits interaction with leukocytes and platelets. (Panel 9) Maintenance of endothelial cell barrier function (orange background) is an active process. Fluid and leukocyte extravasation is prevented by tight junctions containing junctional adhesion molecules and claudins and adherens junctions containing VE-cadherin. Activation of Tie2 by Angpt-1 or the sphingosine 1-phosphate receptor 1 (S1PR1) by sphingosine 1-phosphate (S1P) maintains cortical actin networks and promotes VE-cadherin adherens junctions at the cell surface. Cytoprotective signaling through EPCR and Tie2 inhibits activation of the inflammatory and prothrombotic transcription factor NF-κB. Laminar blood flow activates transcription factors KLF2 and KLF4, which promote maintenance of vascular tone via expression of nitric oxide synthase and C-natriuretic peptide (CNP) and reduce inflammation and thrombosis by increasing Tie2 and TM expression. Tonic bradykinin (BK) signaling through the constitutive bradykinin receptor 2 (B2R) and Angiotensin1-7 (Ang1-7) activation of the Mas1 receptor promotes vascular tone via NO synthase and suppressing inflammation. ACE, angiotensin-converting enzyme.
Antithrombotic properties
The endothelial membrane provides a potent antithrombotic surface. It expresses thrombomodulin and EPCR, which convert the vasculature's most potent prothrombotic enzyme, thrombin (factor IIa), into a producer of a potent anticoagulant, activated protein C (Figure 1, panel 3).10 Endothelium also produces tissue factor pathway inhibitor (TFPI), including TFPIα, which is secreted into the circulation, and TFPIβ, which binds to a GPI anchor (Figure 1, panel 4).11,12 In addition, endothelium secretes tPA and uPA and expresses its receptor, providing potent fibrinolytic capacity (Figure 1, panel 5). Resting endothelium is also protected from complement deposition by inhibitors of the complement cascade (Figure 1, panel 6). The quiescent endothelium prevents platelet adhesion by elaborating PGI2 and NO, which suppresses platelet activation (Figure 1, panel 7).
Glycocalyx
The endothelium secretes a glycocalyx consisting of proteoglycans and glycosaminoglycans that extend out >400 nm from the cell surface and impede entry of plasma proteins and decrease water permeability (Figure 1, panel 8).13 The glycocalyx also mechanotransduces shear forces from flowing blood to activate cytoprotective intracellular signaling. By extending beyond the length of leukocyte-adhesion receptors, the glycocalyx serves a secondary function of inhibiting immune activation. Heparan sulfates within the glycocalyx bind and activate antithrombin, thereby limiting thrombosis. Thus, permeability, flow, inflammation, and thrombosis are all interdependently affected by the glycocalyx.
Barrier function
Endothelial barrier function relies on cell-cell junctions including endothelial-specific vascular endothelial (VE)-cadherin, which comprises adherens junctions, and junctional adhesion molecules, claudins, and occludins present in tight junctions (Figure 1, panel 9).14 Breaching the integrity of these adhesive junctions is an essential part of immune surveillance and is typically limited to postcapillary venules, where tight junctions are minimal.
SARS-CoV-2 infection of endothelium?
Among the early controversies regarding the role of the endothelium in COVID-19 was whether or not endothelial cells are directly infected by SARS-CoV-2. Endotheliopathy in COVID-19 was shown early in the pandemic.15,16 However, whether this endothelial damage resulted from direct endothelial infection by SARS-CoV-2 or the inflammatory response to the virus has not been entirely resolved. Previous studies evaluating the distribution of ACE2, the most thoroughly studied SARS-CoV-2 receptor, showed little expression on endothelial cells compared with expression in the epithelium of the airways.17 Although some reports showed evidence of viral inclusion bodies or viral-like particles in endothelium of postmortem lung samples,18,19 other studies of postmortem samples failed to demonstrate virus in endothelium,20,21 and the similarity of viral-like structures to cross-sections of endoplasmic reticulum, clathrin-coated pits, or multivesicular bodies were cited.22,23 Studies of cultured endothelial cells demonstrated relatively little ACE2 expression and have failed to demonstrate incorporation of either pseudovirus constructs or SARS-CoV-2 isolates upon coculture.24,25
It is possible that although there is far less ACE2 on endothelium than on airway epithelium, there is sufficient expression to support SARS-CoV-2 entry. Furthermore, additional receptors have been shown to mediate SARS-CoV-2 viral entry, including neuropilin-1,26,27 CD147,28 and CD26,29 which are all expressed on endothelium. A single-cell atlas of different organs from COVID-19 patients showed that viral RNA was enriched in myeloid cells but was also elevated in endothelial cell subsets.30 Although intriguing, the finding of viral RNA does not necessarily represent replicating virus and could result from engulfment or attachment of infected cells to endothelial cells.30 Others have successfully infected cultured endothelial cells with isolates of SARS-CoV-2,31 contradicting negative studies and raising the possibility that the virus can replicate in endothelium in vivo. Overall, however, studies published since the initial suggestions of direct viral infection of endothelium by SARS-CoV-2 have failed to convincingly prove that endothelium can support SARS-CoV-2 replication.
Yet, even if SARS-CoV-2 cannot replicate in endothelium, it remains possible that viral components stimulate endothelial activation. Lei et al found that a noninfectious pseudovirus that expressed SAR-CoV-2 spike protein (S protein) was sufficient to cause endothelial dysfunction.32 S protein–induced endothelial damage was observed both in cultured cells and following injection of pseudovirus into Syrian hamsters.32 A second study showed that the S1 subunit is sufficient to elicit microvascular endothelial damage characterized by cytokine release, complement activation, and thrombosis.33 The nucleocapsid protein (N protein) of SARS-CoV-2 stimulates transcriptional activation of several proinflammatory genes in cultured human lung microvascular endothelial cells and upregulates endothelial adhesion proteins.34 Viral pathogen-associated molecular patterns (PAMPs) such as S protein, N protein, viral RNA, and other SARS-CoV-2 components are recognized by innate immune receptors on endothelium.35 Such findings suggest that viral entry and replication are not necessary to cause endothelial cell damage. In addition to viral PAMPs, the elaboration of cytokines by immune cells also contributes substantially to endothelial dysfunction in the setting of COVID-19.
Vascular inflammation induced in COVID-19
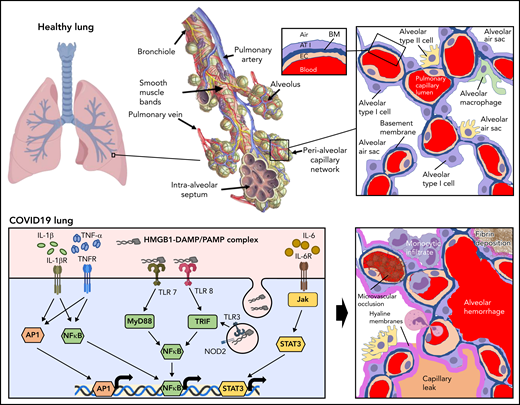
The endothelium both orchestrates and reacts to the progressive inflammatory response elicited by SARS-CoV-2 infection. The functional unit of the lung is the alveolar-capillary interface (Figure 2, healthy lung). The pulmonary capillary network constitutes the largest vascular bed in the body, encompassing 50% of its capillary surface.36 This delicate interface is under constant immune surveillance by patrolling alveolar macrophages and neutrophils. Prior to infection, constitutive signaling through cytoprotective receptors (eg, Tie2) inhibits proinflammatory pathways mediated by NF-kB, MAPK, STAT3, and MyD88.37 The resting endothelium expresses cytokine receptors to enable coordination with resident macrophages. These mechanisms provide for the constitutive clearance of low-level infection agents and particulates that the alveolar-capillary unit encounters with normal breathing.

Inflammatory response to inhalation of COVID-19 in the pulmonary vasculature. Healthy lung: Depicted is a healthy lung (left) and a bronchiole (center) within the lung emphasizing the vasculature and alveolar sacs. A schematic of a histological cross section through an alveolar-capillary unit showing alveolar air sacs (white) lined by alveolar type 1 cells (purple) and separated from capillaries by a basement membrane (blue). The alveolar sac is studded with alveolar type 2 cells (yellow) and alveolar macrophages (green). The inset highlights the air-blood interface that is separated by the alveolar type 1 cell, basement membrane, and the capillary, from top to bottom. COVID lung: The left panel depicts inflammatory signaling through cytokine receptors (IL-1βR, TNFR, and IL-6R) and receptors of innate immunity (TLRs and NOD2) following SARS-CoV-2 infection. The right panel demonstrates sequelae of SARS-CoV-2 infection at the level of the alveolus. Deposition of debris and inflammatory products results in the formation of a hyaline membrane that lines the alveolar sac and impairs exchange (dark pink). Expression of leukocyte receptors results in monocytic infiltrates (purple) and neutrophil extravasation (light pink). Proliferation of alveolar type 2 cells is observed (yellow). Loss of barrier function results in capillary leak (tan). Loss of vascular integrity results in alveolar hemorrhage (red). Microvascular occlusion (red brown) and extravascular fibrin formation (brown) occur. Endothelial swelling and sluffing ensues. TLRs, toll-like receptors; TNFR, tumor necrosis factor receptor.
Inflammatory response to inhalation of COVID-19 in the pulmonary vasculature. Healthy lung: Depicted is a healthy lung (left) and a bronchiole (center) within the lung emphasizing the vasculature and alveolar sacs. A schematic of a histological cross section through an alveolar-capillary unit showing alveolar air sacs (white) lined by alveolar type 1 cells (purple) and separated from capillaries by a basement membrane (blue). The alveolar sac is studded with alveolar type 2 cells (yellow) and alveolar macrophages (green). The inset highlights the air-blood interface that is separated by the alveolar type 1 cell, basement membrane, and the capillary, from top to bottom. COVID lung: The left panel depicts inflammatory signaling through cytokine receptors (IL-1βR, TNFR, and IL-6R) and receptors of innate immunity (TLRs and NOD2) following SARS-CoV-2 infection. The right panel demonstrates sequelae of SARS-CoV-2 infection at the level of the alveolus. Deposition of debris and inflammatory products results in the formation of a hyaline membrane that lines the alveolar sac and impairs exchange (dark pink). Expression of leukocyte receptors results in monocytic infiltrates (purple) and neutrophil extravasation (light pink). Proliferation of alveolar type 2 cells is observed (yellow). Loss of barrier function results in capillary leak (tan). Loss of vascular integrity results in alveolar hemorrhage (red). Microvascular occlusion (red brown) and extravascular fibrin formation (brown) occur. Endothelial swelling and sluffing ensues. TLRs, toll-like receptors; TNFR, tumor necrosis factor receptor.
Infection of type 2 epithelial cells and alveolar macrophages by SARS-CoV-2 results in activation of an antiviral cascade. Timed exposures to SARS-CoV-2 in rhesus macaques shows robust activation of interferons (IFNα and IFNγ), tumor necrosis factor-α (TNF-α), and interleukins in bronchioalveolar samples within the first day of exposure.38 These signals lead to massive recruitment of macrophages and stimulation of a proinflammatory response in endothelium. The pulmonary endothelium facilitates leukocyte recruitment by increased expression of selectins (P-selectin and E-selectin) and adhesion proteins (ICAM-1 and VCAM-1), primarily in postcapillary venules.39 If the virus is not initially controlled, cytoprotective endothelial signaling is either reduced or converted to proinflammatory pathways such as those mediated by thrombin activation of PAR1. Dysregulation of the alveolar epithelial cells and pulmonary capillaries results in massive infiltration of lymphocytes. CD8+ and CD4+ effector cells are subsequently recruited.
The pulmonary endothelium responds to specific components of the inflammatory milieu via an array of receptors and intracellular signaling pathways (Figure 2, COVID-19 lung). PAMPs produced by widespread viral infection include single-stranded viral RNA and viral proteins. Damage-associated molecular patterns (DAMPs) produced from ensuing cell damage and death include ATP, DNA, and HMGB1.40,41 These danger signals activate endothelial pattern recognition receptors, including toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (Figure 2).35 Cytokine receptors respond to TNF-α and interleukins released by activated macrophages and lymphocytes (Figure 2). These signals activate proinflammatory pathways such as NF-kB, STAT3, MAPKs, and MyD88 that stimulate upregulation of transcripts for leukocyte adhesion molecules and cytokines (Figure 2). Endothelial-derived cytokines released in the setting of COVID-19 include IL-6, IL-8, TNF-α, IL-18, and IL-1β. Assembly of the NRLP3 inflammasome contributes to maturation of IL-18 and IL-1β as well as to the generation of caspase, which can stimulate pyroptosis (lytic programmed cell death) in some endothelial cells.35 The inflammation resulting from SARS-CoV-2 infection also results in increased production of reactive oxygen species secondary to activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and downregulation of endothelial nitric oxide synthetase. These endothelial changes promote the switch from a controlled resolution of an infectious agent to an uncontrolled immune response.
The sequelae of this intense inflammatory response on the endothelium are profound. Loss of antithrombotic properties (detailed in “Prothrombotic transformation of the endothelium in COVID-19”) and of barrier function (detailed in “Endothelial barrier function and vascular tone in COVID-19”) ensue. Loss of endothelial barrier function enables leakage of plasma proteins and fluid into the interstitial space and alveoli (Figure 2). Inflammatory cytokines, secretion of proteases, and a reduction in laminar flow lead to glycocalyx shedding and/or inhibition of its production.42 Evidence of loss of adhesion proteins and receptors during endothelial inflammation, of factors that control coagulation on the endothelium, and of glycocalyx degradation products can be observed in the plasma from patients with COVID-19, and these protein have been used as biomarkers to monitor COVID-19 severity (Table 1). High concentrations of proinflammatory mediators from contents leaked with loss of barrier function further compromise endothelial integrity, enabling lymphatic infiltration into the perivascular space.43 Inflammatory injury to the endothelium also promotes alveolar hemorrhage (Figure 2).43 Viral infection and ensuing inflammation leads to loss of pericytes,44 which express ACE2 and are subject to direct viral infection.45 Cytokine stimulation along with pericyte dropout stimulates upregulation of proangiogenic transcripts and evidence of pulmonary intussusceptive angiogenesis, a mechanism of microvascular expansion requiring minimal endothelial proliferation, which may be preferential in severe hypoxia.19 The endothelium itself can become swollen35 and slough off (Figure 2), as indicated by the increased number of circulating endothelial cells, which are associated with increased mortality.46
Prothrombotic transformation of the endothelium in COVID-19
COVID-19 is associated with a high rate of clinical thrombosis. Both microvascular thrombosis causing small vessel occlusion and macrovascular thrombosis are common. The extent to which COVID-19–associated coagulopathy differs from disseminated intravascular coagulation and sepsis-induced coagulopathy remains a topic of debate. However, some differences are apparent. Thrombosis in COVID-19 is associated with high (as opposed to reduced) fibrinogen levels and less reduction of platelet counts. In addition, the PT/aPTT is not typically as elevated in COVID-19, although there is more activation of the complement system.47,48 Antiphospholipid antibodies were initially thought to be associated with thrombosis in COVID-19, but subsequent studies did not substantiate early observations.49-51 The mechanisms leading to the distinct characteristics of COVID-19–associated thrombosis are not fully understood. However, an underlying theme in COVID-19 thrombotic complications is involvement of the endothelium, which undergoes a prothrombotic transformation involving loss of glycocalyx, cytoprotective signaling, and antithrombotic effectors to instead promote fibrin formation, platelet adhesion, and complement activation.
Glycocalyx shedding
The highly hydrated surface created by the glycocalyx is extremely anticoagulant. Shedding could reduce the activity of antithrombin, which relies on heparan sulfate as a co-factor (Figure 3A, panel 1). A phase 2-3 study to assess the effects of protecting the glycocalyx using sulodexide in COVID-19 supported its effectiveness (clinicaltrials.gov #NCT04483830).44
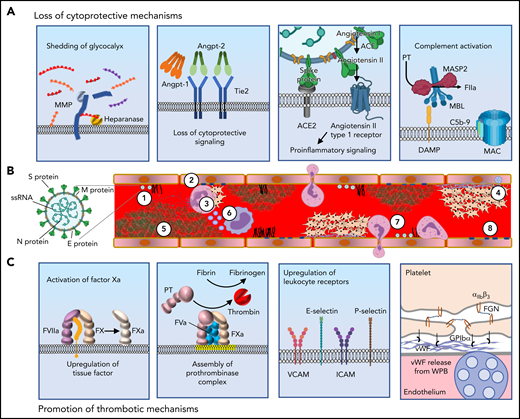
Mechanisms by which SARS-CoV-2 induces the prothrombotic transformation of the endothelium. (A) In severe systemic SARS-CoV-2 infection, endothelilal cytoprotective mechanisms are lost. The glycocalyx is degraded by metalloproteases (MMPs) and heparanases (panel 1). Several cytoprotective signaling pathways are downregulated. Featured in panel 2 is loss of Tie2 signaling, which occurs following the release of angiopoietin-2 from endothelial stores. High concentrations of angiopoietin-2 displace angiopoietin-1, with loss of clustering and decreased Tie2 phosphorylation resulting in loss of cytoprotective signaling. The spike protein of SARS-CoV-2 binds to ACE2, resulting in its endocytosis and loss from the cell surface. This prevents the cleavage of angiotensin II into Ang(1-7). The cytoprotective signaling of Ang(1-7) is therefore lost, and instead, proinflammatory signaling through the angiotensin II type 1 receptor prevails (panel 3). Complement deposition in the setting of COVID-19 results in assembly of the MAC, which permeabilizes the endothelial cell membrane (panel 4). (B) SARS-CoV-2 contains (1) S protein, which binds ACE2 facilitating endocytosis, (2) membrane protein (M protein) and envelope protein (E protein), which reside in the viral membrane and (3) nucleocaspid protein that encapsulates SARS-CoV-2 single stranded RNA (ssRNA). SARS-CoV-2 elicits a prothrombotic transformation of the endothelium that results in occlusion of microcirculation by several mechanisms: (1) Loss of glycocalyx integrity provides SARS-CoV-2 better access to endothelial cell receptors. (2) Decreased cytoprotective signaling results in loss of barrier fortification and increased endothelial damage, dysfunction, and phosphatidylserine exposure (yellow outline). (3) Neutrophil-platelet aggregates are found in the circulation in COVID-19 and facilitate the release of NETs from neutrophils. (4) Activation of platelets and vWF release results in formation of platelet-rich thrombi. (5) Fibrin formation on the endothelium results from activation of both the intrinsic and extrinsic pathways. (6) Generation of TF-bearing microparticles (small purple circles) from activated macrophages also contributes to fibrin formation. (7) Endothelial-leukocyte interactions and loss of barrier function enable arrest and transmigration of leukocytes from the endothelium. (8) Permeabilization of membranes by complement deposition results in endothelial cell damage. (C) Several molecular processes contribute to the prothrombotic transformation of the endothelium. Upregulation of tissue factor in activated endothelium along with downregulation of TM and EPCR results in the activation of factor X to factor Xa (panel 1). Externalization of plasma membrane phosphatidylserine (yellow) facilitates assembly of the prothrombinase complex, resulting in the generation of thrombin from prothrombin (panel 2). Upregulation of leukocyte adhesion molecules including VCAM, ICAM, E-selectin, and P-selectin promotes the association of leukocytes with endothelium and facilitates their transmigration into surrounding tissue (panel 3). Endothelial dysfunction interferes with production of NO and PGI2, and secretion of vWF from Weibel-Palade bodies (WPB, blue) facilitates recruitment of platelets to the endothelial surface. Platelets bind to vWF primarily via GPIbα of the GPIbα-GPIbβ-IX-V complex. Platelet-platelet aggregation is mediated largely through the binding of fibrinogen by integrin αIIbβ3 (panel 4). vWF, von Willebrand factor; NETs, neutrophil extracellular traps.
Mechanisms by which SARS-CoV-2 induces the prothrombotic transformation of the endothelium. (A) In severe systemic SARS-CoV-2 infection, endothelilal cytoprotective mechanisms are lost. The glycocalyx is degraded by metalloproteases (MMPs) and heparanases (panel 1). Several cytoprotective signaling pathways are downregulated. Featured in panel 2 is loss of Tie2 signaling, which occurs following the release of angiopoietin-2 from endothelial stores. High concentrations of angiopoietin-2 displace angiopoietin-1, with loss of clustering and decreased Tie2 phosphorylation resulting in loss of cytoprotective signaling. The spike protein of SARS-CoV-2 binds to ACE2, resulting in its endocytosis and loss from the cell surface. This prevents the cleavage of angiotensin II into Ang(1-7). The cytoprotective signaling of Ang(1-7) is therefore lost, and instead, proinflammatory signaling through the angiotensin II type 1 receptor prevails (panel 3). Complement deposition in the setting of COVID-19 results in assembly of the MAC, which permeabilizes the endothelial cell membrane (panel 4). (B) SARS-CoV-2 contains (1) S protein, which binds ACE2 facilitating endocytosis, (2) membrane protein (M protein) and envelope protein (E protein), which reside in the viral membrane and (3) nucleocaspid protein that encapsulates SARS-CoV-2 single stranded RNA (ssRNA). SARS-CoV-2 elicits a prothrombotic transformation of the endothelium that results in occlusion of microcirculation by several mechanisms: (1) Loss of glycocalyx integrity provides SARS-CoV-2 better access to endothelial cell receptors. (2) Decreased cytoprotective signaling results in loss of barrier fortification and increased endothelial damage, dysfunction, and phosphatidylserine exposure (yellow outline). (3) Neutrophil-platelet aggregates are found in the circulation in COVID-19 and facilitate the release of NETs from neutrophils. (4) Activation of platelets and vWF release results in formation of platelet-rich thrombi. (5) Fibrin formation on the endothelium results from activation of both the intrinsic and extrinsic pathways. (6) Generation of TF-bearing microparticles (small purple circles) from activated macrophages also contributes to fibrin formation. (7) Endothelial-leukocyte interactions and loss of barrier function enable arrest and transmigration of leukocytes from the endothelium. (8) Permeabilization of membranes by complement deposition results in endothelial cell damage. (C) Several molecular processes contribute to the prothrombotic transformation of the endothelium. Upregulation of tissue factor in activated endothelium along with downregulation of TM and EPCR results in the activation of factor X to factor Xa (panel 1). Externalization of plasma membrane phosphatidylserine (yellow) facilitates assembly of the prothrombinase complex, resulting in the generation of thrombin from prothrombin (panel 2). Upregulation of leukocyte adhesion molecules including VCAM, ICAM, E-selectin, and P-selectin promotes the association of leukocytes with endothelium and facilitates their transmigration into surrounding tissue (panel 3). Endothelial dysfunction interferes with production of NO and PGI2, and secretion of vWF from Weibel-Palade bodies (WPB, blue) facilitates recruitment of platelets to the endothelial surface. Platelets bind to vWF primarily via GPIbα of the GPIbα-GPIbβ-IX-V complex. Platelet-platelet aggregation is mediated largely through the binding of fibrinogen by integrin αIIbβ3 (panel 4). vWF, von Willebrand factor; NETs, neutrophil extracellular traps.
Loss of cytoprotective signaling
In the setting of inflammation, angiopoietin-2 is released from endothelial cell Weibel-Palade bodies and displaces angiopoietin-1 from Tie2, resulting in dephosphorylation of Tie2 and inhibition of antithrombotic signaling (Figure 3A, panel 2). The binding of SARS-CoV-2 to and endocytosis of ACE2 results in its endocytosis and increased ACE/ACE2 ratio, resulting in decreased Ang(1-7) formation and decreased Mas1 signaling, promoting a thrombotic phenotype (Figure 3A, panel 3). A potent Ang(1-7) analog is being evaluated for its ability to correct the Ang(1-7) deficiency (clinicaltrials.gov #NCT04419610). Although cleavage of PAR1 by APC promotes cytoprotective signaling, activation of prothrombin at the endothelial cell surface leads to thrombin-mediated cleavage of PAR1, which promotes prothrombotic and proinflammatory signaling. Evidence that these prothrombotic mechanisms occur in COVID-19 patients is derived from plasmas that show increased angiopoietin-2 (Table 1), which correlated with worse clinical outcomes and increased soluble Tie2, which results from shedding of Tie2 by metalloprotease activity.52-55 Tie2 activation by AKB-9778 in COVID-19 patients is a potential therapeutic approach (clinicaltrials.gov #NCT04511650).
Complement activation
SARS-CoV-2 elicits activation of complement via the classical pathway and the lectin pathway (Figure 3A, panel 4). Secreted N protein dimers may directly activate MASP-2, an enzyme of the lectin pathway that is capable of cleaving prothrombin to form thrombin.56 The MAC induces endothelium to actively secrete vWF and assemble the prothrombinase complex,57,58 and C5a induces endothelial P-selectin expression.59 Complement has been associated with microvascular occlusion in the setting of severe COVID-19 based on autopsy findings.60 Unlike patients with active autoimmune diseases, COVID-19 patients do not typically have reduced circulating levels of complement proteins. However, elevation of C3a levels at admission correlate with disease severity,61 as do decreases in serum C3 and C4 concentrations, which also associate with prothrombin time.62 Likewise, C4 levels are inversely correlated with d-dimer levels.62 The C5 inhibitor eculizamab and the C3 inhibitor AMY-101 have been used in COVID-19 patients.63,64 However, a study evaluating the long-acting monoclonal antibody directed at C5, ravulizumab, in severe COVID-19 patients was paused owing to lack of efficacy.65 The lectin pathway inhibitor narsoplimab (an antibody against MASP-2) has shown efficacy in a small study of COVID-19 patients.66
NET formation
As discussed in “Vascular Inflammation induced by COVID-19,” activation of the endothelium results in the upregulation of several leukocyte adhesion molecules, including E-selectin, P-selectin, ICAM-1, and VCAM-1.39 These receptors recruit macrophages and neutrophils. Activation of macrophages results in the expression of TF both on the activated macrophage and on microparticles elaborated by the macrophage (Figure 3B). Interference with leukocyte recruitment using crizanlizumab, which targets P-selectin, has been evaluated (clinicaltrials.gov #NCT04435184). In addition to secreting cytokines, activated neutrophils release their DNA, creating NETs, which stimulate activation of the contact pathway (Figure 3B). Activated neutrophils also express TF.67 In clinical studies, plasma myeloperoxidase-DNA levels correlated with severity of COVID-19, and neutrophils from COVID-19 patients demonstrate NETs at baseline.67,68
Fibrin formation
Early in the pandemic, investigators noted that marked elevations of d-dimer were associated with thrombotic events and poor outcomes.69,70 Loss of the anticoagulant mechanisms, upregulation of tissue factor (Figure 3C, panel 1), phosphatidylserine exposure with assembly of the prothrombinase complex (Figure 3C, panel 2), leukocyte recruitment (Figure 3C, panel 3) with subsequent generation of macrophage extracellular vesicles, and NET formation all contribute to fibrin generation. Plasma from patients with severe COVID-19 induces downregulation of genes encoding for thrombomodulin, EPCR, TFPI, and TFPI and upregulation of TF transcript levels.54 Evaluation of plasma levels in severe COVID-19 patients show increased levels of soluble thrombomodulin and TFPI, suggesting shedding (Table 1).54,71 Both tPA and PAI-1 levels are elevated in severe COVID-19 and associated with poor outcome (Table 1).72-74 Nonetheless, recombinant tPA is being evaluated as a therapeutic in severe COVID-19 (clinicaltrials.gov #NCT04640194, #NCT04356833).
Platelet accumulation and vWF release
Platelets continually signal to the endothelium, and the effect of this interaction is context-dependent. Bioactive factors released from platelets can stabilize the endothelial barrier or promote prothrombotic and proinflammatory endothelial changes.75 Effects of SARS-CoV-2 infection on platelet number and function are well-documented (reviewed in65) and several lines of evidence demonstrate a hyperreactive platelet phenotype in patients with severe COVID-19.76-79 Specifically, platelets from patients with COVID-19 demonstrated increased levels of MRP8/14, which when secreted, leads to a proinflammatory endothelial phenotype and weakens barrier function.80 Conversely, inflamed endothelium promotes platelet activation. Platelet inhibitory NO and PGI2 release are reduced in systemic infection.65 Furthermore, stimulation by cytokines, PAMPs, DAMPs, and proteases in the setting of COVID-19 activates the endothelium to release vWF from Weibel-Palade bodies. vWF binds GPIbα on platelets (Figure 3C, panel 4), endothelial αVβ3, which binds fibrinogen, and CD40, which binds platelet CD40 ligand. These mechanisms facilitate the adhesion of platelets to activated endothelium and contribute to microvascular occlusion as has been observed in autopsy specimens of lungs from patients who died of COVID-19.81 To this end, extensive evidence suggests that elevated levels of vWF, including high molecular weight multimers and a relative deficiency ADAMTS13, are powerful biomarkers predictive of adverse outcomes in severe COVID-19.82
Endothelial barrier function and vascular tone in COVID-19
The critical endothelium barrier between the blood compartment and the extracellular space is compromised following severe SARS-CoV-2 infection (Figure 4A). COVID-19 pneumonia is characterized by diffuse, bilateral ground-glass opacities on computed tomography scanning,83 a radiographic finding suggesting widespread fluid filling of alveoli. The presence of pulmonary edema implies a breakdown of endothelial barrier integrity and in more extreme cases leads to diffuse alveolar damage characteristic of severe COVID-19 pathology.84
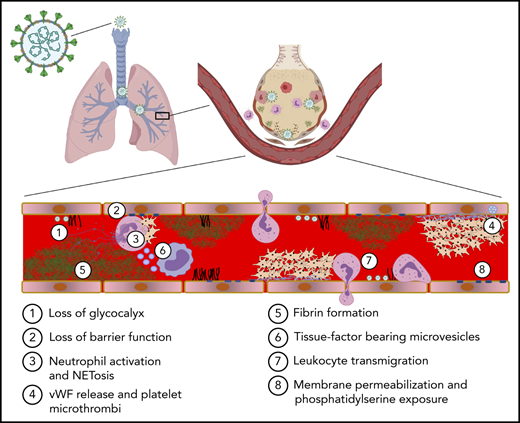
![Mechanism of endothelial barrier function and disruption in COVID-19. (A) The alveolar-capillary interface regulates gas exchange, performs essential barrier functions, maintains blood flow and hemostasis, and controls leukocyte trafficking. Noxious stimuli including cell death, leukocyte activation, protease release, thrombosis, and hypoxia induce endothelial inflammation, which promotes fluid and cellular extravasation that when unchecked can lead to pulmonary edema and ventilation/perfusion mismatch. (B) Severe COVID-19 is associated with cleavage of endothelial cell receptors from the cell surface. Proteases such as matrix metalloproteinase (MMPs), disintegrin and metalloproteinases (ADAMs), and serine proteases promote loss of barrier function directly via cleavage of junctional molecules such as VE-cadherin or indirectly by cleaving cytoprotective receptors such as Tie2 or thrombomodulin (TM) from the cell surface. Transendothelial migration of neutrophils and monocytes is a major source of these proteases. Cleavage of the glycocalyx may induce vascular leak via generation of hyaluronic acid fragments which activate CD44 to promote endothelial permeability. (C) Vasoactive molecules upregulated in COVID-19 may compromise endothelial barrier function and dysregulate vascular tone. Thrombin cleavage of PAR1 results in phosphorylation of myosin light chain (MLC), leading to reorganization of the actin cytoskeleton into contractile stress fibers. Endothelial barrier destabilization is further promoted by activation of integrin β1 by Angpt-2 secreted from Weibel-Palade bodies (WPB). Induction of bradykinin receptor 1 (B1R) during inflammation, combined with excessive kallikrein-kinin activation and the persistence of bradykinin and its breakdown products (eg, Des-Arg[9]-BK) due to ACE2 deficiency may result in excessive vasodilation and edema. (D) Cytokines such as IL-1β, IL-6, and TNFα, DAMPs, PAMPs, and VEGF present in plasma during severe COVID-19 result in reorganization of VE-cadherin away from junctional sites, promoting vascular leak. VEGF induces VE-cadherin phosphorylation, targeting the protein for internalization. Cytokine activation of NF-kB and JAK/STAT3 results in upregulation of leukocyte adhesion proteins such as E-selectin, VCAM-1, and ICAM-1, which mediate leukocyte recruitment and transendothelial migration. (E) Microvascular thrombosis further impairs tissue oxygenation, which activates hypoxia-inducible factor (HIF) to express VEGF, which directly promotes vascular permeability, and Angpt-2 and VE-PTP, which synergize to block Tie2 signaling. Loss of laminar flow and high shear stress from thrombosis or dysregulation of vascular tone inhibits cytoprotective KLF2/4 signaling, which further suppresses the vasodilatory capacity of the endothelium by inhibiting NO synthase and C-natriuretic peptide and decreasing Tie2 mRNA. AT1R, Angiotensin receptor 1; CNP, C-type natriuretic peptide; mRNA, messenger RNA; TEM, transendothelial migration; VEGF, vascular endothelial growth factor.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/3/10.1182_blood.2021012250/5/m_bloodbld2021012250cf4.png?Expires=1765132459&Signature=Qdu4NE5Ks2qnRXGl~2XTj5rhyjZ60CSfpTdaIGGxP80gn5~FhUCvJO-VlrsFGs8nWi8BltpGdMfxxCPMNzmL3RYNtbZtU7p8dvTogGBbPPJ4IOy8zcylkd8lQ5-3b61n9NzCubghkVinGuW-xa0nWaDngg11mQYdFyk-qdoBT3gncO6VhmR~p-Q6j-SgQKQcj6Vvs53DfGacan36wBtMVgZhYDKbQJLH0ybLZGs6TAznoAU2Xmytc9yUbCCBer9L0D6q~bWHM7v8L4-Xn65rklgWomWZdOrGDRDeDqCBFuohlxThQJc0G02m2mhgB3gVTY1CPJxuVuAM2nvZHuMeFQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Mechanism of endothelial barrier function and disruption in COVID-19. (A) The alveolar-capillary interface regulates gas exchange, performs essential barrier functions, maintains blood flow and hemostasis, and controls leukocyte trafficking. Noxious stimuli including cell death, leukocyte activation, protease release, thrombosis, and hypoxia induce endothelial inflammation, which promotes fluid and cellular extravasation that when unchecked can lead to pulmonary edema and ventilation/perfusion mismatch. (B) Severe COVID-19 is associated with cleavage of endothelial cell receptors from the cell surface. Proteases such as matrix metalloproteinase (MMPs), disintegrin and metalloproteinases (ADAMs), and serine proteases promote loss of barrier function directly via cleavage of junctional molecules such as VE-cadherin or indirectly by cleaving cytoprotective receptors such as Tie2 or thrombomodulin (TM) from the cell surface. Transendothelial migration of neutrophils and monocytes is a major source of these proteases. Cleavage of the glycocalyx may induce vascular leak via generation of hyaluronic acid fragments which activate CD44 to promote endothelial permeability. (C) Vasoactive molecules upregulated in COVID-19 may compromise endothelial barrier function and dysregulate vascular tone. Thrombin cleavage of PAR1 results in phosphorylation of myosin light chain (MLC), leading to reorganization of the actin cytoskeleton into contractile stress fibers. Endothelial barrier destabilization is further promoted by activation of integrin β1 by Angpt-2 secreted from Weibel-Palade bodies (WPB). Induction of bradykinin receptor 1 (B1R) during inflammation, combined with excessive kallikrein-kinin activation and the persistence of bradykinin and its breakdown products (eg, Des-Arg[9]-BK) due to ACE2 deficiency may result in excessive vasodilation and edema. (D) Cytokines such as IL-1β, IL-6, and TNFα, DAMPs, PAMPs, and VEGF present in plasma during severe COVID-19 result in reorganization of VE-cadherin away from junctional sites, promoting vascular leak. VEGF induces VE-cadherin phosphorylation, targeting the protein for internalization. Cytokine activation of NF-kB and JAK/STAT3 results in upregulation of leukocyte adhesion proteins such as E-selectin, VCAM-1, and ICAM-1, which mediate leukocyte recruitment and transendothelial migration. (E) Microvascular thrombosis further impairs tissue oxygenation, which activates hypoxia-inducible factor (HIF) to express VEGF, which directly promotes vascular permeability, and Angpt-2 and VE-PTP, which synergize to block Tie2 signaling. Loss of laminar flow and high shear stress from thrombosis or dysregulation of vascular tone inhibits cytoprotective KLF2/4 signaling, which further suppresses the vasodilatory capacity of the endothelium by inhibiting NO synthase and C-natriuretic peptide and decreasing Tie2 mRNA. AT1R, Angiotensin receptor 1; CNP, C-type natriuretic peptide; mRNA, messenger RNA; TEM, transendothelial migration; VEGF, vascular endothelial growth factor.
Mechanism of endothelial barrier function and disruption in COVID-19. (A) The alveolar-capillary interface regulates gas exchange, performs essential barrier functions, maintains blood flow and hemostasis, and controls leukocyte trafficking. Noxious stimuli including cell death, leukocyte activation, protease release, thrombosis, and hypoxia induce endothelial inflammation, which promotes fluid and cellular extravasation that when unchecked can lead to pulmonary edema and ventilation/perfusion mismatch. (B) Severe COVID-19 is associated with cleavage of endothelial cell receptors from the cell surface. Proteases such as matrix metalloproteinase (MMPs), disintegrin and metalloproteinases (ADAMs), and serine proteases promote loss of barrier function directly via cleavage of junctional molecules such as VE-cadherin or indirectly by cleaving cytoprotective receptors such as Tie2 or thrombomodulin (TM) from the cell surface. Transendothelial migration of neutrophils and monocytes is a major source of these proteases. Cleavage of the glycocalyx may induce vascular leak via generation of hyaluronic acid fragments which activate CD44 to promote endothelial permeability. (C) Vasoactive molecules upregulated in COVID-19 may compromise endothelial barrier function and dysregulate vascular tone. Thrombin cleavage of PAR1 results in phosphorylation of myosin light chain (MLC), leading to reorganization of the actin cytoskeleton into contractile stress fibers. Endothelial barrier destabilization is further promoted by activation of integrin β1 by Angpt-2 secreted from Weibel-Palade bodies (WPB). Induction of bradykinin receptor 1 (B1R) during inflammation, combined with excessive kallikrein-kinin activation and the persistence of bradykinin and its breakdown products (eg, Des-Arg[9]-BK) due to ACE2 deficiency may result in excessive vasodilation and edema. (D) Cytokines such as IL-1β, IL-6, and TNFα, DAMPs, PAMPs, and VEGF present in plasma during severe COVID-19 result in reorganization of VE-cadherin away from junctional sites, promoting vascular leak. VEGF induces VE-cadherin phosphorylation, targeting the protein for internalization. Cytokine activation of NF-kB and JAK/STAT3 results in upregulation of leukocyte adhesion proteins such as E-selectin, VCAM-1, and ICAM-1, which mediate leukocyte recruitment and transendothelial migration. (E) Microvascular thrombosis further impairs tissue oxygenation, which activates hypoxia-inducible factor (HIF) to express VEGF, which directly promotes vascular permeability, and Angpt-2 and VE-PTP, which synergize to block Tie2 signaling. Loss of laminar flow and high shear stress from thrombosis or dysregulation of vascular tone inhibits cytoprotective KLF2/4 signaling, which further suppresses the vasodilatory capacity of the endothelium by inhibiting NO synthase and C-natriuretic peptide and decreasing Tie2 mRNA. AT1R, Angiotensin receptor 1; CNP, C-type natriuretic peptide; mRNA, messenger RNA; TEM, transendothelial migration; VEGF, vascular endothelial growth factor.
Breakdown of barrier function occurs secondary to loss of both glycocalyx and endothelial cell-cell adhesion complexes. Loss of glycocalyx, as described above, is an early defining feature of endothelial dysfunction in sepsis42 and in COVID-19.85-89 Cleavage of glycocalyx promotes entry of plasma proteins and increases water permeability, and hyaluronic acid fragments may signal through CD44 to activate Rho/Rho-associated kinase (ROCK) and promote cytoskeletal reorganization (Figure 4B).90 Elevation of the angiopoietin-2/angiopoietin-1 ratio promotes barrier destabilization by activation of integrin β1 (Figure 4C).91 In severe COVID-19, there is marked elevation of angiopoietin-2, which scales with disease severity and may predict survival (Table 1).16,52-54,71,92-94 In the quiescent state, activation of S1P receptors also converges to stabilize VE-cadherin and cortical actin.95 Serum S1P levels were inversely correlated with COVID-19 severity and a predictor of intensive care unit (ICU) admission and survival.96 In contrast to barrier-protective Tie2 and S1P signaling, VEGF receptor ligation by VEGF-A induces rapid and reversable phosphorylation of VE-cadherin, which triggers it for endocytosis and loss of barrier function (Figure 4D). VEGF-A levels are increased in COVID-19 and correlate with disease severity.85,97,98 The anti-VEGF antibody bevacizumab improved oxygenation in a small study of patients with COVID-19 acute respiratory distress syndrome (ARDS)83 and is being investigated in larger trials (clinicaltrials.gov #NCT04822818).
As a result of this dysfunctional signaling, loss of barrier function in postcapillary venules, which is required for normal immune surveillance, goes unchecked and extends to the capillary bed, resulting in catastrophic fluid extravasation. In cultured endothelium, exposure to SARS-CoV-2 spike protein results in a decrease in endothelial cell adhesion proteins, including VE-cadherin and JAM-A,99 and loss of transendothelial electrical resistance, an integrated in vitro assay of endothelial barrier function.100,101 Plasma from patients with severe COVID-19 also reduced transendothelial electrical resistance on cultured endothelial monolayers.102 Using an alveolus-on-a-chip model, infection of pulmonary epithelial cells induced a robust inflammatory response that damaged endothelial cells and their intercellular junctions.103
The use of ACE2 by the SARS-CoV-2 virus to gain entry into host cells has led to extensive speculation that COVID-19 is associated with dysregulation of the renin-angiotensin and kallikrein-kinin pathways. Relative or local ACE2 deficiency at the site of SARS-CoV-2 entry COVID-19 may result in persistence of bradykinin and bradykinin breakdown products such as Des-Arg(9)-BK.104 Combined with inflammation-induced expression of B1R, this potentiation of bradykinin can promote excessive vasodilation and edema105 (Figure 4C). Bronchioalveolar fluid from patients with COVID-19 demonstrated marked downregulation mRNA encoding C-1 inhibitor, which normally blocks plasma kallikrein activation, and upregulation of B1R mRNA.106 C1 esterase inhibitor, B2R antagonist icatibant, and plasma kallikrein inhibitor lanadelumab are being investigated in clinical trials for prevention of severe COVID-19 pneumonia (clinicaltrials.gov #NCT05010876, #NCT04530136, #NCT04978051, and #NCT04422509).
Microvascular thrombosis may indirectly compromise barrier function and vascular tone. Loss of laminar flow from vessel obstruction or occlusion may decrease expression of cytoprotective KLF2, which maintains endothelial cell quiescence and barrier function (Figure 4E).107 Increased thrombin production activates PAR1, resulting in phosphorylation of myosin light chain, and a contractile endothelial cell phenotype that promotes vascular leak (Figure 4C).108
Clinical manifestations of COVID-19 vasculopathy
Patients with preexisting vascular disease including hypertension, diabetes, and coronary artery disease are more likely to develop severe disease following SARS-CoV-2 infection and, not surprisingly, are more likely to suffer cardiovascular complications as a result. The profound vascular dysfunction of COVID-19 may explain unique aspects of COVID-19 lung disease and its identity as a multiorgan syndrome (Figure 5).109 As the lungs are the initial point of viral entry, the pulmonary vasculature bears the brunt of the initial inflammation. However, damage to the pulmonary vasculature and other vascular beds may persist long after the infection has cleared. Therefore, vasculopathy is closely linked not only to acute COVID-19 but also has implications for residual “long COVID-19” symptoms that can persist for months or more.

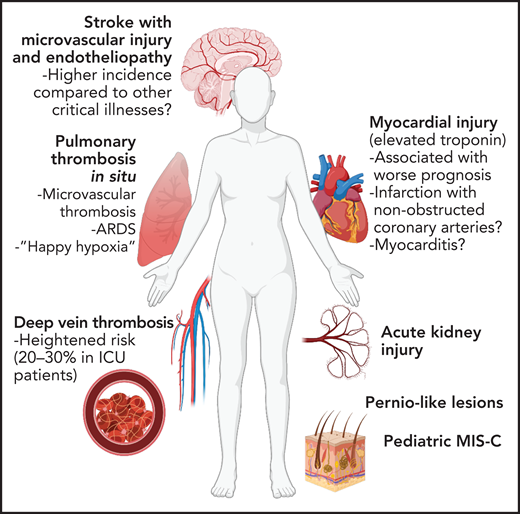
Vascular complications of COVID-19. Due to widespread endothelial dysfunction and thrombosis, COVID-19 has the ability to affect nearly any organ. Pulmonary disease, specifically ARDS, may be driven in large part by vascular dysfunction and microvascular thrombosis. Increased thrombotic events, most notably venous thromboembolism but also myocardial infarction and stroke, have been reported. Vascular dysfunction may contribute neurologic, renal, and dermatologic manifestations of COVID-19, as well as post-COVID sequelae such as multisystem inflammatory syndrome in children (MIS-C).
Vascular complications of COVID-19. Due to widespread endothelial dysfunction and thrombosis, COVID-19 has the ability to affect nearly any organ. Pulmonary disease, specifically ARDS, may be driven in large part by vascular dysfunction and microvascular thrombosis. Increased thrombotic events, most notably venous thromboembolism but also myocardial infarction and stroke, have been reported. Vascular dysfunction may contribute neurologic, renal, and dermatologic manifestations of COVID-19, as well as post-COVID sequelae such as multisystem inflammatory syndrome in children (MIS-C).
Lung complications
COVID-19 pulmonary disease is characterized by ARDS, which is broadly defined by bilateral, multifocal noncardiogenic pulmonary edema and severe hypoxemia and has several infectious and noninfectious etiologies. It is a matter of debate whether COVID-19 ARDS differs from ARDS of other causes.110-112 The lower expression of ACE2 receptors, compared with α2,6 sialic acid residue influenza virus receptors, for example, may result in slower SARS-CoV-2 pulmonary spread compared with influenza and therefore more heterogeneous lung pathology and a more protracted disease course.113 Asynchronous pulmonary involvement may increase the total duration of cytokine exposure a patient experiences113 and increase risk of vascular dysfunction, which may have both pulmonary and systemic ramifications. Diffuse microvascular dysfunction may result in profound ventilation/perfusion mismatch in absence of synchronous airway-alveolar involvement and may contribute to the phenomenon of silent or “happy” hypoxia reported in patients.114 Although the final endpoint of diffuse alveolar damage of COVID-19 may not differ from that of other forms of ARDS,115 microvascular dysfunction with or without thrombosis likely contributes to early hypoxemic respiratory failure and a vicious cycle of hypoxia-inflammation that potentiates vasculopathy and thrombosis.116
Thrombosis
Venous thromboembolism
The incidence of venous thromboembolism (VTE) reported in hospitalized patients is highly variable depending on the severity of illness and method of study. Several biases may have been introduced, such as whether patients were screened for VTE vs a work-up driven by symptoms or studies where only patients who had imaging were included. Despite these caveats, VTE risk does appear heightened in patients hospitalized with COVID-19, at ∼10% to 15% incidence overall, 5% to 8% in non-ICU patients, and 20% to 30% in ICU patients.117-120 In patients with COVID-19 who do not require hospitalization, however, there is no signal that VTE risk is elevated.121,122
Myocardial infarction, stroke, and other arterial thrombotic events
Among patients hospitalized with COVID-19, several studies have demonstrated that elevations in circulating cardiac enzymes are associated with a worse prognosis and are more prevalent in patients with underlying cardiovascular disease.123,124 Whether this myocardial injury is due to myocarditis, myocardial infarction (from atherosclerotic plaque rupture, oxygen supply-demand mismatch with fixed coronary stenosis, or microvascular dysfunction), or stress-induced or critical illness cardiomyopathy is often difficult to ascertain in these studies. Mechanisms of cardiac dysfunction in COVID-19 are multifactorial and incompletely understood. Direct viral infection of vascular pericytes, or perhaps endothelial cells, is predicted to cause extensive vascular dysfunction, allowing extravasation of leukocytes and fluid into the myocardium (ie, myocarditis) or promoting thrombosis in coronary arteries or the microvasculature (ie, myocardial infarction). The cytokine milieu may also lead to direct myocardial toxicity (ie, stress cardiomyopathy). Furthermore, right ventricular dysfunction or failure (cor pulmonale) may result from elevated pulmonary vascular pressures due to pulmonary embolism or diffuse microvascular thrombosis and ARDS.65
The incidence of SARS-CoV-2 myocarditis is unclear given lack of biopsy, imaging, or autopsy data to distinguish inflammatory infiltrate from sterile myocardial damage. Myocarditis most commonly presents concurrently with respiratory disease but may be present several weeks following pulmonary symptoms or, in rare cases, as isolated myocardial involvement.125 Myocardial infarction (cardiac enzyme elevation with associated ischemic ECG changes) in patients with COVID-19 is often accompanied by a lack of a culprit epicardial stenosis on coronary angiogram.126,127 This finding suggests that in COVID-19, bland thrombus, vasospasm, or microvascular disease are more frequently responsible for myocardial infarction compared with classical atherosclerotic plaque rupture or erosion observed in traditional acute coronary syndromes.
Acute viral infection is known to increase the incidence of myocardial infarction and stroke.128 Despite this expected increased risk, the incidence of ischemic stroke may be higher among patients hospitalized with COVID-19 compared with those hospitalized with influenza.129 Reports have documented younger patients without known cardiovascular risk factors experiencing stroke in the setting of COVID-19.130,131 More recent and larger analyses suggest that the incidence of stroke in patients hospitalized with COVID-19 to be ∼1% to 2% and is associated with the presence of established stroke risk factors such as hypertension, diabetes, hyperlipidemia, atrial fibrillation, and congestive heart failure.121,127,129,132 Although its correlation with symptoms is unclear, postmortem analysis of brains from COVID-19 patients demonstrated microvascular injury including weakening of endothelial basal lamina, vascular occlusion, and plasma protein extravasation.133 Furthermore, patients presenting with stroke in the setting of COVID-19 demonstrate evidence of endotheliopathy compared with non–COVID-19 stroke controls.134
Other manifestations
Pernio-like lesions on the hands and feet (aka “COVID toes”) have gained much attention; however, the association with SARS-CoV-2 infection is controversial, and additional causes (eg, lockdown-related cold exposure) may contribute.135,136 Endothelial inflammation and microvascular thrombosis have been found on biopsy of these lesions, however.135,137 Acute kidney injury is a common morbidity associated with severe COVID-19 and portends a worse prognosis.138 It is likely that endothelial dysfunction and microvascular thrombosis play a role in some proportion of COVID-19 renal injury.138 Thrombosis in the mesenteric and portal vasculature have been reported, leading to bowel ischemia.139
“Long COVID,” MIS-C, and vascular involvement
A significant number of patients report persistent or new symptoms despite clearance of SARS-CoV-2 infection. Postacute COVID-19, or so-called “long COVID,” has multiple manifestations including dyspnea, fatigue, exercise intolerance, myocardial inflammation, headaches, malaise, myalgias, and difficulty concentrating.140 The cause of these symptoms is poorly understood, but data are emerging that vasculopathy may persist well into the recovery phase of COVID-19. Elevated D-dimer levels are found in plasma from convalescent patients months after initial COVID-19 symptoms.141,142 Continued immune activation and elevated levels of circulating endothelial cells suggest a persistent endotheliopathy,143 and evidence of procoagulant endothelial dysfunction has also been described.144 Whether these alterations correlate with postacute COVID-19 symptoms has yet to be determined. In one study, young patients without underlying vascular disease demonstrate impaired endothelial physiologic function and increased arterial stiffness despite clearance of SARS-CoV-2 infection.145
In the pediatric population, SARS-CoV-2 infection is usually mild or asymptomatic. However, some develop a subsequent hyperimmune-dysregulatory condition known as MIS-C. This syndrome has elements similar to toxic shock syndrome and Kawasaki disease, including cardiac dysfunction and coronary artery aneurysm formation, and is thought to be mediated in part by endothelial injury and microvascular dysfunction.146
Conclusion
The vasculopathy of COVID-19 is a critical driver of the disease process. Many of the mechanisms that provoke endothelial dysfunction have been deduced based on analogy to previously studied viral infections. Others have been inferred from evaluation of COVID-19 patient plasmas and in vitro testing of SARS-CoV-2, its components, and plasmas in cultured endothelium. Whether the vascular changes in COVID-19 differ in significant ways from those of other forms of severe sepsis has been questioned. It is clear that there is substantial overlap with sepsis induced by other respiratory viruses. However, some aspects of the vascular response and thrombotic tendency are either more pronounced in COVID-19 or unique to the disease. That several of the aforementioned endothelial mechanisms of vasculopathy are common in sepsis, regardless of etiology, makes them no less important in COVID-19 and has directed the use of approved and experimental agents to alleviate the damaging vascular effects of COVID-19.
Acknowledgments
The authors would like to acknowledge the many publications relevant to the area of vasculopathy in COVID-19 that they were not able to include in this review owing to space limitations. R.F. has received support from the National Heart, Lung, and Blood Institute (R35HL135775, R01HL125275) and the Foundation for Women's Wellness. A.A.S. and R.F. received support from the Massachusetts Consortium for Pathogen Readiness Evergrande COVID-19 Response Fund. A.A.S. received support from the John S. LaDue Memorial Fellowship for Cardiovascular Research from Harvard Medical School and from the North American Thrombosis Forum COVID-19 Research Initiative.
Funding support for this article was provided by the National Heart, Lung, and Blood Institute (R35HL135775 and R01HL125275).
Authorship
Contribution: R.F., K.E., and A.A.S. developed conceptual framework for the manuscript; R.F. and A.A.S. wrote the manuscript; and R.F., K.E., and A.A.S. edited the manuscript.
Conflict-of-interest disclosure: R.F. has financial interests in and is a founder of PlateletDiagnostics. His interests are reviewed and managed by Beth Israel Deaconess Medical Center in accordance with their conflict-of-interest policies. The remaining authors declare no competing financial interests.
Correspondence: Robert Flaumenhaft, Division of Hemostasis and Thrombosis, Department of Medicine, Beth Israel Deaconess Medical Center, 330 Brookline Avenue, Boston, MA 02215; e-mail: rflaumen@bidmc.harvard.edu.
Send data sharing requests via e-mail to the corresponding author.



![Mechanism of endothelial barrier function and disruption in COVID-19. (A) The alveolar-capillary interface regulates gas exchange, performs essential barrier functions, maintains blood flow and hemostasis, and controls leukocyte trafficking. Noxious stimuli including cell death, leukocyte activation, protease release, thrombosis, and hypoxia induce endothelial inflammation, which promotes fluid and cellular extravasation that when unchecked can lead to pulmonary edema and ventilation/perfusion mismatch. (B) Severe COVID-19 is associated with cleavage of endothelial cell receptors from the cell surface. Proteases such as matrix metalloproteinase (MMPs), disintegrin and metalloproteinases (ADAMs), and serine proteases promote loss of barrier function directly via cleavage of junctional molecules such as VE-cadherin or indirectly by cleaving cytoprotective receptors such as Tie2 or thrombomodulin (TM) from the cell surface. Transendothelial migration of neutrophils and monocytes is a major source of these proteases. Cleavage of the glycocalyx may induce vascular leak via generation of hyaluronic acid fragments which activate CD44 to promote endothelial permeability. (C) Vasoactive molecules upregulated in COVID-19 may compromise endothelial barrier function and dysregulate vascular tone. Thrombin cleavage of PAR1 results in phosphorylation of myosin light chain (MLC), leading to reorganization of the actin cytoskeleton into contractile stress fibers. Endothelial barrier destabilization is further promoted by activation of integrin β1 by Angpt-2 secreted from Weibel-Palade bodies (WPB). Induction of bradykinin receptor 1 (B1R) during inflammation, combined with excessive kallikrein-kinin activation and the persistence of bradykinin and its breakdown products (eg, Des-Arg[9]-BK) due to ACE2 deficiency may result in excessive vasodilation and edema. (D) Cytokines such as IL-1β, IL-6, and TNFα, DAMPs, PAMPs, and VEGF present in plasma during severe COVID-19 result in reorganization of VE-cadherin away from junctional sites, promoting vascular leak. VEGF induces VE-cadherin phosphorylation, targeting the protein for internalization. Cytokine activation of NF-kB and JAK/STAT3 results in upregulation of leukocyte adhesion proteins such as E-selectin, VCAM-1, and ICAM-1, which mediate leukocyte recruitment and transendothelial migration. (E) Microvascular thrombosis further impairs tissue oxygenation, which activates hypoxia-inducible factor (HIF) to express VEGF, which directly promotes vascular permeability, and Angpt-2 and VE-PTP, which synergize to block Tie2 signaling. Loss of laminar flow and high shear stress from thrombosis or dysregulation of vascular tone inhibits cytoprotective KLF2/4 signaling, which further suppresses the vasodilatory capacity of the endothelium by inhibiting NO synthase and C-natriuretic peptide and decreasing Tie2 mRNA. AT1R, Angiotensin receptor 1; CNP, C-type natriuretic peptide; mRNA, messenger RNA; TEM, transendothelial migration; VEGF, vascular endothelial growth factor.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/3/10.1182_blood.2021012250/5/m_bloodbld2021012250cf4.png?Expires=1765327484&Signature=OOtoCbPnOUPw8yIhkvQxiFZ0t0tE6GPQj7GEg9szaNpHkbQSN4DJlJoRc1X3io7zCdz~lffYy~krhXRfo9EOV7pGzg2t4ZmCHPGS5x6bzyRBYnusIG3~OpKTxmarxOK4rPqeGPyPVM6Nt4~jE1KFq3iQ57rKFkAbrTrH0bqyFqR6hTF7qBQNj3ZUFoT4epQYA-7tmi-lsXepeU8Ra4YHy4aV4tEn3HoEjEcKZR6Ebg3TvOk2XelZG34D8juW2vU7HXj4vHLbGY23585nvfvNqcnrt8TaDlPt~6cQpx7w5vg8YWyvG4QaWdQ-FggFlNhgPjTrQPzTVgsFA7rRlyEt3w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)