

H3K36 tri-methylation by SETD2 has been linked to a plethora of pathways critical for the regulation of a multitude of cellular processes, including transcription, DNA replication and DNA repair after damage, yet the functional implications of perturbed H3K36 tri-methylation by SETD2 mutations or non-genomic loss of function in leukemia are still unclear. We have previously reported that the HMC-1.1 and -1.2 mast cell leukemia (MCL) cell lines and many patients (pts) with advanced systemic mastocytosis (advSM) display H3K36Me3 deficiency as a result of non-genomic loss of function of SETD2. Proteasome inhibition restored SETD2 protein expression and H3K36me3, suggesting that a functional protein is produced but rapidly degraded. To understand the mechanisms underlying this phenomenon, we used an in silico approach to identify candidate SETD2-interacting proteins, followed by experimental confirmation by co-immunoprecipitation. We found that, after proteasomal inhibition, SETD2 co-immunoprecipitates with Aurora Kinase A (AKA). We also found that AKA was overexpressed and hyper-activated in pts with advSM compared to ISM pts and to a pool of healthy donors, and that AKA can phosphorylate SETD2. Both pharmacological inhibition by Danusertib and siRNA-mediated knock-down of AKA rescued SETD2 expression and activity, raising the hypothesis that phosphorylation by AKA might be implicated in proteasome-mediated degradation of SETD2. The new standard of therapy in advSM is midostaurin, that inhibits the activity of both wild‐type and D816V mutant KIT, as well as of various other kinases including AKA. Therefore we investigated if midostaurin effects may be addressed to AKA inhibition and consequent SETD2/H3K36me3 rescue. To this purpose, HMC-1 cells were treated with 5 µM midostaurin for 24 h and AKA, SETD2 and H3K36me3 expression were evaluated by western blotting. Treatment with midostaurin was able to inhibit AKA activity by about 60%, partially restoring SETD2 expression and H3K36Me3. Moreover, midostaurin treatment of HMC-1 cells at micromolar doses induced cytostatic but not cytotoxic effects as shown by cell growth curves performed in liquid medium and as confirmed by annexin V/PI staining and subsequent cytofluorimetric analysis of apoptotic cell death. Our observations in cell lines were confirmed in neoplastic mast cells collected from six patients with advSM before and after three months of midostaurin treatment. Western blotting showed that midostaurin treatment in vivo indeed results in a rescue of SEDT2 expression and activity, associated with a partial de-phosphorylation of AKA.
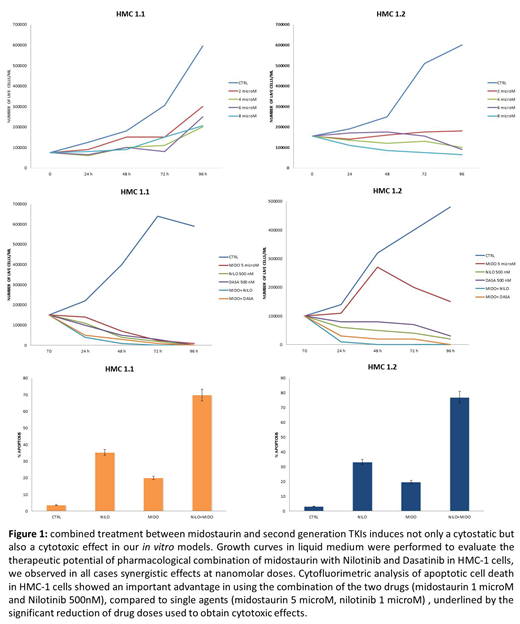
Our subsequent experimental step was therefore to hypothesize and test the possibility of a combined treatment between midostaurin and second generation TKIs to induce not only a cytostatic but also a cytotoxic effect both in our in vitro models and in primary cells obtained from bone marrow samples of advSM patients. We performed growth curves in liquid medium and clonogenic assays to evaluate the therapeutic potential of pharmacological combination of midostaurin with Nilotinib and Dasatinib in HMC-1 cells and in neoplastic mast cells from 3 patients with advSM and we observed in all cases synergistic effects at nanomolar doses. Moreover, cytofluorimetric analysis of apoptotic cell death in HMC-1 cells showed an important advantage in using the combination of the two drugs, compared to single agents, underlined by the significant reduction of drug doses used to obtain cytotoxic effects (Figure 1).
Our results suggest that AKA-mediated post-translational modifications contribute to SETD2 non-genomic loss of function in advSM. Inhibiting AKA and c-Kit activity by midostaurin in combination with a second generation TKI is a promising therapeutic strategy in patients with SETD2/H3K36Me3 deficiency.
Acknowledgments: study supported by AIRC (project code 16996) and AIL (Associazione Italiana contro le Leucemia, Linfomi e Mieloma).
Papayannidis:Incyte: Honoraria; Shire: Honoraria; Novartis: Honoraria; Teva: Honoraria; Amgen: Honoraria; Pfizer: Honoraria. Bonifacio:Pfizer: Honoraria; Amgen: Honoraria; Novartis: Honoraria; Incyte: Honoraria; BMS: Honoraria. Albano:Incyte: Membership on an entity's Board of Directors or advisory committees; Novartis: Membership on an entity's Board of Directors or advisory committees. Elena:Novartis: Consultancy; Pfizer: Consultancy. Valent:Deciphera: Honoraria, Research Funding; Blueprint: Research Funding; Pfizer: Honoraria; Celgene: Honoraria; Novartis: Consultancy, Honoraria, Research Funding. Cavo:celgene: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Other: travel accommodations, Speakers Bureau; janssen: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Other: travel accommodations, Speakers Bureau; bms: Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; sanofi: Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; novartis: Honoraria; takeda: Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; amgen: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; AbbVie: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees. Soverini:Incyte: Consultancy.

