Key Points
A risk-adapted, MRD-driven transplant strategy is a feasible approach for the treatment of younger adults with AML.
Pretransplant MRD positivity should not contraindicate delivery of an allogeneic stem cell transplant.

Abstract
We designed a trial in which postremission therapy of young patients with de novo acute myeloid leukemia (AML) was decided combining cytogenetics/genetics and postconsolidation levels of minimal residual disease (MRD). After induction and consolidation, favorable-risk patients (FR) were to receive autologous stem cell transplant (AuSCT) and poor-risk patients (PR) allogeneic stem cell transplant (AlloSCT). Intermediate-risk patients (IR) were to receive AuSCT or AlloSCT depending on the postconsolidation levels of MRD. Three hundred sixty-one of 500 patients (72%) achieved a complete remission, 342/361 completed the consolidation phase and were treatment allocated: 165 (48%) to AlloSCT (122 PR, 43 IR MRD-positive) plus 23 rescued after salvage therapy, for a total of 188 candidates; 150 (44%) to AuSCT (115 FR, 35 IR MRD-negative) plus 27 IR patients (8%) with no leukemia-associated phenotype, for a total of 177 candidates. Overall, 110/177 (62%) and 130/188 (71%) AuSCT or AlloSCT candidates received it, respectively. Two-year overall (OS) and disease-free survival (DFS) of the whole series was 56% and 54%, respectively. Two-year OS and DFS were 74% and 61% in the FR category, 42% and 45% in the PR category, 79% and 61% in the IR MRD-negative category, and 70% and 67% in the IR MRD-positive category. In conclusion, AuSCT may still have a role in FR and IR MRD-negative categories. In the IR MRD-positive category, AlloSCT prolongs OS and DFS to equal those of the FR category. Using all the available sources of stem cells, AlloSCT was delivered to 71% of the candidates.This trial was registered at www.clinicaltrials.gov as #NCT01452646 and EudraCT as #2010-023809-36.
Introduction
Despite the continuously growing knowledge about the genetic and molecular landscape of acute myeloid leukemia (AML),1,,,,-6 the paradigm of treatment of young adults with AML is still largely based on the “one-size-fits-all” approach, with postremission strategies still depending on donor availability rather than on the actual risk of disease relapse.7 In the short term, this has led to satisfactory rates of complete remission (CR) (70%-80%), but in the long-term survival, estimates are still disappointing, with <30% to 40% of patients becoming long-term survivors.8,9
Indeed, dealing with the high propensity for relapse and the considerable genetic heterogeneity of AML requires either development of new agents or adoption of modern, risk-adapted therapeutic programs. Risk-adapted approaches may consist of integrating pretreatment prognosticators, such as cytogenetics and molecular genetics, with posttreatment parameters, such as assessment of minimal (or measurable) residual disease (MRD).10
Even though in AML cytogenetic is a historical and robust determinant of outcome, the modern stratification of the patients in “favorable-risk" (FR), “intermediate-risk” (IR), or “adverse-risk” categories relies ever more increasingly on the baseline molecular pattern.11,12 Based on this, FR patients achieve overall survival (OS) and disease-free survival (DFS) rates of 50% to 60% at 3 to 5 years with standard chemotherapy, whereas those with adverse risk show OS and DFS rates of 5% to 20% at 3 to 5 years if not submitted to allogeneic stem cell transplantation (AlloSCT).7,11,13 Therefore, it appears that in FR and adverse-risk patients, the sole genetic/cytogenetic profile, regardless of the MRD levels, is helpful enough to guide decisions for the delivery of AlloSCT in the postremission phase. On the other hand, there are no accepted criteria to direct the decision-making process after consolidation for patients in the IR category: for these patients, evaluation of the MRD status appears appropriate to extrapolate those at high (MRD positive) or low (MRD negative) risk of relapse, for whom differentiated treatments may be adopted.
Although MRD assessment in AML is prognostic,14,,,-18 still <50% of relapses are detected by MRD; thus, the false negative rate is still high, resulting in low specificity. Moreover, MRD is assessed exploiting disparate flow cytometry or molecular protocols so that its use for treatment decisions in AML is still at an early stage. Depending on the technical platforms and targets, a sensitivity of 10−3 to 10−6 is reported.19 In particular, we observed that the integrated evaluation of baseline prognosticators and MRD improves risk assessment and helps optimizing postremission therapy.20 In fact, directing MRD-positive patients toward intensified therapy like AlloSCT while sparing those MRD-negative patients the procedure-related morbidity and mortality may be highly beneficial in terms of toxicity minimization.10
Considering all the above, the Gruppo Italiano Malattie EMatologiche dell’Adulto (GIMEMA) Foundation has developed a risk-adapted, MRD-oriented, prospective clinical trial, the strategy of which consisted of the prognostic integration of pretreatment cytogenetics and genetics with postconsolidation MRD, as detected by multiparametric flow cytometry (MFC). Based on this strategy, patients were to receive postconsolidation autologous stem cell transplantation (AuSCT) or AlloSCT, respectively, depending on their risk profile. We report here the final analysis of this multicenter study.
Patients and methods
Patients
Previously untreated patients with a diagnosis of de novo AML according to the World Health Organization diagnostic criteria21 were recruited to the GIMEMA AML1310 Study (EudraCT #2010-023809-36; www.clinicaltrials.gov Identifier #NCT01452646) provided they met the criteria for eligibility (see supplemental Material, available on the Blood Web site). The study was approved by the ethics committees of the participating hospitals/academic institutions and was conducted in accordance with the Declaration of Helsinki. All participants gave their informed consent.
Study design
The main objective of the study was to verify whether the delivery of a postremission therapy, the intensity of which was risk driven, improved the outcome of adult patients with AML in terms of increased antileukemic efficacy. The primary endpoint of the study was OS at 24 months from treatment start; for comparative purposes, we included a historical control consisting of patients recruited to the previous LAM99P GIMEMA trial.1 Secondary endpoints were CR or complete remission incomplete (CRi) rate after induction, DFS, and cumulative incidence of relapse (CIR) from CR. Upfront evaluation included bone marrow (BM) aspirate for morphology, cytogenetics, molecular genetics, and MFC analysis. The baseline MFC assessment was a necessary step, not only for diagnostic purposes but also to identify leukemia-associated immunophenotypes (LAIPs). Identification of baseline LAIPs was the essential requirement for monitoring MRD after therapy; at the established time point, BM MRD was determined by a high-sensitivity 8-color MFC assay. Based on several retrospective validations in the context of former European Organization for the Research and Treatment of Cancer/GIMEMA protocols,22 the threshold for discriminating MRD-negative from MRD-positive cases was set at 3.5 × 10−4 residual leukemic cells, and the selected time point was the postconsolidation phase, once the hematologic recovery was complete. Patients were studied at diagnosis for the presence of RUNX1-RUNX1T1 or CBFβ/MYH11 rearrangements, defining core binding factor (CBF) leukemias, and for NPM1, FLT3, and c-KIT mutations. In CBF or NPM1-positive AML, MRD was investigated as reported elsewhere.14,23,24 Molecular analysis, LAIPs assessment, and postconsolidation MRD determinations were centralized at Laboratorio di Diagnostica Integrata Oncoematologica “OPPO”, at Tor Vergata University Hospital of Rome, whereas conventional karyotype was carried out at local institutions. Response to treatment was assessed on BM and peripheral blood, according to the recommendations of an international working group.25 Patients who did not achieve CR/CRi or PR after the first induction course or CR/CRi after 2 induction courses were considered treatment failures. The AML1310 trial was designed at a time when European LeukemiaNet 2010/2017 and National Comprehensive Cancer Network (NCCN) 2018 recommendations were not yet published. Therefore, when the trial regulatory path was concluded, we started recruiting and stratifying patients according to contemporary classification, that was the NCCN 2009, version 1.26 For the purpose of our study, 4 categories of risk were identified (Table 1): favorable-risk (NCCN-FR) or poor-risk (NCCN-PR) patients, who were submitted to AuSCT or AlloSCT, respectively; intermediate-MRD negative (NCCN-IR-Neg) or positive (NCCN-IR-Pos) patients, who were to receive AuSCT or AlloSCT, respectively. Moreover, we enucleated a fifth group of patients belonging to the IR category, in whom we failed to identify any LAIP (NCCN-IR-no-LAIP category); these patients were allocated to the AuSCT postconsolidation option. AlloSCT and AuSCT were to be performed within 3 months of the end of the consolidation course.
Treatment
Induction consisted of IV daunorubicin 50 mg/m2 daily on days 1, 3, and 5; IV etoposide 50 mg/m2 daily on days 1 to 5; and IV cytarabine 100 mg/m2 as a daily continuous infusion, days 1 to 10. All patients in CR/CRi, after 1 to 2 induction cycles, received 1 consolidation course consisting of IV daunorubicin 50 mg/m2 daily on days 4,·5, and 6 and IV cytarabine 500 mg/m2 every 12 hours on days 1 to 6. In patients belonging to NCCN-FR and NCCN-IR categories, peripheral blood stem cell collection was attempted by initiating, on day 20 from the start of consolidation therapy, granulocyte colony-stimulating factor until completion of stem cell collection. In the case of failure to collect a sufficient number of peripheral blood stem cells, BM was used as a source. In the case of poor BM harvest, instead of AuSCT, patients were to receive a second consolidation course with high-dose cytarabine (HDARAC). Postconsolidation therapy was based on risk allocation: NCCN-FR patients were to receive AuSCT; NCCN-PR patients were to receive AlloSCT; NCCN-IR patients were to receive AuSCT or AlloSCT depending on the levels of BM MRD as measured by MFC, after consolidation therapy. Allocation to AlloSCT required the procedure to be performed whatever the source of stem cells (HLA-identical sibling, HLA-identical unrelated donor, cord blood, HLA-haploidentical sibling). Salvage therapy consisted of 1 or 2 courses of IV fludarabine 30 mg/m2 daily, on days 1 to 5; cytarabine 2000 mg/m2 daily, on days 1 to 5; idarubicin 8 mg/m2 daily, on days 1 to 3. Whatever the original NCCN risk category of assignment, patients with resistant disease after 1 to 2 cycles of induction therapy were considered PR and allocated to the AlloSCT procedure once CR/CRi was achieved.
Statistical analysis and sample size calculation
The primary objective was the percentage of OS at 2 years. An estimated number of 213 subjects were initially required to accomplish this primary objective. This sample size was to achieve a 90% power to detect a difference of 10% between the null hypothesis that OS at 2 years is 50% and the alternative hypothesis that OS is 60%, using a single-stage phase 2 design with a 5% significance level (based on data of the historic control group GIMEMA LAM99P).1 Based on the historical control group, we also considered that ∼70% of the observed patients would have been classified as IR, therefore allowing attainment of the figure of 150 patients available for MRD-driven treatment allocation. However, after 173 subjects were enrolled, only 56 belonged to the IR category (32% vs 70% expected). Therefore, to reach the target of 150 subjects belonging to the IR category, an amendment to the protocol was adopted in 2013, and the sample size was adjusted to 515 subjects to recruit. The efficacy analysis was performed as per treatment received, including individuals who commenced induction therapy and censoring patients at the time when they received a nonassigned treatment. OS (time elapsed from treatment start to death) and DFS (time from CR to relapse or death in remission) were calculated using the Kaplan-Meier product limit estimator. Differences in terms of OS and DFS were evaluated by means of Log-Rank test in univariate analysis and by means of Cox regression model in multivariate analysis, after assessment of proportionality of hazards. All variables with a P value < .15 in univariate analysis were considered in the multivariate models. The influence of the transplant (AuSCT and AlloSCT) on the survival outcome was evaluated in the Cox model by means of a time-dependent covariate. CIR was estimated by cumulative incidence curves using the proper nonparametric method. Patients’ and disease characteristics were summarized by means of cross-tabulations for categorical variables or by quintiles for continuous variables. Differences between categorical variables or response rates in subgroups were tested by the χ2 or Fisher exact tests, as appropriate. Confidence intervals (CIs) were calculated at 95% level, and all tests were 2 sided, accepting P ≤ .05 as indicating a statistically significant difference. All analyses were performed using the SAS (version 9.4) and R (R Foundation for Statistical Computing, Vienna, Austria) system software. Study data were collected and managed using the REDCap20 electronic data capture tools hosted at GIMEMA Foundation.
Results
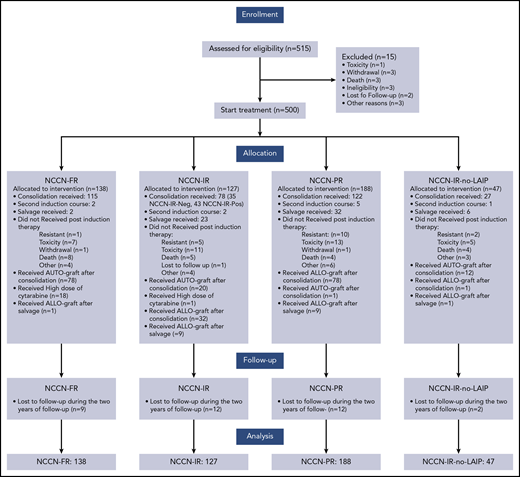
Between January 2012 and May 2015, 515 patients with de novo AML, seen at 55 GIMEMA institutions, were registered to the trial. Fifteen patients did not commence induction because of pretherapy death, infections, or ineligibility, and 500 started treatment and were available for the analysis. Demographic characteristics are summarized in Tables 2 and 3. Median age was 49 (18-60.9) years, and 52% were men. For 429 evaluable patients, cytogenetic distribution was favorable, intermediate, and poor in 11%, 73%, and 16%, respectively. Among 500 cases, RUNX1/RUNX1T1 was detected in 27 (5%) with 12 (44%) also c-KIT mutated; CBFβ/MYH11 was positive in 37 (7%) with 4 (11%) also c-KIT mutated, and FLT3-ITD and NPM1 mutations were detected in 46 (9%) and in 107 (21%), respectively. Finally, concomitant mutations of NPM1 and FLT3-ITD were observed in 80 cases (16%). We found no instances of FLT3 mutations in CBF-positive AML. Based on these data, patients’ distribution within the risk categories was as follows: 138 (28%) were NCCN-FR, 127 (25%) were NCCN-IR, 47 (9%) were NCCN-IR-no-LAIP, and 188 (38%) were NCCN-PR. Patients’ disposition is illustrated in Figure 1. After the first induction cycle, 333 (67%) and 21 (4%) patients achieved a CR and CRi, respectively. A second induction course was delivered to 10 of 13 patients in PR, with 7 entering CR. Therefore, after 1 to 2 cycles of induction, 361 (72%) patients obtained a CR: 88% in the NCCN-FR category, 65% and 69% in the NCCN-IR and NCCN-PR category, respectively (P < .001). Eighty-four (17%) patients had a refractory AML and 63 of them received a salvage therapy; 23 of these 63 (37%) achieved a CR. Three hundred forty-two of 361 (95%) patients started the consolidation phase and were treatment allocated: 177 (52%) to AuSCT (115 [65%] NCCN-FR, 35 [20%] NCCN-IR-Neg, 27 [15%] NCCN-IR-no-LAIP) and 165 (48%) to AlloSCT (122 [74%] NCCN-PR, 43 [26%] NCCN-IR-Pos). Of the 177 AuSCT candidates, 110 (62%) were transplanted (78 [71%] NCCN-FR, 20 [18%] NCCN-IR-Neg, 12 [11%] NCCN-IR-no-LAIP). Of the 165 AlloSCT candidates, 110 (67%) were transplanted (78 [71%] NCCN-PR, 32 [29%] NCCN-IR-Pos). If we include also the 23 patients who achieved a CR after salvage therapy, the group of AlloSCT candidates enlarges to 188. Because 20 of these 23 patients were given AlloSCT, the number of AlloSCT candidates who received it was 130/188 (71%). For the 78 patients belonging to the NCCN-PR category, the source of stem cells was a HLA-identical sibling in 26, a HLA-identical unrelated donor in 34, umbilical cord blood in 1, and HLA-haploidentical sibling in 17; for the 32 belonging to the NCCN-IR-Pos category, the source of stem cells was a HLA-identical sibling in 12, a HLA-identical unrelated donor in 9, umbilical cord blood in 1, and HLA-haploidentical sibling in 10. By physicians’ decision, 1 patient belonging to the NCCN-PR category received AuSCT and 1 belonging to NCCN-FR category received AlloSCT.

OS, DFS, and CIR
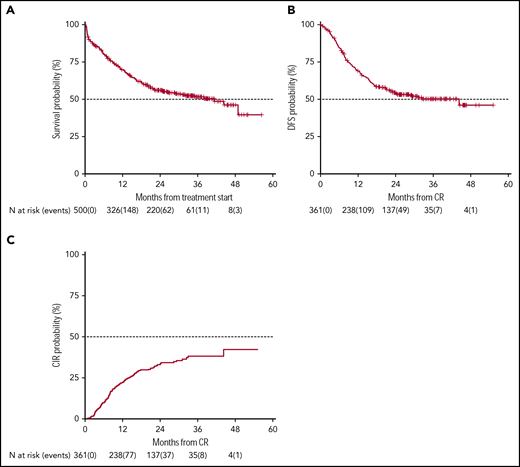
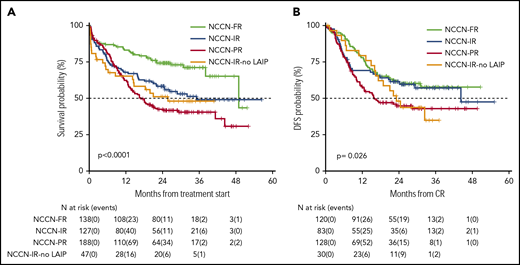
OS and DFS rates at 24 months of our historical control were 49% (95% CI 47-52) and 55% (95% CI 52-59), respectively.1 In the present trial, after a median follow-up of 28.8 months, 2-year OS was 56% (95% CI 52-61) with a median duration of 38 months (Figure 2), and DFS was 54% (95% CI 49-60) with a median duration of 32.4 months (Figure 2). The estimated OS at 24 months of 56% was less than the alternative hypothesis of 60%. However, the upper value of 95% CI included also the alternative hypothesis of 60% 2-year survival. Therefore, we considered the trial as not conclusive with regards to the primary endpoint. CIR, considering death in CR as a competing risk, was 33% (95% CI 28-38) (Figure 2). When splitting the survival analysis according to the identified categories of risk, 2-year OS was 42% (95% CI 36-50) for NCCN-PR patients, 58% (95% CI 50-68) for NCCN-IR patients, 74% (95% CI 67-82) for NCCN-FR patients, and 50% (95% CI 37-67) for NCCN-IR-no LAIP patients (P < .0001) (Figure 3). Two-year DFS was 45% (95% CI 37-55) for NCCN-PR patients, 61% (95% CI 52-73) for NCCN-IR patients, 61% (95% CI 52-71) for NCCN-FR patients, and 48% (95% CI 33-70) for those belonging to the NCCN-IR-no LAIP category (P = 0·026) (Figure 3). Using this risk-adapted approach, DFS duration of NCCN-FR and NCCN-IR was superimposable, whereas the NCCN-IR-no LAIP one was the shortest. When we focused on the NCCN-IR patients, whose postconsolidation choice was MRD-driven, no significant differences were observed in terms of 2-year OS between those MRD negative (79% [95% CI 66-94]) and MRD positive (70% [95% CI 57-86]) (P = .713) (Figure 4). The same was observed regarding the 2-year DFS (MRD negative = 61% [95% CI 47-80]; MRD positive = 67% [95% CI 53-83]) (P = .773) (Figure 4). The multivariate analysis confirmed the independent role of risk category in affecting CR rate, duration of OS, and DFS. The transplant procedure (AuSCT plus AlloSCT), analyzed as a time-dependent variable, affected independently the duration of OS. Age affected independently the duration of OS and DFS, whereas WBCc achievement of CR (supplemental Table 2).
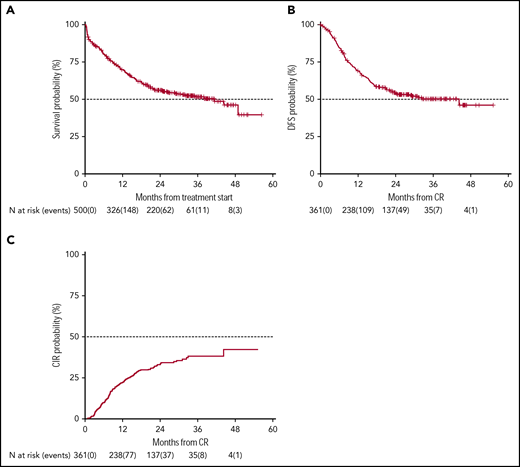
Survival estimates and CIR of the whole patient population. With a median follow-up of 28.8 months, 2-year-OS (A) and DFS (B) were 56% (median duration 38 months) and 54% (median duration 32.4 months), respectively. (C) CIR, considering death in CR as a competing risk, was 33%.
Survival estimates and CIR of the whole patient population. With a median follow-up of 28.8 months, 2-year-OS (A) and DFS (B) were 56% (median duration 38 months) and 54% (median duration 32.4 months), respectively. (C) CIR, considering death in CR as a competing risk, was 33%.
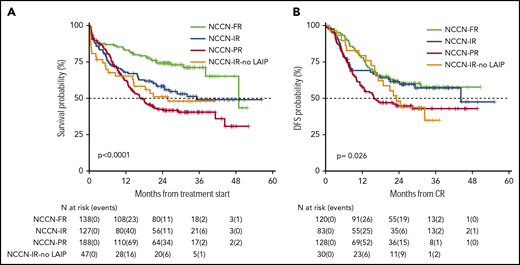
Survival estimates plotted by NCCN categories of risk. (A) Two-year OS was 42%, 58%, 74%, and 50%, respectively, for patients belonging to the NCCN-PR, NCCN-IR, NCCN-FR, and NCCN-IR-no LAIP categories. (B) Two-year DFS was 45%, 61%, 61% and 48%, respectively, for patients belonging to the NCCN-PR, NCCN-IR, NCCN-FR and NCCN-IR-no LAIP categories.
Survival estimates plotted by NCCN categories of risk. (A) Two-year OS was 42%, 58%, 74%, and 50%, respectively, for patients belonging to the NCCN-PR, NCCN-IR, NCCN-FR, and NCCN-IR-no LAIP categories. (B) Two-year DFS was 45%, 61%, 61% and 48%, respectively, for patients belonging to the NCCN-PR, NCCN-IR, NCCN-FR and NCCN-IR-no LAIP categories.
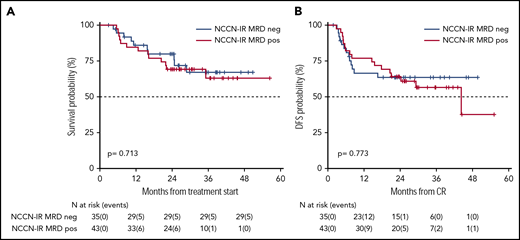
Survival estimates of NCCN-IR category, plotted by the status of MRD after consolidation therapy. (A) Two-year OS of NCCN-IR MRD negative and MRD positive patients were 79% and 70%, respectively. (B) Two-year DFS were 61% and 67%, respectively.
Survival estimates of NCCN-IR category, plotted by the status of MRD after consolidation therapy. (A) Two-year OS of NCCN-IR MRD negative and MRD positive patients were 79% and 70%, respectively. (B) Two-year DFS were 61% and 67%, respectively.
MFC and molecular integrated evaluation of MRD
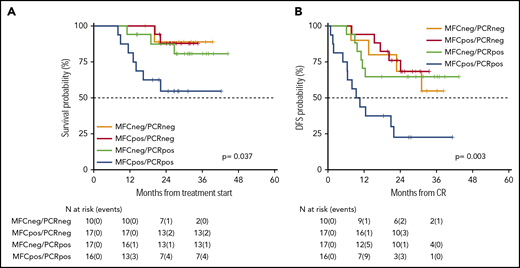
As an ancillary activity of the protocol, of 251 patients whose AML was characterized by the presence of a molecular marker useful for MRD assessment, we received 112 BM samples (RUNX-RUNX1 = 9, CBFB-MYH11 = 9, and NPM1 = 94) at the postconsolidation time point. In 60 of these, we had the opportunity to combine the postconsolidation results of MFC and reverse transcription quantitative polymerase chain reaction (RT-qPCR) MRD studies. This integrated analysis identified 4 categories of patients: double negative (MFCneg/PCRneg), double positive (MFCpos/PCRpos), and single positive (MFCpos/PCRneg or MFCneg/PCRpos). Patients who were double negative had a 2-year OS and DFS of 89% (95% CI 71-100) and 69% (95% CI 44-100), respectively. Patients who were MFCpos/PCRneg had a 2-year OS and DFS of 88% (95% CI 73-100) and 76% (95% CI 58-100), respectively. Patients who were MFCneg/PCRpos had a 2-year OS and DFS of 87% (95% CI 72-100) and 65% (95% CI 45-92), respectively. Finally, patients who were double positive had a 2-year OS and DFS of 55% (95% CI 34-87) and 22% (95% CI 9-58), respectively (Figure 5A-B; P = .037 and .003, respectively).
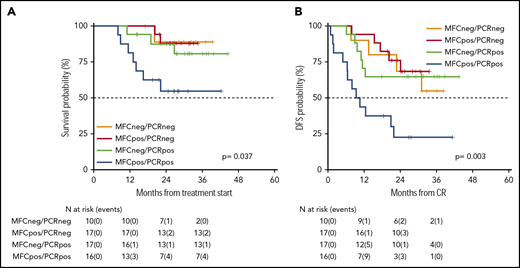
Survival estimates of 60 patients whose MRD was analyzed integrating MFC and RT-qPCR. By integrating MFC and RT-qPCR, 4 categories of distinct MRD status were identified with different duration of OS (A) and DFS (B). Patients who were double negative had a 2-year OS and DFS of 89% and 69%, respectively. Patients who were MFCpos/PCRneg had a 2-year OS and DFS of 88% and 76%, respectively. Patients who were MFCneg/PCRpos had a 2-year OS and DFS of 87% and 65%, respectively. Finally, patients who were double positive had a 2-year OS and DFS of 55% and 22%, respectively.
Survival estimates of 60 patients whose MRD was analyzed integrating MFC and RT-qPCR. By integrating MFC and RT-qPCR, 4 categories of distinct MRD status were identified with different duration of OS (A) and DFS (B). Patients who were double negative had a 2-year OS and DFS of 89% and 69%, respectively. Patients who were MFCpos/PCRneg had a 2-year OS and DFS of 88% and 76%, respectively. Patients who were MFCneg/PCRpos had a 2-year OS and DFS of 87% and 65%, respectively. Finally, patients who were double positive had a 2-year OS and DFS of 55% and 22%, respectively.
AuSCT vs HDARAC consolidation
As per protocol, 19 patients (18 NCCN-FR and 1 NCCN-IR) received HDARAC, because they did not have enough stem cells collected. Figure 6 shows OS and DFS of these patients compared with those who were submitted to AuSCT. OS was 83% (95% CI 67-100) and 85% (95% CI 78-93), respectively (P = .753); DFS was 68% (95% CI 50-93) and 63% (95% CI 54-73), respectively (P = .595). Of these 19 patients, 15 were NCCN-FR MFCneg/PCRneg, MFCpos/PCRneg, or MFCneg/PCRpos, 3 were NCCN-FR MFCpos/PCRpos, and 1 was NCCN-IR MRD negative.
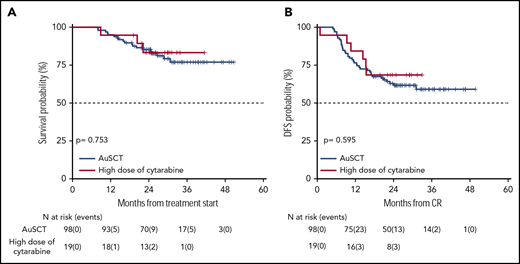
Survival estimates of the 19 patients who received high dose of cytarabine versus those who received AuSCT. (A) Patients who received high dose of cytarabine or an autologous stem cell transplant had an OS of 83% and 85%, respectively. (B) DFS was 68% and 63%, respectively.
Survival estimates of the 19 patients who received high dose of cytarabine versus those who received AuSCT. (A) Patients who received high dose of cytarabine or an autologous stem cell transplant had an OS of 83% and 85%, respectively. (B) DFS was 68% and 63%, respectively.
Discussion
The role of molecular and cytogenetic abnormalities in predicting response to therapy and survival in patients with AML has been extensively documented.24,-26 Indeed, genetic/cytogenetic abnormalities are powerful prognosticators, so that obtaining information about their presence is essential for an optimal decision-making process. The clinical implication is that, based on their genetic status, patients would benefit from more or less aggressive postconsolidation strategy such as AuSCT and AlloSCT or, in a more modern vision, from targeted new agents. However, prognostic models barely based on pretreatment covariates, such as genetic status, have a limited predictive ability.11,27,-29 This highlights not only the need to expand further our knowledge about the genetic and molecular pattern of AML but also the potential role of “factors after diagnosis,” such as MRD monitoring. Therefore, integrating baseline factors and monitoring of MRD appears a promising tool to refine and possibly customize our outcome prediction ability in AML. This philosophy was at the basis of the GIMEMA AML1310 protocol in which, deviating from the classical “one-size-fits-all” approach, we applied a risk-adapted and MRD-driven approach. AlloSCT is generally recommended when the risk of relapse exceeds 35% to 40% if the procedure is not performed.7,10 In this view, NCCN-PR category represents a priority and, in these patients, AlloSCT should be performed as soon as CR is achieved. However, an HLA-identical sibling is available for <30% of the patients,30 and, in reality, even <30% receive it, due to disease recurrence.11 In our study, utilization of any available source of stem cells resulted in 71% of AlloSCT candidates receiving it. Adoption of this strategy also translated to a 2-year OS and DFS of 42% and 45%, respectively, for the NCCN-PR category (Figure 2). Such figures compare very favorably with the 2-year OS and DFS of 20% to 30% currently reported for this category.27,29 Based on our study design, patients belonging to the NCCN-FR category were given AuSCT as a postconsolidation therapy. The role of AuSCT is controversial; in 1 randomized study, it provided better DFS and similar OS as conventional consolidation chemotherapy.31,32 In our NCCN-FR category, 2-year OS and DFS were 74% and 61%, respectively (Figure 3). We believe there is still a role for AuSCT; indeed, this option has the advantage of sparing patients multiple courses of postconsolidation chemotherapy (usually high-/intermediate-dose cytarabine). In fact, the recently revised European LeukemiaNet classification suggests that limiting AuSCT to MRD-negative AML might improve the results.11 Based on our limited experience, HDARAC might represent the choice for patients with very “high-quality” CR such as those NCCN-FR MFCneg/PCRneg (Figure 6). Management of patients belonging to the NCCN-IR category is still controversial. For these patients, the relapse rate after AuSCT can be as high as 50% to 55%,10 so that this option appears as a suboptimal approach. Indeed, AlloSCT is recommended for patients within this category. However, in selected patients with MRD-negative CR, there might still be a room for AuSCT.10 In the present study, we planned AlloSCT or AuSCT for NCCN-IR patients, based on the level of MRD after the postconsolidation course. By making this choice, we observed that the 2-year OS and DFS were 58% and 61%, respectively (Figure 3). This figure compares very favorably with recent analyses showing, for these patients, a 2-year OS and DFS of ∼35% and 50%.27,29 Using this strategy, we also noted that the 2-year DFS of NCCN-IR patients was prolonged to equal that of NCCN-FR patients (Figure 3). Finally, within the NCCN-IR category, we focused on outcome as influenced by the postconsolidation MRD status. By delivering AuSCT to NCCR-IR-Neg and AlloSCT to NCCN-IR-Pos patients, we observed no difference in terms of 2-year OS or DFS (Figure 4). The stratification role of MRD determination in IR patients has been recently suggested in a prospective survey of the NCRI-AML17 trial.33 According to the authors, an MRD-positive finding helps selecting patients who can benefit from AlloSCT. An indirect confirmation of the importance of MRD determination in IR category was that our 47 NCCN-IR-no-LAIP patients who were submitted to AuSCT had the shortest duration of 2-year OS and DFS (Figure 2). A reasonable explanation is that these patients harbored significant postchemotherapeutic levels of MRD, meaning that AlloSCT would have been the most appropriate choice. Our results highlight the potent antileukemic effect exerted by AlloSCT in NCCN-IR-Pos patients and the minimization of toxicity after AuSCT in NCCN-IR-Neg ones. This interpretation can be extended to include the overall population we had under investigation; indeed, generating the maximum antileukemic effort in high-risk patients (NCCN-PR + NCCN-IR-Pos) and preserving from excess of toxicity those who are at low risk (NCCN-FR + NCCN-IR-Neg) appears a very plausible goal. In this view, the integration of different techniques for MRD monitoring may offer the chance to improve even further our capability to discriminate prognostically discrete subsets of patients, directing treatment more precisely. Combining MFC and RT-qPCR for cases carrying a molecular signature, we demonstrated that double-positive patients had the worst prognosis. For these patients, a front-line intensified program appears a reasonable option (Figure 5). Although there is evidence that AlloSCT is not able to reverse the unfavorable long-term impact of MRD positivity,34,,-37 we believe that a pretransplant MRD-positive status should not be a contraindication for performing it.38,-40 In the study by Walter35 and Araki,37 patients who were MRD positive before the transplant had an outcome comparable to the one of patients with active disease. However, these studies were retrospective; the patient population was heterogeneous in terms of age and conditioning regimens received, and there was a concentration of adverse karyotype and secondary AML in the group of MRD-positive patients. Our experience takes advantage of a prospective and homogeneous context in terms of therapy delivered and risk stratification. A recent, retrospective analysis of 547 patients enrolled in the Hemato-Oncology Foundation for Adults in the Netherlands and the Swiss Group for Clinical Cancer Research protocols indicates that, although all categories benefit from AlloSCT, the absolute benefit was greater in pretransplant MRD-positive than MRD-negative patients.41 Our present experience adds a piece of information favoring the use of AlloSCT in MRD-positive patients, and future trials should possibly explore the prognostic role of different levels of pretransplant MRD38 and the value of posttransplant maintenance.
In conclusion, we recognize that the study suffers from some intrinsic limitations due to the changes occurring over the time (more modern biologic knowledge, new AML classifications, and an ever more frequent MRD monitoring) that make the historical control and the study population not fully superimposable. However, this is one of the first attempts to apply a prospective program of risk-adapted, MRD-driven therapy, integrating upfront genetics and postconsolidation MRD status, in AML of adults. In the NCCN-FR category, AuSCT guarantees the same survival expectation as multiple courses of cytarabine. In the NCCN-IR category, AlloSCT can be avoided if MRD is not measurable; if MRD is positive, AlloSCT can prolong OS and raise the DFS duration to the level of NCCN-FR patients. Finally, using all the available sources of stem cells allowed AlloSCT to be delivered to a large proportion of the candidates, emphasizing the feasibility of the trial transplant policy.
Individual participant data will not be shared.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the following contributors: Giorgia Giuliani and Mariangela Iodice, for data collection at the GIMEMA Data Center; Laura Di Donato, for pharmacovigilance activity (GIMEMA); and Stefano Soddu, for assistance in statistical analysis (GIMEMA). The authors thank the following centers for their contribution to patient recruitment: Ospedale A. Businco, Cagliari (La Nasa Giorgio), Azienda Ospedaliera Pugliese Ciaccio, Catanzaro (Molica Stefano), Ospedale Santa Maria Goretti, Latina (De Blasio Angelo), Azienda Ospedaliera Universitaria Federico II, Napoli (Pane Fabrizio), A.U. Policlinico Paolo Giaccone, Palermo (Siragusa Sergio), CTMO Ospedale San Salvatore, Pesaro (Visani Giuseppe), Policlinico A. Gemelli, Roma (De Stefano Valerio), Azienda Ospedaliera San Camillo Forlanini, Roma (Pierelli Luca), Dipartimento di Medicina Clinica e Sperimentale, Sassari (Fozza Claudio), IRCCS AOU San Martino IST, Genova (Angelucci Emanuele), CTMO Universita' degli Studi di Parma, Parma (Aversa Franco), C.T.M.O. Istituti Ospitalieri, Cremona (Molteni Alfredo), Ospedali Riuniti, Foggia (Capalbo Silvana Franca), Ospedale S. Luigi Gonzaga, Orbassano (Guerrasio Angelo), Azienda Ospedaliera SS. Antonio e Biagio e Cesare Arrigo, Alessandria (Ladetto Marco), Azienda Ospedaliera Pisana, Pisa (Petrini Mario), Cancer Center Humanitas, Rozzano (Santoro Armando), Universita' degli Studi di Padova, Padova (Semenzato Pietro), A. Tortora di Pagani, Pagani (SA) (Califano Catello), I.F.O. Istituto Nazionale Tumori Regina Elena, Roma (Mengarelli Andrea), Ist. Nazionale Tumori, Milano (Corradini Paolo), Azienda Ospedaliera Ospedali Riuniti Papardo Piemonte, Messina (Mannina Donato), Ospedale Binaghi, Cagliari (La Nasa Giorgio), Ospedale Ca' Foncello, Treviso (Gherlinzoni Filippo), Ospedale Infermi, Rimini (Tosi Patrizia), A.O. S.Anna e S.Sebastiano, Caserta (Frigeri Ferdinando), Azienda Ospedaliera Pia Fondazione di Culto e di Religione Card. G.Panico, Tricase (LE) (Pavone Vincenzo), ASL Le/1 P.O. Vito Fazzi, Lecce (Di Renzo Nicola), Ospedale Monsignor Raffaele Dimiccoli, Barletta (Tarantini Giuseppe), Istituto Scientifico Romagnoli per lo Studio e la Cura dei Tumori IRST, Meldola (Martinelli Giovanni), AUSL Ospedale G. da Saliceto, Piacenza (Vallisa Daniele), Ospedale Civile, Civitanova Marche (Centurioni Riccardo).
This study was conducted thanks to the partial contribution of the Associazione Italiana Contro le Leucemie-Linfomi e Mieloma.
This work is dedicated to the memory of Francesco Lo-Coco, a dear friend and a valuable colleague.
Authorship
Contribution: A.V., A.P., P.F., M.V., F.L.-C., W.A., and S.A. created the concept and design; A.V., A. Candoni, L. Melillo, V.C., R.C., P.d.F., G.S., P.S., F.L., G.M., M.L., P.M., M.P.M., A. Cuneo, F.A., F.F., A. Tafuri, A. Chierichini, A. Tieghi, N.S.F., D.C., R.F., C.A., L. Maurillo, F.B., M.I.D.P., W.A., and S.A. provided the study materials or patients; A.V., A.P., A. Candoni, L. Melillo, V.C., R.C., P.d.F., G.S., P.S., F.L., G.M., M.L., P.M., M.P.M., A. Cuneo, F.A., F.F., A. Tafuri, A. Chierichini, A. Tieghi, N.S.F., D.C., R.F., C.A., E.L.S., L. Maurillo, F.B., M.I.D.P., M.I.-C., T.O., S.L., and M.T.V. collected and assembled the data; A.V., A.P., E.L.S., L. Maurillo, F.B., M.I.D.P., M.T.V., F.L.-C., W.A., and S.A. analyzed and interpreted the data; L. Maurillo, F.B., M.I.D.P., M.I.-C., T.O., S.L., and M.T.V. performed the laboratory testing and monitoring; and all authors contributed to the writing of the manuscript and to the final approval of the manuscript.
Conflict-of-interest disclosure: A.V. reports personal fees from Pfizer, Celgene, Novartis, Daiichi-Sankyo, and Jazz Pharmaceuticals outside the submitted work. S.A. reports personal fees from Amgen, Celgene, Novartis, and Daiichi-Sankyo outside of the submitted work. R.F. reports personal fees from Roche, Genentech, Janssen, Gilead, Celgene, Novartis, Ariad, and Amgen outside of the submitted work. M.L. reports grants from Novartis and MSD, personal fees from Novartis, Abbvie, and Gilead outside of the submitted work. The remaining authors declare no competing financial interests.
Francesco Lo-Coco died on 3 March 2019.
Correspondence: Adriano Venditti, Fondazione Policlinico Tor Vergata, Viale Oxford 81-00133, Rome, Italy; e-mail: adriano.venditti@uniroma2.it.