Key Points
Constitutively active AKT is present in high-risk CLL and RT, and active Akt transforms murine CLL toward aggressive lymphoma.
Akt orchestrates development of RT via induction of Notch1 signaling in B cells, fueled by Dll1-expressing T cells.

Abstract
Richter’s transformation (RT) is an aggressive lymphoma that occurs upon progression from chronic lymphocytic leukemia (CLL). Transformation has been associated with genetic aberrations in the CLL phase involving TP53, CDKN2A, MYC, and NOTCH1; however, a significant proportion of RT cases lack CLL phase–associated events. Here, we report that high levels of AKT phosphorylation occur both in high-risk CLL patients harboring TP53 and NOTCH1 mutations as well as in patients with RT. Genetic overactivation of Akt in the murine Eµ-TCL1 CLL mouse model resulted in CLL transformation to RT with significantly reduced survival and an aggressive lymphoma phenotype. In the absence of recurrent mutations, we identified a profile of genomic aberrations intermediate between CLL and diffuse large B-cell lymphoma. Multiomics assessment by phosphoproteomic/proteomic and single-cell transcriptomic profiles of this Akt-induced murine RT revealed an S100 protein-defined subcluster of highly aggressive lymphoma cells that developed from CLL cells, through activation of Notch via Notch ligand expressed by T cells. Constitutively active Notch1 similarly induced RT of murine CLL. We identify Akt activation as an initiator of CLL transformation toward aggressive lymphoma by inducing Notch signaling between RT cells and microenvironmental T cells.
Introduction
During the clinical course of chronic lymphocytic leukemia (CLL), ∼2% to 10% of patients develop an aggressive lymphoma, termed Richter’s Transformation (RT).1 RT typically displays histomorphologic characteristics similar to those of diffuse large B-cell lymphoma (DLBCL), with limited treatment options.2-4 Transformation to RT has been shown to associate with somatic genetic events acquired in the CLL phase, including TP53, CDKN2, MYC, EGR2, or NOTCH1, while observed to be genetically distinct from de novo DLBCL, notably lacking recurrent mutations in genes such as CD79B and BCL2.5,6 However, a significant proportion of cases cannot be attributed to CLL phase–associated somatic genetic events, indicating that they are required but not sufficient for driving the histologic shift.7,8 Previous CLL phase–associated treatment, germline genetics, and aspects of CLL phase biology have also been observed to be RT risk factors, whereas the role of the tumor microenvironment (TME) in the transformation from CLL to RT has not been studied. Moreover, it has yet to be elucidated whether a central mechanism connecting these risk factors exists.
A key determinant in the pathogenesis of CLL is B-cell receptor (BCR) signaling, which mediates disease heterogeneity via either proliferation or anergy.9,10 In the CLL phase, BCR-associated events correlated with transformation to RT include unmutated IGHV (IGHV-U) status and BCR stereotypy,11 as well as the expression of CD49d, CD38, and ZAP70. The BCR induces tonic phosphoinositol-3-kinase (PI3K)/AKT signaling believed to contribute to malignant transformation.12,13 PI3K phosphorylates the secondary messenger PIP2 to PIP3 that activates the membranal kinase PDK-1 to phosphorylate PIP3-bound AKT at the activating residue Thr308.14,15 Downstream effects of AKT include protein synthesis, cell survival, proliferation, and glucose metabolism.15 We therefore hypothesized that AKT activation might be the missing link in the transformation of CLL to RT.
The current study found that: (1) AKT is activated in high-risk CLL and in >50% of patients with RT; (2) constitutively active Akt promotes CLL transformation toward RT in Eµ-TCL1 mice in vivo; (3) Notch1 is activated via constitutive Pi3k/Akt in RT cells; and (4) RT-intrinsic Notch signaling is induced by Dll1-expressing Cd4+ T cells from the TME.
Materials and methods
Mouse strains and patient material
Experiments were performed according to human (medical faculty of the University of Cologne [reference no. 13-091]) and murine (LANUV 84-02.04.2014.A146 and 84-02.04.2019.A009) ethical approvals. Eµ-TCL1 mice and Cd19-Cre mice were described previously and were on the C57/BL6 background.16 To generate the R26-fl-Akt-C strain, a SERCA targeting vector containing a myristoylation TAG Akt-C was introduced into the ROSA26 locus, as previously described.17 Finally, Eµ-TCL1 mice were intercrossed with Cd19-CreAkt-C mice. Furthermore, we examined Cd19-CreFoxO1ADA mice,18 Cd19-CreNotch1-IC mice,19 and Gsk3βS9A/wt mice.20
Cell preparation, immunophenotyping, and multiomics profiling
Murine samples were isolated from total splenocytes, with total splenocytes used for immunophenotyping and single-cell RNA sequencing (scRNA-Seq; 10X Genomics), Cd19+ MACS sorting conducted for whole-exome sequencing (WES), and phosphoproteomic/proteomic analysis. Immunophenotyping was conducted by using a standard panel by flow cytometry. WES and scRNA-Seq experiments were conducted at the Cologne Center for Genomics using standardized protocols. Phosphoproteomic/proteomic analysis was conducted as previously described.21 Notch pathway member expression profiling was conducted via flow cytometry (MACSQuant 10 and VYB).
Immunofluorescence analysis
Murine spleens were fixed in 4% paraformaldehyde. Paraffin-embedded samples were deparaffinized and stained after retrieval. Hematoxylin and eosin staining was performed (Mayer’s Hematoxylin Solution #MHS32-1L and Eosin Y Solution Aqueous #HT110232-1L; Sigma-Aldrich) according to standard protocol and imaged with AxioVision 4.2 (Carl Zeiss MicroImaging). Human sections were deparaffinized and stained with primary antibody anti-pAKT Ser473 (#3787; Cell Signaling). The sections were counterstained with 4′6-diamidino-2-phenylindole.
Immunoblot analysis
Immunoblots of murine and human samples were performed by using standard techniques with the following antibodies: anti-pAKT Ser473 (#4060 and #9271; Cell Signaling), anti-pAKT Thr308 (#4056; Cell Signaling), anti-panAKT (#4685 and #2920; Cell Signaling), pAKT (#4075; Cell Signaling), anti-pGSK3b (#9315; Cell Signaling), anti-calnexin (#208880; EMD Millipore), and anti-actin (#MAB1501; Millipore).
Further details on sample preparation, murine intercrossing, experimental conditions, and bioinformatics analysis are provided in the supplemental Methods (available on the Blood Web site).
Results
Active AKT is increased in high-risk CLL and RT
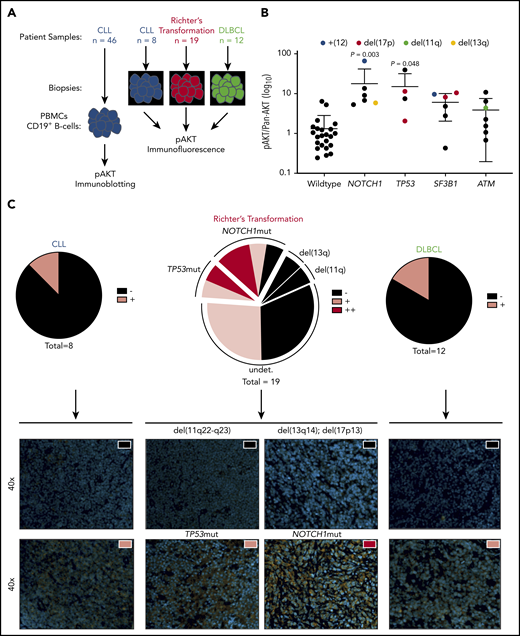
AKT activation in CLL cells isolated from blood of patients is heterogeneous (supplemental Figure 1A-B). To investigate whether AKT activation was associated with high-risk CLL, we screened 46 CLL patient samples with complementary targeted re-sequencing on CLL driver genes for Ser473-phosphorylation of AKT by immunoblot (Figure 1A-B). NOTCH1mut and TP53mut CLL samples had significantly increased AKT activation compared with wild type, whereas no such increase was observed in SF3B1mut and ATMmut cases. Because both NOTCH1 and TP53 have been associated with transformation to RT, we hypothesized that increased AKT activity could be a common mechanism of transformation. To test this theory, we immunohistochemically investigated tumor biopsy specimens from CLL (n = 8), RT (n = 19), and DLBCL (n = 12) for Ser473-phosphorylation of AKT (Figure 1C; supplemental Figure 1C). Here, increased frequency and intensity of AKT activation in RT samples were observed compared with CLL and DLBCL (CLL = 12.5%, DLBCL = 16.7%, RT = 52.6%). Furthermore, we identified that almost 50% of positively stained RT cases with complete genetic characterization carried mutations in TP53 and NOTCH1. These data therefore show that active AKT is often observed in RT, both with and without somatic genetic events gained in the CLL phase.
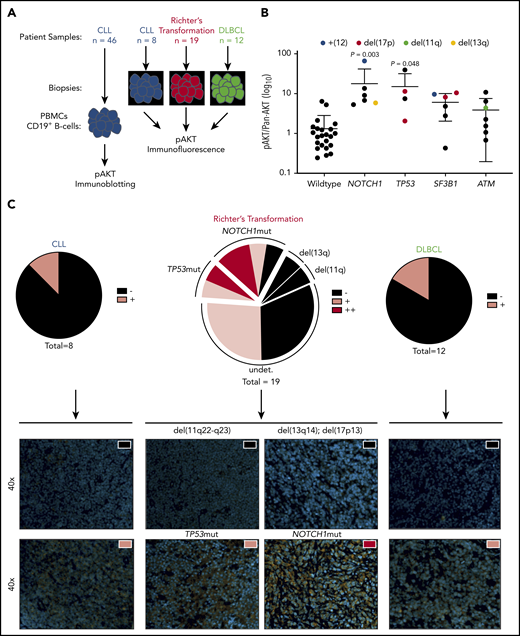
Increased AKT activation in CLL subsets associated with RT and increased frequency in patients with RT. (A) Flow diagram of AKT activation screening setup. (B) Histogram showing pAKT (Ser473)/panAKT expression (AKT activation) from freshly isolated peripheral B cells, in a cohort of patients with CLL stratified according to mutations in NOTCH1, TP53, SF3B1, and ATM (n = 46). Colored dots represent chromosomal aberrations identified via fluorescence in situ hybridization (blue = trisomy12; red = 17p deletion; green = 11q deletion; yellow = 13q deletion). (C) Immunofluorescence imaging of CLL (n = 8), RT (n = 19), and DLBCL (n = 12) for pAKT (Ser473). Proportion of cases defined as negative (black), positive (pink), or double positive (red) shown as pie charts for each entity (upper panel). Representative images from positive and negative cases from each entity (lower panel). All images shown at ×40 magnification. Nuclear staining (4′6-diamidino-2-phenylindole) and pAKT (Ser473) staining shown in orange. Significant differences in AKT activation between wild-type and TP53/NOTCH1/SF3B1/ATM mutated CLL peripheral blood mononuclear cells (PBMCs) by immunoblotting assessed by using one-way analysis of variance, with Tukey multiple comparison correction.
Increased AKT activation in CLL subsets associated with RT and increased frequency in patients with RT. (A) Flow diagram of AKT activation screening setup. (B) Histogram showing pAKT (Ser473)/panAKT expression (AKT activation) from freshly isolated peripheral B cells, in a cohort of patients with CLL stratified according to mutations in NOTCH1, TP53, SF3B1, and ATM (n = 46). Colored dots represent chromosomal aberrations identified via fluorescence in situ hybridization (blue = trisomy12; red = 17p deletion; green = 11q deletion; yellow = 13q deletion). (C) Immunofluorescence imaging of CLL (n = 8), RT (n = 19), and DLBCL (n = 12) for pAKT (Ser473). Proportion of cases defined as negative (black), positive (pink), or double positive (red) shown as pie charts for each entity (upper panel). Representative images from positive and negative cases from each entity (lower panel). All images shown at ×40 magnification. Nuclear staining (4′6-diamidino-2-phenylindole) and pAKT (Ser473) staining shown in orange. Significant differences in AKT activation between wild-type and TP53/NOTCH1/SF3B1/ATM mutated CLL peripheral blood mononuclear cells (PBMCs) by immunoblotting assessed by using one-way analysis of variance, with Tukey multiple comparison correction.
Constitutive activation of Akt via B-cell–specific Akt-C expression in Eµ-TCL1 mice reduces life span, despite similar CLL progression
To investigate whether Akt activity is sufficient to transform CLL to RT, we generated a Rosa26 mouse model in which a loxP-flanked stop cassette precedes an N-terminal myristoylation tagged Akt-C version (supplemental Figure 2A-B). Upon Cd19-Cre–mediated recombination of the loxP-flanked stop cassette, Akt-C expression leads to constitutive B-cell–specific Akt-C activity due to its membranal localization in which Pdk1 activates Akt. We intercrossed Eµ-TCL1tg/wt (Eµ-TCL1) mice with Cd19-Cretg/wt; R26-fl-Akt-C (Cd19-CreAkt-C) to obtain Eµ-TCL1tg/wt; Cd19-Cretg/wt and R26-fl-Akt-C mice (Eµ-TCL1Akt-C) (Figure 2A). Akt-C expression induced a significant increase in pAKT activity in Eµ-TCL1Akt-C vs Eµ-TCL1 mice (supplemental Figure 2C), regardless of the fact that TCL1 is a known interaction partner and promoter of Akt in this mouse model. We followed the disease progression of Eµ-TCL1Akt-C mice in the peripheral blood, observing no significant differences in the number of Cd19+/Cd5+ cells compared with Eµ-TCL1 mice (Figure 2B; supplemental Figure 2D). However, at ∼7 months of age, Eµ-TCL1Akt-C mice quickly developed symptoms reaching euthanization criteria, whereas >80% of Eµ-TCL1 mice remained alive at this time point (median overall survival Eµ-TCL1Akt-C vs Eµ-TCL1, 7.6 vs 11.6 months; P < .001) (Figure 2C). These data suggest that Akt-C leads to a dramatic reduction in overall survival, mimicking the clinical picture observed in patients with RT.
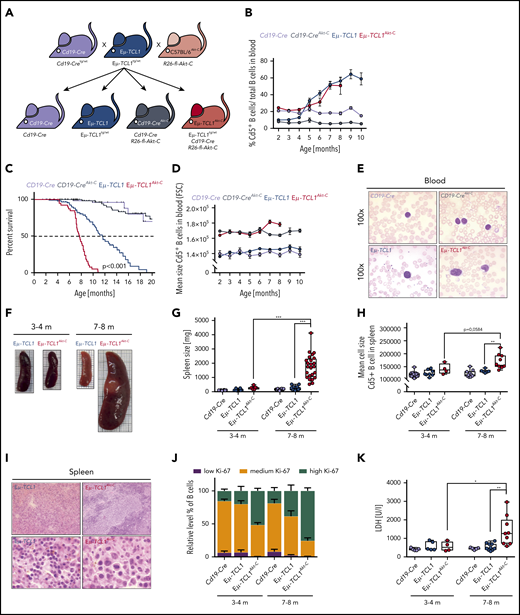
B-cell–specific constitutive activation of Akt in the Eµ-TCL1 mouse model induces aggressive lymphoma with a diffuse large B-cell phenotype in vivo. (A) Breeding scheme required to generate requisite genotypes. (B) Relative quantification of Cd5+ B cells from serial blood samples from Cd19-Cre (black), Cd19-CreAkt-C (gray), Eµ-TCL1 (blue), and Eµ-TCL1Akt-C (red) mice. Blood samples were measured between 2 months and 10 months of age with 1-month intervals. (C) Pairwise Kaplan-Meier overall survival analysis. Significance between groups was tested by using the pairwise log-rank P values. (D) Mean cell size (FSC value) of Cd5+ B cells from peripheral blood measured between 2 and 10 months of age with 1-month intervals. (E) Representative hematoxylin and eosin staining of blood samples. (F) Representative images of spleens from Cd19-Cre (black; top left), Cd19-CreAkt-C (gray; bottom left), Eµ-TCL1 (blue; top right), and Eµ-TCL1Akt-C (red; bottom right) mice aged 3 to 4 months and 7 to 8 months, respectively. (G) Box plot of spleen weight for mice aged 3 to 4 months and 7 to 8 months. (H) Box plot showing the mean cell size (FSC value) within the spleen for mice aged 3 to 4 months and 7 to 8 months. (I) Representative hematoxylin and eosin staining of spleen preparations. (J) Bar graph displaying relative levels of B cells showing low, medium, and high amounts of Ki-67 in mice aged 3 to 4 months and 7 to 8 months. (K) Box plot showing the lactate dehydrogenase (LDH) levels for mice aged 3 to 4 months and 7 to 8 months. *P < .05, **P ≤ .01, ***P ≤ .001 (unpaired, 2-sided Student t test).
B-cell–specific constitutive activation of Akt in the Eµ-TCL1 mouse model induces aggressive lymphoma with a diffuse large B-cell phenotype in vivo. (A) Breeding scheme required to generate requisite genotypes. (B) Relative quantification of Cd5+ B cells from serial blood samples from Cd19-Cre (black), Cd19-CreAkt-C (gray), Eµ-TCL1 (blue), and Eµ-TCL1Akt-C (red) mice. Blood samples were measured between 2 months and 10 months of age with 1-month intervals. (C) Pairwise Kaplan-Meier overall survival analysis. Significance between groups was tested by using the pairwise log-rank P values. (D) Mean cell size (FSC value) of Cd5+ B cells from peripheral blood measured between 2 and 10 months of age with 1-month intervals. (E) Representative hematoxylin and eosin staining of blood samples. (F) Representative images of spleens from Cd19-Cre (black; top left), Cd19-CreAkt-C (gray; bottom left), Eµ-TCL1 (blue; top right), and Eµ-TCL1Akt-C (red; bottom right) mice aged 3 to 4 months and 7 to 8 months, respectively. (G) Box plot of spleen weight for mice aged 3 to 4 months and 7 to 8 months. (H) Box plot showing the mean cell size (FSC value) within the spleen for mice aged 3 to 4 months and 7 to 8 months. (I) Representative hematoxylin and eosin staining of spleen preparations. (J) Bar graph displaying relative levels of B cells showing low, medium, and high amounts of Ki-67 in mice aged 3 to 4 months and 7 to 8 months. (K) Box plot showing the lactate dehydrogenase (LDH) levels for mice aged 3 to 4 months and 7 to 8 months. *P < .05, **P ≤ .01, ***P ≤ .001 (unpaired, 2-sided Student t test).
Akt-C in Eµ-TCL1 mice drives RT
To evaluate whether Eµ-TCL1Akt-C mice had undergone transformation from CLL to RT, we examined the morphology of the malignant B cells in the peripheral blood in monthly intervals, as well as in tumor sections of spleens from mice at an age at which 100% of mice were still at risk in the Kaplan-Meier survival analysis (3-4 months) or at the median survival time of the Eµ-TCL1Akt-C mouse model (7-8 months). Although the relative cell size (FSC; forward scatter) of Cd5+ blood-derived B cells was enlarged in Eµ-TCL1Akt-C mice compared with Eµ-TCL1 mice at all time points, an additional increase in relative cell size was observed at months 7 and 8 (Figure 2D). Cytomorphologic evaluation of blood smears of Eµ-TCL1Akt-C mice revealed pleomorphic blastoid cells with large degranulated nuclei and increased cytoplasm, whereas blood-derived cells in Eµ-TCL1 mice showed round and condensed nucleoplasm (Figure 2E). Strikingly, Eµ-TCL1Akt-C mice exhibited splenomegaly at 7 to 8 months of age, accompanied by the appearance of large Cd19+/Cd5+ blastoid cells as well as higher lactate dehydrogenase levels and reduced thrombocyte counts (Figure 2F-H,K; supplemental Figure 2E). In Eµ-TCL1Akt-C mice, hematoxylin and eosin staining revealed large blastoid cells with pleomorphic nuclei and prominent nucleoli predominating while containing frequent mitotic figures and high Ki-67 positivity (Figure 2I-J; supplemental Table 1). Importantly, these results were replicated when using Cγ1-Cretg/wt mice as an alternative B-cell–specific Cre-allele to express Akt-C in Eµ-TCL1 cells, recapitulating the RT phenotype with all features observed in Eµ-TCL1Akt-C mice (supplemental Figure 3A-F). Taken together, our data unequivocally show that constitutively active Akt in Eµ-TCL1Akt-C mice transforms CLL toward an aggressive DLBCL-type lymphoma defined as RT with clinical features concordant with observations in human patients.
Eµ-TCL1Akt-C mutational landscape represents an intermediary state between CLL and DLBCL
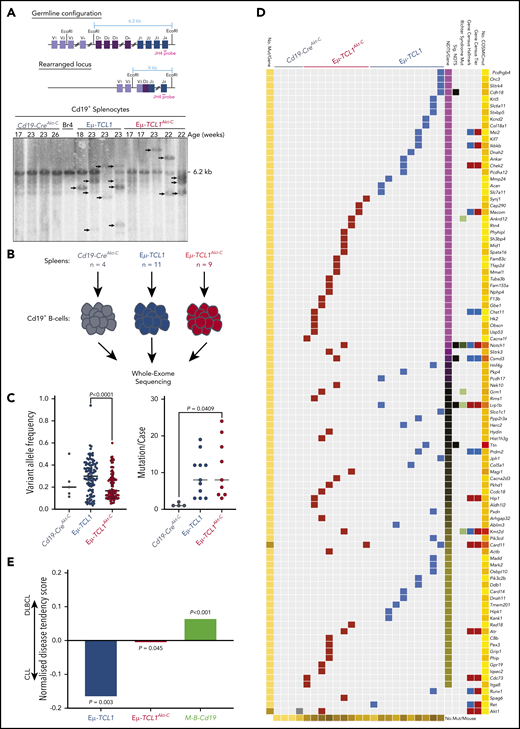
To ascertain whether Akt-C drives changes at the genomic level, we analyzed IgH gene rearrangements by Southern blot, revealing similar oligo-clonal characteristics in Eµ-TCL1Akt-C compared with Eµ-TCL1 mice (Figure 3A). To investigate the impact of Akt-C, we analyzed B cells from spleens of 23 mice (Eµ-TCL1Akt-C, n = 9 [aged 8-9 months]); Eµ-TCL1, n = 10 [aged 10-12 months], Cd19-CreAkt-C, n = 4) by WES (Figure 3B). Eµ-TCL1Akt-C were composed of mutations with significantly reduced variant allele frequency compared with Eµ-TCL1 mice (P < .0001) while sharing a similar mutation burden (Figure 3C). In concordance with previous WES studies on the Eµ-TCL1 mouse model,22 we observed a distinct lack of recurrently mutated genes, which was also the case for Eµ-TCL1Akt-C mice (Figure 3D; supplemental Table 3.1). To elucidate the potential functional relevance of these individual gene mutations, we assessed the tendency of these genes to be mutated in human CLL and DLBCL using the cancer browser of the Catalogue Of Somatic Mutations In Cancer.23 As an internal control, mutation data were included from a recently described murine ABC-DLBCL model (Cd19-Cre–driven combined Myd88 and Bcl2 aberrations [Cd19-Cretg/wt;Rosa26LSL.BCL2.IRES.GFP/wt;Myd88c-p.L252P/wt], hereafter M-B-Cd19).24 Eµ-TCL1–mutated genes were significantly enriched for genes mutated in CLL (P = .003), whereas the M-B-Cd19 model was significantly enriched for genes mutated in DLBCL (P < .001). Strikingly, gene mutations in Eµ-TCL1Akt-C mice exhibited an intermediary profile between CLL and DLBCL (Figure 3E). Collectively, despite the absence of recurrent mutations, we observed an altered mutational profile in Eµ-TCL1Akt-C mice intermediary between CLL and DLBCL.
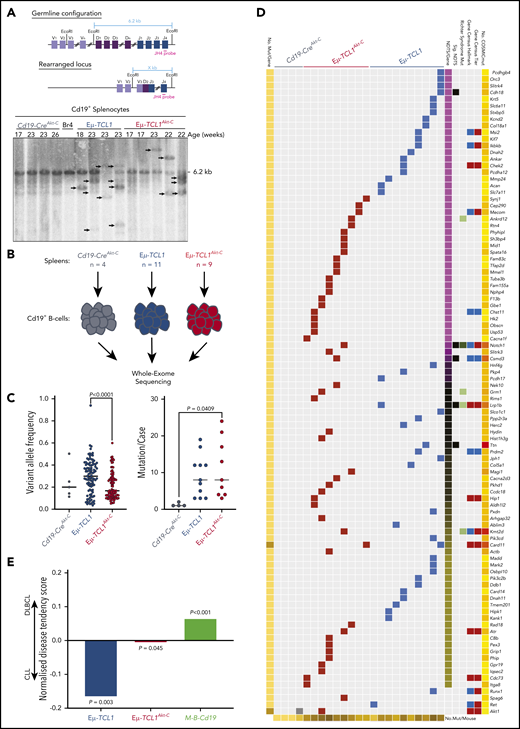
Catalogue Of Somatic Mutations In Cancer (COSMIC)-annotated Eµ-TCL1Akt-C–mutated genes define the model as a genetically intermediary form between CLL and DLBCL. (A) Schematic strategy to identify clonal VDJ rearrangements. Using the JH4 probe on EcoRI-digested genomic DNA results in a 6.2 kb germline configuration band in Southern blot analysis. Clonal VDJ rearrangements indicative of B-cell leukemia and lymphoma can be identified by clear bands other than 6.2 kb (upper panel). Southern blot analysis of Cd19+ MACS-purified B cells derived from indicated mice using the aforementioned strategy. (B) Flow diagram of WES experimental setup. (C) Distribution of variant allele frequencies (left panel) and mutations per case (right panel) between Cd19-CreAkt-C (n = 4), Eµ-TCL1 (n = 10), and Eµ-TCL1Akt-C (n = 9) mice. (D) Waterfall plot of COSMIC-annotated mutations from Cd19-CreAkt-C (n = 4), Eµ-TCL1 (n = 10), and Eµ-TCL1Akt-C (n = 9) mice. Mutations clustered according to normalized disease tendency scores of COSMIC CLL and DLBCL mutation data (pink = CLL-associated; yellow = DLBCL-associated; black = equal distribution). Mutations were annotated for occurrence in COSMIC Cancer Gene Census Tiers (blue = animal 1; red = animal 2), COSMIC Cancer Gene Census Hallmark classification (blue = across all cancers; red = leukemia/lymphoma-specific), observed to be mutated in RT. (E) Bar graph representing the mean normalized disease tendency scores per genotype (blue = Eµ-TCL1; red = Eµ-TCL1Akt-C; green = M-B-Cd19). P values were generated via binomial testing, presuming equal chances of each gene being mutated in either CLL or DLBCL (0.5).
Catalogue Of Somatic Mutations In Cancer (COSMIC)-annotated Eµ-TCL1Akt-C–mutated genes define the model as a genetically intermediary form between CLL and DLBCL. (A) Schematic strategy to identify clonal VDJ rearrangements. Using the JH4 probe on EcoRI-digested genomic DNA results in a 6.2 kb germline configuration band in Southern blot analysis. Clonal VDJ rearrangements indicative of B-cell leukemia and lymphoma can be identified by clear bands other than 6.2 kb (upper panel). Southern blot analysis of Cd19+ MACS-purified B cells derived from indicated mice using the aforementioned strategy. (B) Flow diagram of WES experimental setup. (C) Distribution of variant allele frequencies (left panel) and mutations per case (right panel) between Cd19-CreAkt-C (n = 4), Eµ-TCL1 (n = 10), and Eµ-TCL1Akt-C (n = 9) mice. (D) Waterfall plot of COSMIC-annotated mutations from Cd19-CreAkt-C (n = 4), Eµ-TCL1 (n = 10), and Eµ-TCL1Akt-C (n = 9) mice. Mutations clustered according to normalized disease tendency scores of COSMIC CLL and DLBCL mutation data (pink = CLL-associated; yellow = DLBCL-associated; black = equal distribution). Mutations were annotated for occurrence in COSMIC Cancer Gene Census Tiers (blue = animal 1; red = animal 2), COSMIC Cancer Gene Census Hallmark classification (blue = across all cancers; red = leukemia/lymphoma-specific), observed to be mutated in RT. (E) Bar graph representing the mean normalized disease tendency scores per genotype (blue = Eµ-TCL1; red = Eµ-TCL1Akt-C; green = M-B-Cd19). P values were generated via binomial testing, presuming equal chances of each gene being mutated in either CLL or DLBCL (0.5).
Phosphoproteomic/proteomic profiling identifies overexpression of S100a4, increased phosphorylation of Hes1, and increased activation of kinases associated with Notch signaling in Eµ-TCL1Akt-C mice
To assess the impact of active Akt on protein amount and phosphorylation, splenic B-cell–derived proteins of Eµ-TCL1Akt-C and Eµ-TCL1 mice were investigated by using proteomics and phosphoproteomics (Figure 4A). The total amount of captured proteins remained unchanged between both genotypes, whereas Eµ-TCL1Akt-C mice showed a significant increase in the abundance of phosphorylated peptides (Figure 4B; supplemental Tables 4.1 and 4.2). The calcium-binding protein S100a4, which has previously been observed to be associated with metastatic cancer progression, was the most significantly upregulated protein in the proteomics dataset.25 Interestingly, we observed downregulation of Nfatc1, a negative transcriptional regulator of S100a4 expression, with significantly increased phosphorylation in Eµ-TCL1Akt-C mice.26-28 In line with this evidence, Nfatc1 deficiency in Eµ-TCL1 mice culminated in an aggressive lymphoma comparable to RT. In the current study, a specific role for downstream Lyn/Syk/Akt/Erk was observed to prevent nuclear Nfatc1 leading to RT.29,30 Consistently, we observed that Lyn was significantly phosphorylated (Figure 4C) and had a marginally increased upstream kinase score in Eµ-TCL1Akt-C mice (Figure 4D; supplemental Table 4.3), suggesting that constitutively active Akt signaling may affect Nfatc1 transcriptional activity. In addition, we observed increased phosphorylation of Hes1, a prominent Notch1 target gene.31 To reduce the dataset to the kinome level, an upstream kinase analysis was conducted by using the PhosphoSitePlus database and a simplified equation inspired by Beekhof et al (supplemental Methods).32 This analysis identified increased activation of Wee1, Cdk4, and Csnk2b, kinases that have previously been shown to be associated with Notch signaling (Figure 4D-E).33,34 Notably, a reciprocal regulation between Notch and Nfatc1 has been reported in numerous cancer types.35,36
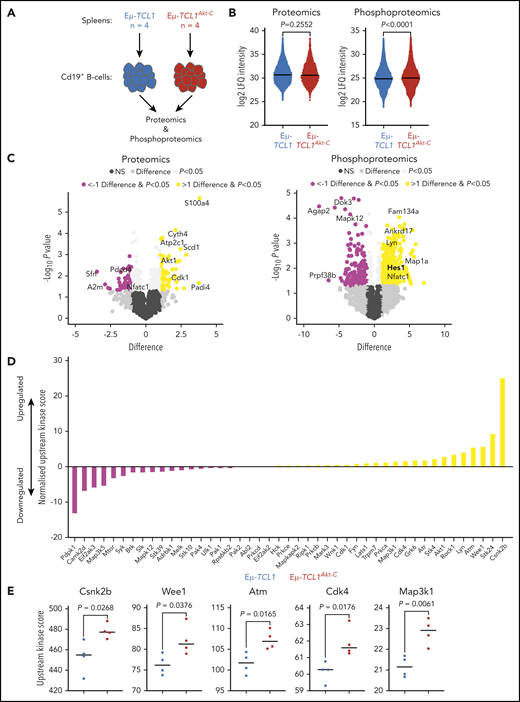
Overexpression of S100a4, increased phosphorylation of Hes1, and increased activation of Cdk4 and Wee1 infer Notch1 activation. (A) Flow diagram of phosphoproteomic/proteomic experimental setup. (B) Log2 label-free quantification (LFQ) intensity values of all proteins captured (left panel) or phosphopeptides (right panel) across all mice per genotype. (C) Volcano plots representing the difference in log2 LFQ intensity of proteins (left panel) and phosphopeptides (right panel) between Eµ-TCL1 (n = 4) and Eµ-TCL1Akt-C (n = 4) mice. Significant DE proteins/phosphopeptides defined as difference >1 and P < .05 (upregulated in Eµ-TCL1Akt-C = yellow; downregulated = pink). (D) Normalized upstream kinase scores for all 45 kinases with available PhosphoSitePlus kinome data. Bars represent the difference in upstream kinase scores between Eµ-TCL1 (n = 4) and Eµ-TCL1Akt-C (n = 4) (upregulated in Eµ-TCL1Akt-C = yellow; downregulated = pink). (E) Upstream kinase scores from the top significantly upregulated kinases in Eµ-TCL1Akt-C mice. Significant differences between groups assessed by using Student t test, significant proteins/peptides defined as P < .05 as well as difference >1 /<1 for phosphoproteomics/proteomics, whereas for upstream kinase analysis. P < .05 kinases were considered significant. NS, not significant.
Overexpression of S100a4, increased phosphorylation of Hes1, and increased activation of Cdk4 and Wee1 infer Notch1 activation. (A) Flow diagram of phosphoproteomic/proteomic experimental setup. (B) Log2 label-free quantification (LFQ) intensity values of all proteins captured (left panel) or phosphopeptides (right panel) across all mice per genotype. (C) Volcano plots representing the difference in log2 LFQ intensity of proteins (left panel) and phosphopeptides (right panel) between Eµ-TCL1 (n = 4) and Eµ-TCL1Akt-C (n = 4) mice. Significant DE proteins/phosphopeptides defined as difference >1 and P < .05 (upregulated in Eµ-TCL1Akt-C = yellow; downregulated = pink). (D) Normalized upstream kinase scores for all 45 kinases with available PhosphoSitePlus kinome data. Bars represent the difference in upstream kinase scores between Eµ-TCL1 (n = 4) and Eµ-TCL1Akt-C (n = 4) (upregulated in Eµ-TCL1Akt-C = yellow; downregulated = pink). (E) Upstream kinase scores from the top significantly upregulated kinases in Eµ-TCL1Akt-C mice. Significant differences between groups assessed by using Student t test, significant proteins/peptides defined as P < .05 as well as difference >1 /<1 for phosphoproteomics/proteomics, whereas for upstream kinase analysis. P < .05 kinases were considered significant. NS, not significant.
scRNA-Seq identifies an aggressive B-cell subpopulation in Eµ-TCL1Akt-C defined by cell–cell interactions and overactivation of Notch
To analyze the transcriptomic profiles of the aggressive subclones arising from indolent CLL in the Eµ-TCL1Akt-C mice, we characterized splenocytes via scRNA-Seq (Figure 5A). After implementing per-sample preprocessing and clustering using standard settings, we integrated all samples, leading to a dataset of 33 224 cells composed of 21 clusters (Figure 5B; supplemental Figure 5.1; supplemental Table 5.1). Genotype-specific cell number increases were observed within clusters, notably in the malignant B cells, in which 5 clusters were observed to be enriched or shared between Eµ-TCL1Akt-C and Eµ-TCL1 (clusters encircled in red), whereas 2 clusters were observed to be enriched in Eµ-TCL1 mice (clusters encircled in blue). To unravel the transcriptional differences between Eµ-TCL1Akt-C and Eµ-TCL1 cells within the enriched/shared clusters (RT-C1 to RT-C5), we conducted differential expression (DE) analysis across all cells, identifying 570 significantly DE genes supplemental Table 5.2). We then tested for gene ontology (GO) enrichment using all 570 significant genes, from which we calculated the disease tendency of each GO term to be composed of genes significantly upregulated in either Eµ-TCL1Akt-C or Eµ-TCL1 mice (supplemental Table 5.3). This analysis identified an enrichment of cell–cell interaction-associated GO terms composed of Eµ-TCL1Akt-C significant genes, with TCR/BCR signaling also being increased in Eµ-TCL1Akt-C mice. We delved deeper into the cell–cell interaction GO terms via network visualization (Figure 5C), identifying a profile of Eµ-TCL1Akt-C upregulated genes associated with S100 signaling (S100a4, S100a9), Notch signaling (Hes1, Cd44, Itga4 [Cd49d], Cd86, Flna, Zmiz1), and Pi3k/Akt signaling (Pik3r1), as well as the immune checkpoint gene Cd274 (Pd-l1) (Figure 5D). Significantly upregulated genes exhibited variable transcription factor binding sites for FoxO1, Nfatc1, and Rbpjκ (Notch1) in their 2 kb upstream promoter region, suggesting a direct transcriptional link between these transcription factors in Akt-C–expressing cells. Importantly, increased S100a4 expression was validated in B cells from Eµ-TCL1Akt-C mice that were dependent on Nfatc1 and Rbjκ/Notch regulation but not on FoxO1 in B cells derived from Gsk3βS9A/+(Nfatc1 nuclear-excluded), Cd19-Cretg/wt; R26-fl-Notch1-IC (Cd19-CreNotch-IC nuclear-active Notch1), and Cd19-Cretg/wt; R26-fl-FoxO1ADA (Cd19-CreFoxO1ADA nuclear FoxO1) (supplemental Figure 5.2A-B). Furthermore, we validated increased expression for Hes1 at the RNA level and for Pd-l1 at the protein level in B cells from Eµ-TCL1Akt-C mice. In conclusion, we identified a specific aggressively transformed cluster in the Eµ-TCL1Akt-C mice appearing among the indolent leukemic B cells defined by Akt-mediated cell–cell interaction associated transcriptional regulation as well as a Notch/S100-protein signature corresponding to the aggressive nature of RT.
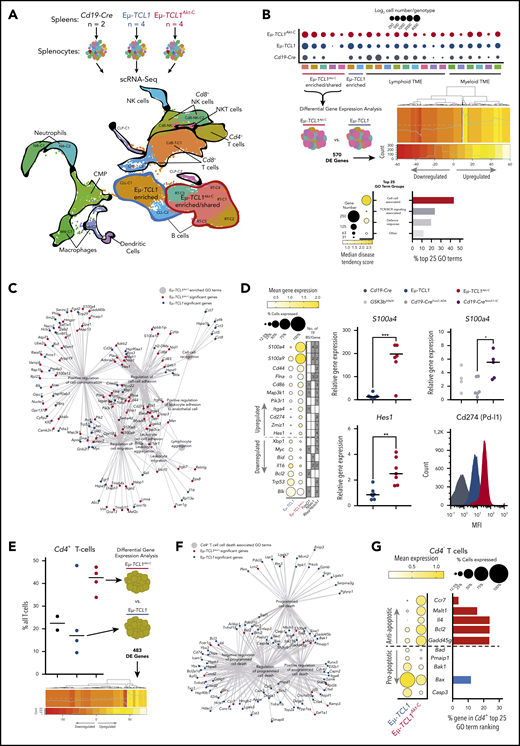
Cell–cell interactions and overexpression of S100a4/Hes1/Cd274(Pd-l1) defines RT gene signature in Eµ-TCL1Akt-C–enriched B-cell clusters. (A) Flow diagram of scRNA-Seq experimental setup (upper panel). Integrated UMAP from Cd19-Cre (n = 2), Eµ-TCL1 (n = 4), and Eµ-TCL1Akt-C (n = 4) mice (n = 33 224 cells). Demarked clusters represent B-cell tumor clusters enriched within either Eµ-TCL1 (blue) or Eµ-TCL1Akt-C (red) and the TME (black). (B) Dot plot representing the number of cells appearing in each cluster per genotype (upper panel). Graphical representation of differential gene expression analysis and heatmap showing the normalized percent cell expression changes of significantly differentially expressed genes (middle panel). Dot plot representing the median disease tendency score of GO term groups with the dot size representing the number of genes associated with all GO terms (lower panel, left graph). Bar chart representing the percentage of GO term groups of the top 25 Eµ-TCL1Akt-C–associated GO terms (lower panel, right graph). (C) Network visualization of all cell–cell associated GO terms from the analysis conducted in B. GO terms denoted as large gray circles, Eµ-TCL1Akt-C and Eµ-TCL1 significant genes denoted as small red and blue circles, respectively, and gray lines represent the association between genes and GO terms. (D) Dot plot of selected cell–cell associated GO term genes with biological links to Notch signaling, Pi3k/Akt signaling, or RT. Size of the dots represents the percentage of cells with a normalized expression value >1, with the color of the dots representing the mean expression of each gene across all cells within the cluster. Additional annotation beside the dot plot represents the number of transcription factor–binding sites for all 25 genes for FoxO1, Nfatc1, and Rbpjκ/Notch1, respectively (left panel). Messenger RNA expression validation in Cd19+ splenocytes for S100a4 and Hes1 genes in Eµ-TCL1 and Eµ-TCL1Akt-C mice, as well as S100a4 in Gsk3βS9A/+ (Nfatc1 nuclear excluded), Cd19-Cretg/wt; R26-fl-Notch1-IC (Cd19-CreNotch1-IC nuclear Notch1), and Cd19-Cretg/wt; R26-fl-FoxO1ADA (Cd19-CreFoxO1ADA nuclear FoxO1) via quantitative polymerase chain reaction. Protein expression validation in CD19+ splenocytes for Cd274 (Pd-l1) in Cd19-Cre, Eµ-TCL1, and Eµ-TCL1Akt-C mice (right panel). (E) Percentage of Cd4+ T cells per mouse per genotype as a function of all T cells (left panel). Graphical representation of differential gene expression analysis of Cd4+ T cells (right panel) and heatmap showing the normalized percent cell expression changes of significantly differentially expressed genes (lower panel panel). (F) Network visualization of all cell death–associated GO terms from the analysis conducted in E. GO terms denoted as large gray circles, Eµ-TCL1Akt-C and Eµ-TCL1 significant genes denoted as small red and blue circles, and gray lines represent the association between genes and GO terms. (G) Dot plot of selected cell death–associated GO term genes. Size of the dots represents the percentage of cells with a normalized expression value >1, with the color of the dots representing the mean expression of each gene across all cells within the cluster (left panel). Bar chart representing in percent terms how often the genes presented occur in the top 25 Eµ-TCL1Akt-C–associated GO terms, with red bars for Eµ-TCL1Akt-C significant genes and blue bars for Eµ-TCL1 significant genes. *P < .05, **P ≤ .01, ***P ≤ .001 (unpaired, 2-sided Student t test). CLP, common lymphoid progenitor; CMP, common myeloid progenitor; MFI, mean fluorescent intensity; NK, natural killer; NKT, natural killer T.
Cell–cell interactions and overexpression of S100a4/Hes1/Cd274(Pd-l1) defines RT gene signature in Eµ-TCL1Akt-C–enriched B-cell clusters. (A) Flow diagram of scRNA-Seq experimental setup (upper panel). Integrated UMAP from Cd19-Cre (n = 2), Eµ-TCL1 (n = 4), and Eµ-TCL1Akt-C (n = 4) mice (n = 33 224 cells). Demarked clusters represent B-cell tumor clusters enriched within either Eµ-TCL1 (blue) or Eµ-TCL1Akt-C (red) and the TME (black). (B) Dot plot representing the number of cells appearing in each cluster per genotype (upper panel). Graphical representation of differential gene expression analysis and heatmap showing the normalized percent cell expression changes of significantly differentially expressed genes (middle panel). Dot plot representing the median disease tendency score of GO term groups with the dot size representing the number of genes associated with all GO terms (lower panel, left graph). Bar chart representing the percentage of GO term groups of the top 25 Eµ-TCL1Akt-C–associated GO terms (lower panel, right graph). (C) Network visualization of all cell–cell associated GO terms from the analysis conducted in B. GO terms denoted as large gray circles, Eµ-TCL1Akt-C and Eµ-TCL1 significant genes denoted as small red and blue circles, respectively, and gray lines represent the association between genes and GO terms. (D) Dot plot of selected cell–cell associated GO term genes with biological links to Notch signaling, Pi3k/Akt signaling, or RT. Size of the dots represents the percentage of cells with a normalized expression value >1, with the color of the dots representing the mean expression of each gene across all cells within the cluster. Additional annotation beside the dot plot represents the number of transcription factor–binding sites for all 25 genes for FoxO1, Nfatc1, and Rbpjκ/Notch1, respectively (left panel). Messenger RNA expression validation in Cd19+ splenocytes for S100a4 and Hes1 genes in Eµ-TCL1 and Eµ-TCL1Akt-C mice, as well as S100a4 in Gsk3βS9A/+ (Nfatc1 nuclear excluded), Cd19-Cretg/wt; R26-fl-Notch1-IC (Cd19-CreNotch1-IC nuclear Notch1), and Cd19-Cretg/wt; R26-fl-FoxO1ADA (Cd19-CreFoxO1ADA nuclear FoxO1) via quantitative polymerase chain reaction. Protein expression validation in CD19+ splenocytes for Cd274 (Pd-l1) in Cd19-Cre, Eµ-TCL1, and Eµ-TCL1Akt-C mice (right panel). (E) Percentage of Cd4+ T cells per mouse per genotype as a function of all T cells (left panel). Graphical representation of differential gene expression analysis of Cd4+ T cells (right panel) and heatmap showing the normalized percent cell expression changes of significantly differentially expressed genes (lower panel panel). (F) Network visualization of all cell death–associated GO terms from the analysis conducted in E. GO terms denoted as large gray circles, Eµ-TCL1Akt-C and Eµ-TCL1 significant genes denoted as small red and blue circles, and gray lines represent the association between genes and GO terms. (G) Dot plot of selected cell death–associated GO term genes. Size of the dots represents the percentage of cells with a normalized expression value >1, with the color of the dots representing the mean expression of each gene across all cells within the cluster (left panel). Bar chart representing in percent terms how often the genes presented occur in the top 25 Eµ-TCL1Akt-C–associated GO terms, with red bars for Eµ-TCL1Akt-C significant genes and blue bars for Eµ-TCL1 significant genes. *P < .05, **P ≤ .01, ***P ≤ .001 (unpaired, 2-sided Student t test). CLP, common lymphoid progenitor; CMP, common myeloid progenitor; MFI, mean fluorescent intensity; NK, natural killer; NKT, natural killer T.
scRNA-Seq identifies that prosurvival signaling drives enrichment of Cd4+ T cells in the TME of Eµ-TCL1Akt-C mice
To further explore the potential biological importance of upregulation of the cell–cell interaction signature in the malignant B cells, we analyzed the TME in the integrated single-cell dataset, identifying an enrichment of Cd4+ T cells in Eµ-TCL1Akt-C mice compared with Eµ-TCL1 and Cd19-Cre mice (Figure 5E). DE gene analysis between Eµ-TCL1Akt-C and Eµ-TCL1 cells was conducted, identifying 483 significantly DE genes; upon GO analysis, we observed a lack of GO terms with a disease tendency score indicative of Eµ-TCL1Akt-C enrichment (B cells = 24% >0.1, Cd4+ T cells = 0.42% >0.1) (supplemental Tables 5.4 to 5.5; supplemental Figure 5.2C). Therefore, we assessed the GO terms enriched for genes significantly upregulated in Eµ-TCL1 cells, identifying programmed cell death–associated GO terms to be a predominant feature in Eµ-TCL1 mice (Figure 5F). However, we observed that the distribution of proapoptotic to antiapoptotic genes differed between the two genotypes, with Eµ-TCL1Akt-C cells proportionally composed of more antiapoptotic genes (supplemental Figure 5.2D). Importantly, these genes included central players in prosurvival signaling, such as Bcl2, as well as other genes involved in T-cell biology (Ccr7 and Il4) (Figure 5G). Finally, these antiapoptotic Eµ-TCL1Akt-C upregulated genes were often found in the GO terms that were indicative of Eµ-TCL1Akt-C enrichment, further supporting their importance in the milieu of upregulated genes in the Cd4+ T cells of the Eµ-TCL1Akt-C TME. In summary, we identified a TME population that may have been elicited to proliferate with increased prosurvival capacity via the RT malignant clone.
Overactivation of Notch signaling in Eµ-TCL1Akt-C RT cells is supported by concomitant Dll1 overexpression on Cd4+ T cells
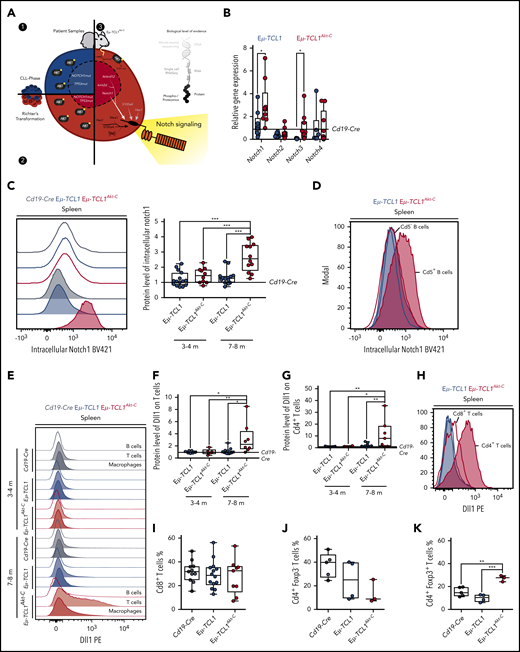
Our experimental data from CLL and RT patient biopsy specimens, in combination with the multiomics assessment of the Eµ-TCL1Akt-C mouse model, provide various lines of evidence that active Akt signaling drives Notch activation in CLL to RT transformation (Figure 6A). To consolidate these findings, we assessed the RNA expression of Notch receptors (Notch1-4) in the malignant B cells, observing significant increases in expression of Notch1 and Notch3 in Eµ-TCL1Akt-C vs Eµ-TCL1 mice as well as Notch target gene regulation (Figure 6B; supplemental Figure 6A). Moreover, significantly higher protein levels of Notch1 were observed in Eµ-TCL1Akt-C mice displaying a fully transformed RT phenotype at 7 to 8 months of age but not in mice at 3 to 4 months (Figure 6C). Importantly, only the Cd5+ RT cells revealed Notch1 expression, whereas Cd5– B cells from the same mouse exhibited levels similar to CLL cells (Figure 6D). Notch1 is activated after ligand-binding presented on adjacent cells, namely Jagged (Jag) and delta-like (Dll) proteins.37-39 To specify the ligand-presenting cells, we analyzed protein levels of Dll1 in splenic cells. Interestingly, we observed significantly increased levels of Dll1 ligand on T cells, but not on other TME cells nor on the RT cells of Eµ-TCL1Akt-C mice displaying a fully transformed RT phenotype with specific expression on Cd4+ T cells (Figure 6E-H; supplemental Figure 6B-C). Because Os et al showed that Cd4+ T cells support the survival and proliferation of CLL cells, we decided to assess the proportion of Cd4+/Cd8+ T cells in both genotypes.40 Importantly, we observed a significantly enriched proportion of Cd4+ Foxp3+ T cells in Eµ-TCL1Akt-C mice, whereas the Cd8+ T-cell population remained unchanged (Figure 6I-K). In summary, these data show that RT cells are supported by Dll1-expressing Cd4+ T cells to induce Notch1 overactivation.
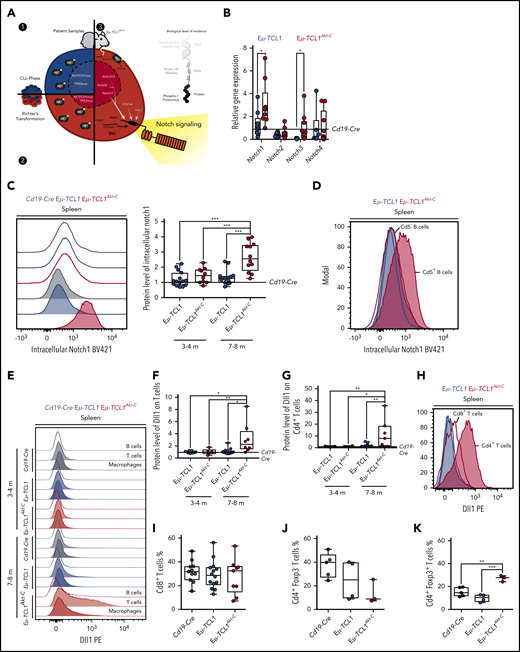
Notch1 activation supported via Dll1 from Cd4+T cells. (A) Graphic representing the identification of Notch1 signaling as a central component of all data presented in Figures 1 to 5. (B) Messenger RNA quantification of Notch receptors and (C) representative blot and relative protein quantification of activated Notch1 in Eµ-TCL1 and Eµ-TCL1Akt-C mice at 3 to 4 months and 7 to 8 months via flow cytometry, respectively. (D) Representative blot of activated Notch1 in Cd5– and Cd5+ B cells via flow cytometry in Eµ-TCL1 and Eµ-TCL1Akt-C mice at 7 to 8 months. (E) Representative blot and relative protein quantification of Dll1 in B cells, T cells, and macrophages in Eµ-TCL1 and Eµ-TCL1Akt-C mice at 3 to 4 months and 7 to 8 months via flow cytometry. (F) Dll1 phycoerythrin (PE) on splenic T cells at 3 to 4 months and 7 to 8 months. (G) Dll1 PE on splenic Cd4+ T cells at 3 to 4 months and 7 to 8 months. (H) Representative blot of Dll1 on splenic Cd4+ and Cd8+ T cells in Eµ-TCL1 and Eµ-TCL1Akt-C mice at 7 to 8 months. (I) Percentage of Cd8+ T cells from Cd19-Cre, Eµ-TCL1, and Eµ-TCL1Akt-C mice. Respective quantification of Cd4+ Foxp3– T cells (J) and of Cd4+ Foxp3+ regulatory T cells (K). *P < .05, **P ≤ 0.01, ***P ≤ .001 (unpaired, 2-sided Student t test).
Notch1 activation supported via Dll1 from Cd4+T cells. (A) Graphic representing the identification of Notch1 signaling as a central component of all data presented in Figures 1 to 5. (B) Messenger RNA quantification of Notch receptors and (C) representative blot and relative protein quantification of activated Notch1 in Eµ-TCL1 and Eµ-TCL1Akt-C mice at 3 to 4 months and 7 to 8 months via flow cytometry, respectively. (D) Representative blot of activated Notch1 in Cd5– and Cd5+ B cells via flow cytometry in Eµ-TCL1 and Eµ-TCL1Akt-C mice at 7 to 8 months. (E) Representative blot and relative protein quantification of Dll1 in B cells, T cells, and macrophages in Eµ-TCL1 and Eµ-TCL1Akt-C mice at 3 to 4 months and 7 to 8 months via flow cytometry. (F) Dll1 phycoerythrin (PE) on splenic T cells at 3 to 4 months and 7 to 8 months. (G) Dll1 PE on splenic Cd4+ T cells at 3 to 4 months and 7 to 8 months. (H) Representative blot of Dll1 on splenic Cd4+ and Cd8+ T cells in Eµ-TCL1 and Eµ-TCL1Akt-C mice at 7 to 8 months. (I) Percentage of Cd8+ T cells from Cd19-Cre, Eµ-TCL1, and Eµ-TCL1Akt-C mice. Respective quantification of Cd4+ Foxp3– T cells (J) and of Cd4+ Foxp3+ regulatory T cells (K). *P < .05, **P ≤ 0.01, ***P ≤ .001 (unpaired, 2-sided Student t test).
B-cell–intrinsic Notch1-IC expression in Eµ-TCL1 mice recapitulates the Eµ-TCL1Akt-C RT phenotype
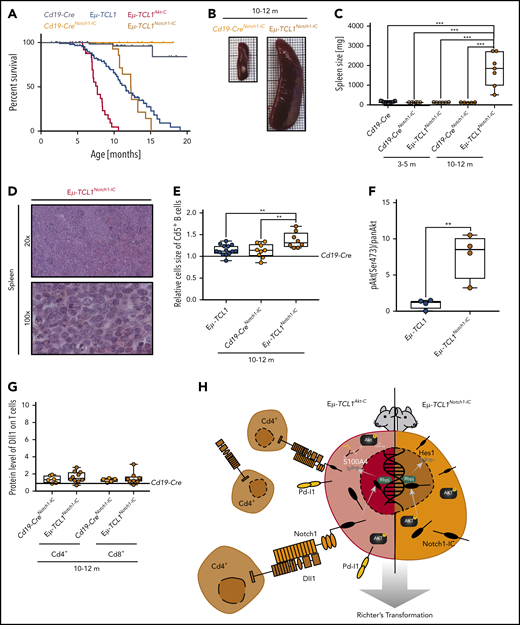
To elucidate whether CLL to RT transformation via Notch1 could be driven intrinsically in the Eµ-TCL1 mouse model, we used a conditional model of constitutively activated Notch1-IC19 intercrossed with Eµ-TCL1 and Cd19-Cre mice (Eµ-TCL1Notch1-IC) (supplemental Figure 7A). The initial disease development of Cd19+/Cd5+ B cells in the blood of Eµ-TCL1Notch1-IC mice was significantly slower than in Eµ-TCL1 mice (supplemental Figure 7B). Mice did not show reduced survival compared with Eµ-TCL1 mice, which could be explained due to altered B-cell differentiation affected by the Notch1-IC allele.41 However, once Cd5+ B cells appeared in the blood, these mice rapidly succumbed to the disease, with a survival trajectory comparable to that of Eµ-TCL1Akt-C mice (Figure 7A). At that age, Eµ-TCL1Notch1-IC mice presented with massive splenomegaly, which was confirmed as RT showing a disrupted splenic architecture with blastoid cells (Figure 7B-D). Furthermore, we observed that the malignant B cells of Eµ-TCL1Notch1-IC mice were significantly increased in size compared with Eµ-TCL1 counterparts (Figure 7E), while also having significantly higher active Akt levels (Figure 7F; supplemental Figure 7C). Finally, Eµ-TCL1Notch1-IC mice did not show concomitant Dll1 overexpression on T cells in the TME, confirming that constitutive activation of Notch1 is sufficient to drive CLL to RT transformation without TME support (Figure 7G-H; supplemental Figure 7D-E). In conclusion, constitutive activation of Notch1 in Eµ-TCL1 mice recapitulates the transformed phenotype of aggressive lymphoma and functionally identifies the Akt and Notch1 signaling axis as key determinants of murine RT. Our model therefore clarifies that what occurs in CLL toward RT in mice must be further validated in human patients.
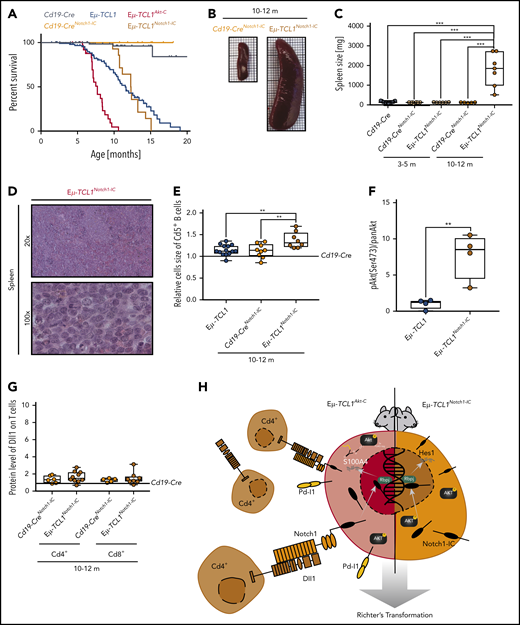
Constitutively active Notch-IC drives RT. (A) Pairwise Kaplan-Meier overall survival analysis for Cd19-CreNotch1-IC (yellow) vs Eµ-TCL1Notch1-IC(orange) mice; for comparison included from Figure 2 were Cd19-Cre (black), Cd19-CreAkt-C (gray), Eµ-TCL1 (blue), and Eµ-TCL1Akt-C (red) mice. (B) Representative images of spleens from Cd19-CreNotch1-IC vs Eµ-TCL1Notch1-IC mice aged 10 to 12 months. (C) Box plot of spleen weight for mice aged 10 to 12 months. (D) Representative hematoxylin and eosin staining of spleen preparations derived from Eµ-TCL1Notch1-IC mice showing DLBCL-like morphology. (E) Relative mean cell size (FSC) of Cd5+ splenic B cells aged 10 to 12 months. (F) Box plot showing pAkt (Ser473)/panAKT quantified from immunoblot of supplemental Figure 7D. (G) Box plot showing Dll1 protein level of Cd4+ T cells from Eµ-TCL1Notch1-IC mice. (H) Graphical abstract displaying Notch1 functional role in RT. **P ≤ .01, ***P ≤ .001 (unpaired, 2-sided Student t test).
Constitutively active Notch-IC drives RT. (A) Pairwise Kaplan-Meier overall survival analysis for Cd19-CreNotch1-IC (yellow) vs Eµ-TCL1Notch1-IC(orange) mice; for comparison included from Figure 2 were Cd19-Cre (black), Cd19-CreAkt-C (gray), Eµ-TCL1 (blue), and Eµ-TCL1Akt-C (red) mice. (B) Representative images of spleens from Cd19-CreNotch1-IC vs Eµ-TCL1Notch1-IC mice aged 10 to 12 months. (C) Box plot of spleen weight for mice aged 10 to 12 months. (D) Representative hematoxylin and eosin staining of spleen preparations derived from Eµ-TCL1Notch1-IC mice showing DLBCL-like morphology. (E) Relative mean cell size (FSC) of Cd5+ splenic B cells aged 10 to 12 months. (F) Box plot showing pAkt (Ser473)/panAKT quantified from immunoblot of supplemental Figure 7D. (G) Box plot showing Dll1 protein level of Cd4+ T cells from Eµ-TCL1Notch1-IC mice. (H) Graphical abstract displaying Notch1 functional role in RT. **P ≤ .01, ***P ≤ .001 (unpaired, 2-sided Student t test).
Discussion
Our analysis showed increased frequency of active AKT in primary RT samples to CLL samples, as well as in CLL samples with high-risk mutations for RT, such as those carrying TP53 and NOTCH1 mutations. This indicates that active AKT signaling represents a key feature in disease progression beyond the immediate contribution of respective oncogenic drivers.
To elucidate the functional impact of active Akt on CLL disease pathogenesis, we crossed the Eµ-TCL1 mouse model with a construct driving constitutive Akt activation specifically in B cells. This method induced an aggressive lymphoma phenotype with very high disease penetrance, mimicking the clinical features of RT, namely slow disease development followed by sudden progression with DLBCL-like morphology. This phenotype was only observed in combination with Eµ-TCL1, which compared with a lack of cancer-associated mutations in Cd19-CreAkt-C mice supports the notion that constitutively active Akt alone is not sufficient to drive leukemia/lymphomagenesis. Although it has been proposed that TCL1 prolongs Akt activation,42 the constitutive activation of Akt provided by the Rosa26 construct used in this study was significantly higher than what can be achieved by TCL1 overexpression alone.
Although genomic analyses of Eµ-TCL1 and Eµ-TCL1Akt-C mice failed to identify recurrent mutated genes, a profile defined by our “disease tendency score” categorized Eµ-TCL1Akt-C mice as having an intermediary mutational signature between CLL and DLBCL. Notwithstanding the preferential occurrence of genes associated with RT in the Eµ-TCL1Akt-C model, including Notch1 does suggest a shared mechanism between murine and human RT.
To trace these respective mechanisms, our multiomics analysis revealed increased Notch signaling and the upregulation of S100-proteins in Eµ-TCL1Akt-C cells by various means. In patients with RT, the most frequently acquired mutation is activating NOTCH1 (30% of all mutations).5,6,43 The evolutionary conserved Notch1 signaling affects proliferation, maturation, and survival of B cells44-46 and is induced by cell–cell interaction in which Dll ligands on one cell type bind to Notch receptors on another to proteolytically process the Notch intracellular domain (NICD) that represents the active moiety to regulate gene expression.37 The S100-protein family has been implicated in various malignancies as mediators of proliferation.25 In particular, S100A4 has been implicated in relapsed/refractory DLBCL47 as well as a potential target in AML,48 with a functional role in metastasis and cytokine release.25
Here, we observed in relation to Notch signaling increased Hes1 expression and phosphorylation and increased activity of Wee1 and Cdk4 in RT cells via phosphoproteomics/proteomics. Hes1 is a bona fide target gene of Notch signaling,31 whereas Wee1 and Cdk4 have been previously observed to be associated with Notch signaling activation.33,34 Furthermore, in the scRNA-Seq analysis, we identified a cell–cell interaction GO term profile in Eµ-TCL1Akt-C enriched/shared clusters, with significantly increased expression of genes involved in S100-protein and Notch signaling, including; S100a4, S100a9, Cd44, Itga4 (Cd49d), Hes1, Flna, and Zmiz1. S100A4 has been shown to be regulated by both Notch and PI3K/AKT signaling in head and neck cancers (10.1158/0008-5472.CAN-10-2350), and S100A9 has been implicated in interleukin-17–induced NOTCH1 activation of oligodendrocyte progenitor cells in demyelinating disease.49 Cd44 and Cd49d have both been implicated as high-risk markers in CLL,50,51 with Cd44 also shown to synergize with Notch1 to drive T-cell acute lymphoblastic leukemia in vivo,52 whereas Cd49d has been associated with trisomy 12 status and NOTCH1mut status, both of which are associated with CLL to RT transformation.53,54 Finally, such Akt-Notch1 interactions have also been previously reported in a drosophila screen and in T-cell acute lymphoblastic leukemia.55,56 Furthermore, an Akt/Notch2-IC synergism promotes B-cell differentiation toward the marginal zone compartment.41 Taken together, all these findings suggest the overactivation of Notch signaling driven by constitutive Akt activation and vice versa.
Clearly, Akt-mediated transcriptional control regulates Notch expression also in nontransformed cells; however, because we were only able to validate Notch1 activation and target gene upregulation in RT cells, we expanded our perspective toward the TME as a potential source of stimulating ligands. Here, we identified a specific increase of Cd4+ Foxp3+ T cells in the Eµ-TCL1Akt-C mice with increased prosurvival capacity, which we later characterized as having increased Dll1 ligand expression. We hypothesize that the constitutive activation of Akt drives a cell–cell interaction program that elicits the expansion of Cd4+ T cells and overexpression of Dll1, which in turn provides Notch1 activation facilitating CLL to RT transformation. This hypothesis is supported by our data that Eµ-TCL1Akt-C mice have sudden disease onset between 6 and 8 months, while already showing increased Notch1 expression but lacking active Notch1-IC pretransformation. Beyond this hypothesis, we show the transformative potential of active Notch1 signaling in Eµ-TCL1Notch1-IC mice displaying complete histomorphologic features of aggressive RT.
Ultimately, our data suggest that aggressive transformation of indolent lymphomas does not only depend on the acquisition of genomic aberrations but may also be initiated by a changed functional status of a potentially oncogenic signaling pathway fueled from the TME. This model will allow further elucidation of targeted RT therapies, including PI3K/AKT and immune checkpoint inhibition approaches. Particularly, we are able to model immune checkpoint inhibition and target TME interactions such as NOTCH1 to combine these approaches to overcome refractory RT.57,58 Finally, pAKT might be considered a new biomarker for high-risk CLL, and combined inhibition of PI3K/AKT and NOTCH1 might represent a potential treatment option for high-risk CLL and RT patients with active AKT.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are indebted to their patients who contributed tissue and blood samples to this study. They are grateful for technical assistance from Cathy Baitzel, Reinhild Brinker, Brigitte Hampel, Patrick Jankowski, Anke Lietzau, Michael Michalik, Yvonne Meyer, Christiane Schäfer, Pia Schol, and Nadine Spenrath. The authors also thank Janine Altmueller and Peter Nuernberg at the Cologne Center for Genomics for outstanding support.
This work was supported by the German Research Foundation (DFG; KFO 286) RP5 (F.T.W. and C.P.P.), RP6 (M.H.), RP1 (M.H.), and CP2 (R.B. and C.H.). C.P.P. was supported by the Foerderprogramm Nachwuchsforschungsgruppen NRW 2015–2021, CAP Program of the Center for Molecular Medicine Cologne, and a research grant by Gilead Sciences.
Authorship
Contribution: C.P.P., S.J.B., and F.T.W. wrote the manuscript; V.K., S.J.B., M.A., M. Hallek, C.P.P., and F.T.W. designed the experiments. V.K., S.J.B., M.A., N.N., M.P., A.R., N.H., G.K., N.R., C.H., J.C.B., C.P.P., and F.T.W. performed experiments and analyzed the data; P.L., C.H., M. Herling, E.M.H., A.R., W.K., R.B., R.M., D.R., R.B., G.G., L.P.F., H.C.R., M. Hallek, S.J.B., and C.P.P. provided clinical samples and analyzed clinical data; S.J.B., M.P., and C.P.P. and performed DNA WES analysis; S.J.B., M.N., M.P., M.F., T.G., and C.P.P. performed scRNA-Seq analysis; V.K., J.L.W., M.K., and S.J.B. performed mass spectrometry analysis; H.C.R., M.P., S.C.S., N.H., N.R., M. Herling, and C.H. provided analytical tools; and all authors read, revised, and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Christian P. Pallasch, Department I of Internal Medicine I, University Hospital of Cologne, Kerpener Str. 62, 50937 Cologne, Germany; e-mail: christian.pallasch@uk-koeln.de; or F. Thomas Wunderlich, MPI for Metabolism Research, Gleueler Str. 5050931 Cologne, Germany; e-mail: thomas.wunderlich@sf.mpg.de.