Key Points
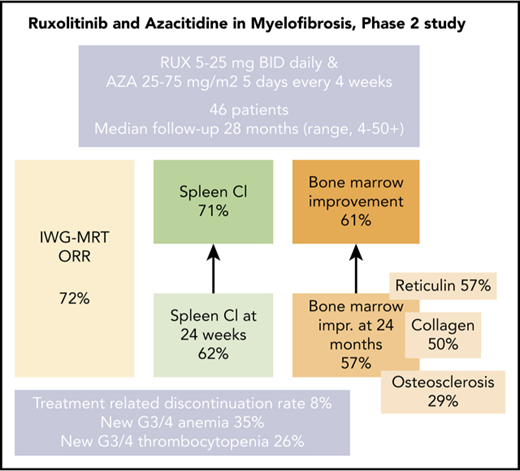
The combination of RUX and AZA was safe with encouraging spleen response rates at 24 weeks and any time on study.
RUX and AZA demonstrated marked improvements in bone marrow fibrosis at 24 months when compared with RUX alone.

Abstract
Ruxolitinib (RUX)-based combinations may provide benefit for patients with myelofibrosis (MF). In this open-label, nonrandomized, prospective phase 2 study, patients with MF initially received RUX twice per day continuously in 28-day cycles for the first 3 cycles. Azacitidine (AZA) 25 mg/m2 (days 1-5) was added starting with cycle 4 and could be subsequently increased to 75 mg/m2 (days 1-5). Forty-six patients were enrolled with a median follow-up of 28 months (range, 4-50+ months). An International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) response was achieved in 33 patients (72%), with a median time to response of 1.8 months (range, 0.7-19.0 months). One-fourth (7 of 33) of the IWG-MRT responses occurred after the addition of AZA. A reduction of >50% in palpable spleen length at 24 weeks and at any time on the study was achieved in 62% and 71% of the evaluable patients, respectively. Among patients who achieved a >50% reduction in spleen length at 24 weeks, 95% had maintained it at 48 weeks. Notably, improvements in bone marrow reticulin fibrosis grade occurred in 57% of the patients at 24 months. Treatment discontinuations as a result of drug-related toxicities occurred in 4 patients (9%), all as a result of cytopenias. New onset grade 3 to 4 anemia and thrombocytopenia occurred in 35% and 26% of patients, respectively. RUX and AZA were safe, with encouraging spleen response rates and improvement in bone marrow fibrosis in patients with MF. This trial was registered at www.clinicaltrials.gov as #NCT01787487.
Introduction
Dysregulation with constitutive activation of the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway is central to the pathobiology of myelofibrosis (MF).1 The JAK1/2 inhibitor ruxolitinib (RUX; INCB018424; Incyte, Wilmington, DE) demonstrated superior reduction in splenomegaly and improvements in MF-related symptoms compared with placebo (COMFORT-I; Controlled Myelofibrosis Study With Oral JAK Inhibitor Treatment-I)2 or best available therapy (COMFORT-II),3 respectively. RUX was approved by the US Food and Drug Administration for the treatment of patients with intermediate- or high-risk MF and by the European Medicine Agency for the treatment of MF-related splenomegaly or symptoms in adult patients with MF. The benefits of using RUX therapy for the spleen, MF symptoms, and quality of life noted in the COMFORT-I and -II trials have been maintained with longer follow-up and include an overall survival (OS) benefit.4,5 Although RUX affords benefits in a majority of patients with MF, activation of the non-JAK pathway could lead to therapeutic resistance, suggesting a need for rationally developed therapeutic combinations.
In the United States and Europe, azacitidine (AZA) is approved for patients with myelodysplastic syndrome6,7 and is commonly used to treat older patients with newly diagnosed acute myeloid leukemia (AML).8,9 In a phase 2 trial in 34 patients with MF, AZA demonstrated an International Working Group (IWG) consensus criteria overall response rate of 24%10,11 but a short response duration of 4 months.
We hypothesized that the combination of AZA and RUX would target distinct clinical and pathological manifestations of MF, resulting in synergistic efficacy. Herein, we report results of a phase 2 clinical trial evaluating the safety and efficacy of RUX with AZA in patients with MF.
Patients and methods
Patient eligibility
This was an open-label, nonrandomized, prospective phase 2 study of RUX with AZA in patients with MF conducted at the University of Texas MD Anderson Cancer Center (MDACC). Patients age 18 years or older with primary myelofibrosis (PMF), post-polycythemia vera MF, or post-essential thrombocythemia MF12 with intermediate-1 or -2 or high-risk score according to the Dynamic International Prognostic Scoring System (DIPSS)13 requiring therapy were eligible. Additional eligibility criteria included an Eastern Cooperative Oncology Group performance status of ≤2, creatinine ≤2.5 mg/dL, bilirubin ≤2 mg/dL, alanine aminotransferase ≤2.5 times the upper limit of normal, absolute neutrophil count (ANC) ≥1.0 × 109/L, and platelet count ≥50 × 109/L. Patients who had received prior therapy with RUX or AZA were not eligible. Standard or investigational therapy for MF within 14 days of starting study therapy was not allowed with the exception of hydroxyurea. This was a single-center study supported by Incyte Corporation (USA), approved by the Institutional Review Board at the University of Texas MDACC, and conducted in accordance with the principles of the Declaration of Helsinki. All patients provided a signed informed consent form.
Baseline and on-study assessments
Pretreatment evaluations included a complete history and physical examination (including spleen and liver measurement by palpation by the treating physician), a transfusion history for 3 months prior to day 1 of protocol therapy, and completion of the Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF) and the MD Anderson Symptom Inventory (MDASI) questionnaire. All patients were tested for the BCR/ABL1 translocation by fluorescence in situ hybridization and/or polymerase chain reaction and for JAK2V617F by reverse transcription polymerase chain reaction assay, as previously described.14 A next-generation sequencing–based analysis for the detection of somatic mutations in 28 additional genes was performed on DNA extracted from the bone marrow (BM), as previously published15 (supplemental Table 1, available on the Blood Web site). Testing for CALR mutations was performed separately.16 Evaluations of BM (histomorphology, cytogenetics, and molecular studies) were performed at baseline, after 6 and 12 cycles of therapy, and then once every 12 cycles. Reticulin fibrosis was graded as proposed by Thiele et al.17 Collagen and osteosclerosis grading were performed in accordance with the 2016 World Health Organization classification of myeloid neoplasms18 (supplemental Methods).
MPN-SAF and MDASI questionnaires were obtained before each cycle for the first 6 cycles and then every 3 cycles for the remainder of the study. Responses per the 2013 International Working Group for Myelofibrosis Research and Treatment (IWG MRT)19 were documented at the end of each cycle for the first 6 cycles and then after every 3 cycles for the remainder of the study.
Treatment schedule
To mitigate myelosuppression and high early discontinuation rates observed in prior RUX combinations in patients with MF,20,21 we implemented a sequential dosing approach with a 3-month run-in phase with single-agent RUX. RUX was given once per day orally continuously in 28-day cycles and was started at 5, 15, or 20 mg twice per day orally in patients with platelet counts of 50 to 100 × 109/L, 100 to 200 × 109/L, and >200 × 109/L, respectively. AZA 25 mg/m2 was introduced on cycle 4 day 1 and was given subcutaneously or intravenously on days 1 to 5 of each cycle. In case of proliferative disease or elevated BM blasts (>10%), AZA could be introduced earlier or the dose could be increased up to 75 mg/m2 (supplemental Table 2).
In case of hematologic toxicity (platelets <25 × 109/L or ANC ≤0.5 × 109/L), RUX was interrupted and could be resumed at 5 mg once per day (platelets >25 × 109/L and ANC >0.5 × 109/L) or at 5 mg twice per day (platelets >50 × 109/L and ANC >1.0 × 109/L). AZA was completely held for the impacted cycles and could be resumed with 1 dose level reduction for subsequent cycles.
The study was initially designed for a maximum of 15 cycles on protocol with continuation of the combination off protocol in patients with clinical benefit (7 patients referred to as the initial patients; supplemental Table 3). The protocol was amended in July 2013 so patients could continue on study indefinitely.
Study design and objectives
This study recruited 46 patients between March 1, 2013, and April 1, 2017. The data cutoff was October 1, 2017. The primary objective was efficacy evaluated as the best objective IWG-MRT response achieved on the study. The secondary objective was safety. Adverse events (AEs) were assessed according to the National Cancer Institute Common Toxicity Criteria for Adverse Events, version 4.03.22 Serious AEs (SAEs) were defined as AEs that caused death or life-threatening complications or that led to a patient’s hospitalization. Exploratory end points included time to response (eg, anemia and transfusion dependence or molecular, cytogenetic, or BM responses), duration of each response, and survival.
Futility and toxicity were monitored simultaneously using the Bayesian approach of Thall et al23,24 and the extension by Thall and Sung.25 The futility stopping rule mandated termination if there was a more than 95% chance that the objective response rate was <50%. The toxicity monitoring rule was invoked if there was a >90% chance that the rate of grade ≥3 clinically relevant nonhematologic AEs or SAEs was ≥20%. The stopping rules for futility and toxicity were applied in cohorts of 5 patients starting from the tenth patient.
Statistical methods
OS was defined as time from study enrollment until death as a result of any cause, time to response was defined as the time from study enrollment to the date of first response, and duration of response was defined as the time from the date of first response to the date of progression.
A univariable logistic regression model was used to evaluate factors associated with an IWG-MRT response. The distributions of all time-to-event end points were estimated using the Kaplan-Meier method. Univariable Cox proportional hazards models were applied to evaluate factors associated with OS. A multivariable Cox proportional hazards model was obtained by first including the factors with P value < .05 in the univariable analysis; a backward elimination was then performed using 0.05 for the significance level needed for a factor to remain in the multivariable model. Statistical analyses were carried out using IBM SPSS Statistics 24 for Windows (SPSS Inc., Chicago, IL).
Results
Patient characteristics and treatments
Patients’ baseline characteristics are provided in Table 1. Twenty-five patients had PMF, 10 had post-polycythemia vera MF, and 11 had post-essential thrombocythemia MF. Three patients (6%) had blasts >10% in their peripheral blood or in BM at enrollment. Thirty-six patients (78%) were intermediate risk and 10 (22%) were high risk per DIPSS.26 Twenty-six patients (57%) had received prior therapy for myeloproliferative neoplasms (eg, hydroxyurea [n = 20], anagrelide [n = 11], thalidomide or pomalidomide [n = 6]).
At data cutoff (October 1, 2017), 17 patients (40%) continued to receive therapy with RUX and AZA, with 13 patients being treated on study (CONSORT diagram, supplemental Figure 1). The median treatment duration for all 46 enrolled patients was 18 months (range, 1-53 months). Thirty-one patients (67%) remain alive with a median follow-up of 28 months (range, 4-50 months). Overall, 33 patients discontinued the study. This included the 7 initial patients, who continued RUX with AZA therapy off protocol for a median of 28 months (range, 4-39 months), with eventual discontinuation of therapy with RUX and AZA in 3 patients (progression to AML in 1 patient and death in 2 patients). Six patients went off therapy as a result of elective allogeneic stem cell transplantation (SCT). The remaining 15 patients discontinued therapy because of progression to AML or accelerated-phase MF (n = 4), myelosuppression (n = 4), uncontrolled proliferation requiring cytotoxic therapy (n = 2), death (n = 1), financial or travel issues (n = 3), and concurrent medical illness (n = 1).
The median number of RUX cycles administered was 24 (range, 1-60 cycles) with 15 cycles (range, 1-52 cycles) administered for AZA. Forty-one patients (89%) received AZA on study; 5 were not able to start AZA because of cytopenia (n = 3) or early study discontinuation (n = 2). Among these 41 patients, 36 patients (86%) received AZA starting with cycle 4 day 1, and 5 started AZA earlier (cycle 1 [n = 4], cycle 3 [n = 1]).
Forty-four patients (91%) started RUX twice-per-day doses: 24 patients started at 20 mg, 18 started at 15 mg, 1 started at 10 mg, and 1 started at 25 mg. Median dose density was similar for RUX as a single agent and in combination with AZA: 15 mg twice per day (range, 5 mg once per day to 25 mg twice per day). The initial dose of RUX needed to be reduced in 28 patients (61%) at some point on study because of anemia (n = 9), neutropenia (n = 2), thrombocytopenia (n = 9), 1 or more cytopenia (n = 4), fatigue (n = 2), pneumonia (n = 1), and elevated liver enzymes (n = 1).
Among the 41 patients treated with AZA (starting dose of 25 mg/m2), 12 patients (26%) required incremental increases in dose to 50 mg/m2 (n = 7) or 75 mg/m2 (n = 5) because of increasing blasts (n = 3), white cell proliferation (n = 2), or worsening splenomegaly or symptoms (n = 7). Overall, 14 patients (34%) had to hold or reduce AZA doses at some point during the study because of cytopenia (n = 4), fatigue (n = 3), infection (n = 5), or unrelated medical issues (n = 2). The median number of AZA cycles per patient was 4 (range, 1-7 cycles).
Efficacy
Objective response.
Protocol-defined IWG-MRT responses (lasting >12 weeks) were documented in 33 (72%) of 46 patients, including partial remission in 2 patients and clinical improvement in 31. The median time to an IWG-MRT response (n = 33) was 1.8 months (range, 0.7-19 months). Response details are summarized in Table 2. Seven (21%) of the 33 responders achieved IWG-MRT responses after the addition of AZA (all started AZA on cycle 4 day 1). Median time to response after the addition of AZA was 4.4 months (range, 0.1-16.5 months).
The only significant factor for achievement of an IWG-MRT response on univariable analysis was the pretherapy BM blast percentage, wherein patients with higher pretherapy BM blasts had a lower response rate (odds ratio, 0.75; 95% confidence interval [CI], 0.58-0.98; P = .03; supplemental Table 4). Because of the lack of predictive factors on univariable analysis, multivariable analysis was not feasible. Among patients with increased blasts >10% (n = 3), only 1 patient achieved IWG-MRT response (clinical improvement in the spleen and improved total symptom score [TSS]) with a reduction in blasts to 2% maintained for 28 months (supplemental Table 5).
Spleen responses.
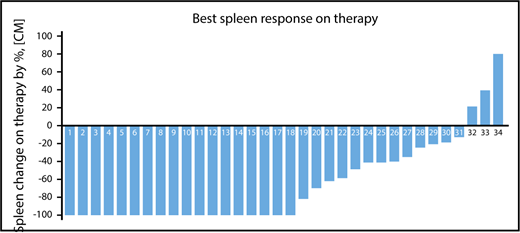
Among 34 patients with splenomegaly >5 cm below costal margin, an IWG-MRT clinical improvement in the spleen was achieved in 21 patients (62%) at any time on study and in 18 patients (53%) by week 24 (Figure 1; supplemental Figure 2). An additional 3 patients with baseline splenomegaly of 5 to 10 cm achieved a >50% reduction, but none had complete resolution of palpable spleen. In total, a >50% reduction in palpable spleen length was achieved in 24 patients (71%) at any time on study and 21 patients (62%) at week 24 on study.
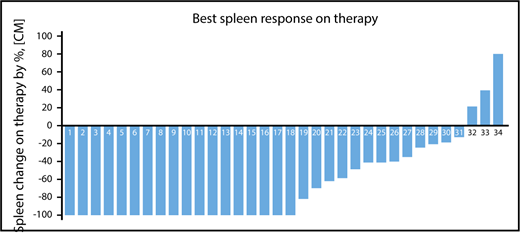
Palpable spleen length changes at any time on study. A waterfall plot showing the best percentage of palpable spleen length changes at any time during therapy among the 34 patients with baseline palpable splenomegaly >5 cm below costal margin.
Palpable spleen length changes at any time on study. A waterfall plot showing the best percentage of palpable spleen length changes at any time during therapy among the 34 patients with baseline palpable splenomegaly >5 cm below costal margin.
Overall, week 24 and week 48 mean percentage reductions in spleen length by palpation were 73% (95% CI, 59%-86%) and 63% (95% CI, 25%-100%), respectively. The median duration of spleen response was not reached (range, 7 months to not reached). Among patients who achieved a >50% reduction in spleen length at 24 weeks, 95% had maintained this at 48 weeks.
Five responses (24%) showing clinical improvement in the spleen occurred after the addition of AZA on cycle 4 day 1, with a median time to response after the introduction of AZA of 1.8 months (range, 1-13.8 months). The median spleen palpable length at the start was 10 cm (range, 8-20 cm), and median dose of RUX was 15 mg twice per day in 3 patients, 20 mg twice per day in 1 patient, or 25 mg twice per day in 1 patient (supplemental Table 6).
Symptom response.
MPN-SAF TSS scores at enrollment were <20 in 17 patients (37%), between 21 and 50 in 21 patients (44%), and >51 in 8 patients (20%). Overall, 25 (54%) of 46 patients achieved a TSS response with a median time to response of 2 months (range, 0.7-16 months). Among the 29 patients with baseline TSS >20, a TSS response was achieved in 17 patients (59%) (supplemental Figure 3). TSS responses occurred after the introduction of AZA in 4 patients (baseline TSS >20 in all 4 patients).
Anemia response.
Five patients (11%) were transfusion dependent at baseline and 1 (20%) achieved transfusion independence. Forty-one patients were transfusion independent at the beginning of the study, and 3 (7%) experienced an increase of ≥2 g/dL in their hemoglobin relative to baseline (IWG-MRT clinical improvement of anemia, 7%). Eleven patients (ie, 27% of 41 previously transfusion-independent patients) required transfusion while receiving therapy (supplemental Table 7).
Cytogenetic response.
Among 15 patients with initial cytogenetic abnormalities (trisomy 8 [n = 3]; deletion 20q [n = 1), 3 achieved complete cytogenetic remission with a median time to response of 12 months. All 3 patients had received both RUX and AZA (supplemental Table 8).
Molecular response.
Among 24 JAK2V617F-mutated patients, 16 (67%) had sequentially evaluable JAK2 allele burdens. Thirteen (81%) of 16 had a decrease in the JAK2 allele burden at 24 weeks, including 3 (19%) with a >50% reduction and 4 (25%) with a 20% to 49% reduction (supplemental Figure 4). Median time to best reduction of JAK2 allele burden was 12 months (range, 6-24 months).
BM response.
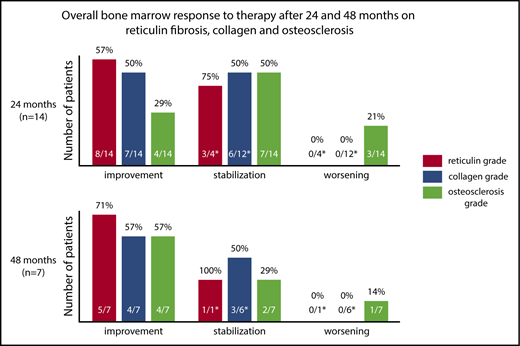
Sequential BM samples were available in 31 patients (67%). Two different pathologists who had limited access to clinical results at the time of analysis assessed the BM morphology and response. Any improvement in BM morphology (minor, major, and complete histomorphologic response) was achieved in 19 patients (61.2%) at any time on study. Figure 2 demonstrates BM changes in reticulin fibrosis, collagen and osteosclerosis at 24 and 48 months. At 24 months, improvements in BM reticulin fibrosis and collagen were observed in 57% and 50% of evaluable patients (n = 14), respectively. No patient experienced a worsening in BM reticulin fibrosis or collagen while on study. Osteosclerosis improvement was observed in 29% of patients; however, 21% of patients (n = 3) showed a worsened osteosclerosis grade among which only 1 patient had improvement in BM reticulin (supplemental Figures 5 and 6). Only 1 patient with improvement in BM morphology did not achieve a concurrent clinical IWG-MRT response (supplemental Figure 7).

BM histomorphologic changes at 24 and 48 months. A bar graph showing the percentage of evaluable patients who had improvement, stabilization, or progression in BM reticulin fibrosis, collagen, and osteosclerosis at 24 and 48 months compared with pretherapy. Changes in BM histomorphology (upper panel) at 24 months and (lower panel) at 48 months. BM response was classified as improvement, stabilization, or worsening. Patients with baseline BM fibrosis grade 3 were excluded from this analysis because progression and stabilization were not defined. Numerator identifies patients with a grade different than that at pretherapy. The denominator identifies eligible patients included in the analysis (eg, excludes patients with BM fibrosis grade 3 for worsening).
BM histomorphologic changes at 24 and 48 months. A bar graph showing the percentage of evaluable patients who had improvement, stabilization, or progression in BM reticulin fibrosis, collagen, and osteosclerosis at 24 and 48 months compared with pretherapy. Changes in BM histomorphology (upper panel) at 24 months and (lower panel) at 48 months. BM response was classified as improvement, stabilization, or worsening. Patients with baseline BM fibrosis grade 3 were excluded from this analysis because progression and stabilization were not defined. Numerator identifies patients with a grade different than that at pretherapy. The denominator identifies eligible patients included in the analysis (eg, excludes patients with BM fibrosis grade 3 for worsening).
Safety
As of September 31, 2017, 4 patients (8%) had discontinued protocol because of drug-related toxicities (neutropenia [n = 2], anemia [n = 1], and thrombocytopenia [n = 1]).
AEs irrespective of attribution on a study occurred in 44 patients (95%), and grade ≥3 AEs occurred in 31 patients (67%) (Table 3). The frequency of AEs was high within the first 12 weeks and beyond 48 weeks on study (53% [n = 23] and 63% [n = 19] of patients at risk) (supplemental Table 9). SAEs occurred at any time on study in 34 patients (74%) and are summarized in supplemental Table 10. The most frequent SAEs occurring in ≥4% of patients were respiratory infections and fevers (non-neutropenic and neutropenic). Nonhematologic grade AEs observed in >10% of patients involved the gastrointestinal system and infections (Table 3).
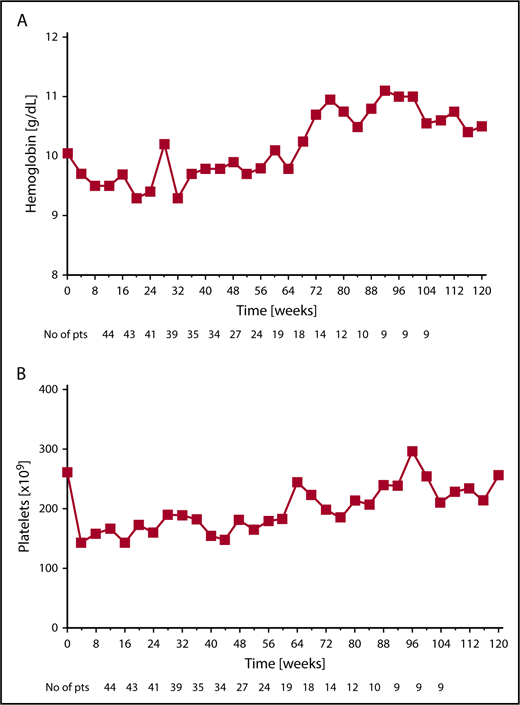
The most common hematologic AEs (all grades) were anemia and thrombocytopenia in 72% and 63% of patients, respectively. New onset anemia, thrombocytopenia, and neutropenia grade ≥3 on study occurred in 35%, 26%, and 24% of patients, respectively. The median hemoglobin declined from a baseline level of 10.3 g/dL to a nadir of 9.4 g/dL at 12 weeks of therapy, and then increased to a new steady state of ∼10 g/dL by week 24 and remained above this level beyond week 24. Median platelet number on study did not decrease below 180 × 109/L at any time (Figure 3A-B).
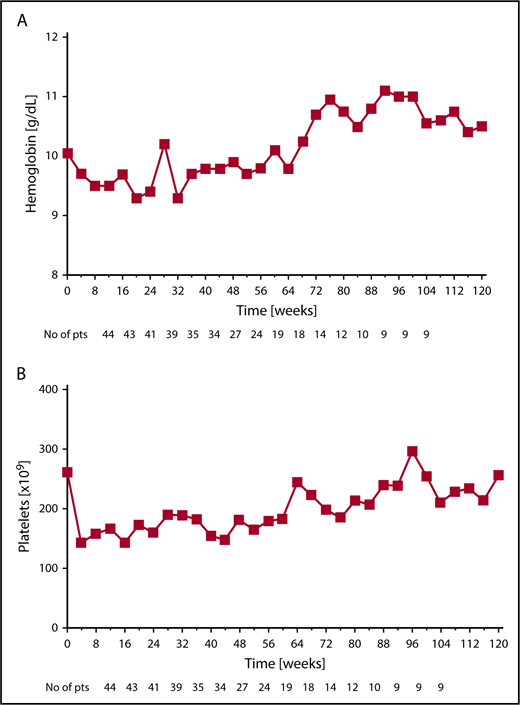
Median hemoglobin and platelet levels for patients receiving combination therapy. Changes over time in (A) median hemoglobin and (B) median platelets for all available patients at the given time point.
Median hemoglobin and platelet levels for patients receiving combination therapy. Changes over time in (A) median hemoglobin and (B) median platelets for all available patients at the given time point.
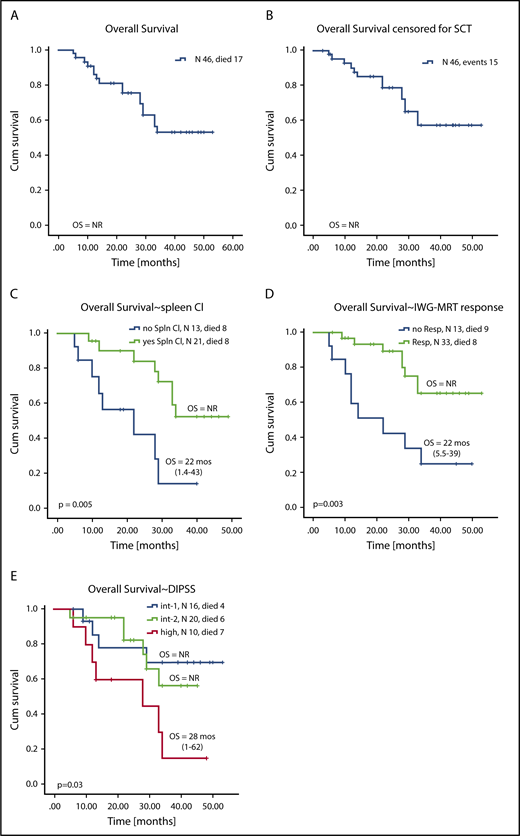
OS
At the time of this analysis, 17 of 46 patients had died after a median follow-up of 22 months (range, 5-50 months). In total, 3 patients died while receiving RUX with AZA therapy, 1 each from sepsis, meningitis, and stage IV melanoma. Among the remaining 14 patients who died after discontinuation of RUX with AZA, the causes of death were known in 12 patients: AML (n = 4), pneumonia (n = 1), acute complications after allogeneic SCT (n = 3), multiorgan failure as a result of sepsis (n = 1), splenic rupture (n = 1), cardiac arrest (n = 1), and complications after splenectomy (n = 1).
Overall, 6 patients have disease that progressed to AML during their follow-up: 3 who were receiving RUX with AZA therapy and 3 after discontinuing therapy (supplemental Table 11) with a median time to progression to AML of 5 months from enrollment (range, 0.9-18.4 months). Overall, 11 patients (24%) underwent allogeneic SCT: 3 patients had early SCT (within <6 months of receiving therapy) and 8 had late SCT (after >6 months of receiving therapy). Three of 11 patients died after SCT, with a median survival of 3 months after SCT (range, 3-34 months) as a result of multiorgan failure (n = 2) or myocardial infarction (n =1).
Median OS (with and without censoring for SCT) was not reached (Figure 4A-B). Factors associated with superior OS on univariable analysis were higher pretherapy hemoglobin levels, lack of baseline palpable splenomegaly, not having high-risk DIPSS, achievement of clinical improvement in the spleen, and achievement of an IWG-MRT response (Figures 3C and 4D; supplemental Table 12). On multivariable analysis, achievement of clinical improvement in the spleen (hazard ratio, 5.6; 95% CI, 1.84-17.3; P = .002) and not having high-risk DIPSS (hazard ratio, 5.1; 95% CI, 1.7-15; P = .003) remained significant for OS. When stratified by DIPSS, median OS was not reached in patients with intermediate-1 and -2 risk and was 28 months (range, 1-62 months) in patients with high-risk DIPSS (Figure 4E-F). Given the relatively short follow-up and that approximately one-third of patients went on to receive SCT, the survival data are preliminary and need longer follow-up to mature.
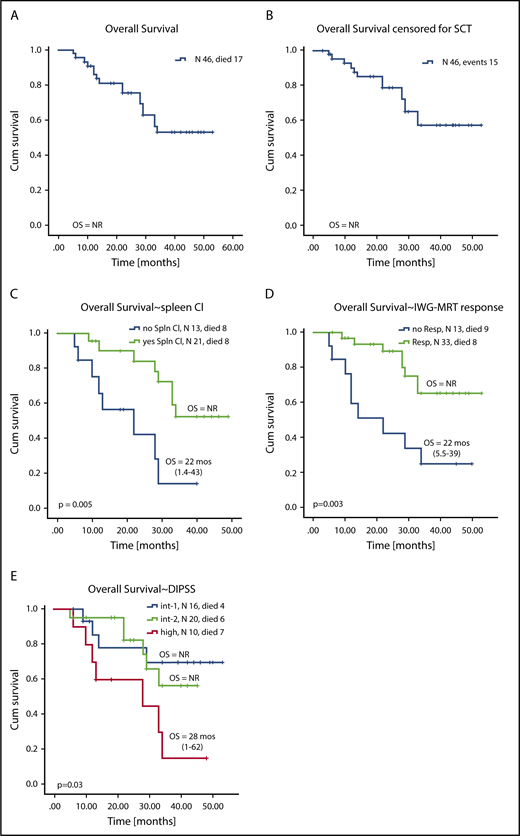
Kaplan-Meier curves showing OS for all patients enrolled on study. OS during study for all patients (A) not censored for allogeneic SCT and (B) censored for allogeneic SCT. OS stratified by (C) achievement of clinical improvement in spleen length, (D) achievement of any IWG-MRT response during therapy, and (E) DIPSS risk score at enrollment.
Kaplan-Meier curves showing OS for all patients enrolled on study. OS during study for all patients (A) not censored for allogeneic SCT and (B) censored for allogeneic SCT. OS stratified by (C) achievement of clinical improvement in spleen length, (D) achievement of any IWG-MRT response during therapy, and (E) DIPSS risk score at enrollment.
Discussion
In our study, RUX with AZA demonstrated an IWG-MRT 2013 response rate of 70% in patients with DIPSS intermediate-1 or -2 or high-risk MF requiring therapy. A reduction of >50% in palpable spleen length in patients with baseline palpable splenomegaly >5 cm was observed in 71% of the evaluable patients at any time on study, in 62% of the patients at week 24, and in 69% of the patients at week 48. Although direct comparisons are not possible, the proportion of patients achieving a spleen response at any time on study or at 24 or 48 weeks of therapy with RUX and AZA compares favorably to proportions noted with single-agent RUX in the COMFORT studies.4,5 In the COMFORT-I study, 59% of patients achieved a spleen response (defined as a ≥35% reduction in spleen volume, which corresponds to a ≥50% reduction in spleen length by palpation) at any time on study, and 42% of patients achieved spleen response at week 24.4 Similar to COMFORT-I in which 67% of the patients maintained the spleen response achieved at 24 weeks at 48 weeks or beyond, the spleen responses in this study were durable, with 95% of patients maintaining the 24-week spleen responses at 48 weeks or beyond. The mean palpable spleen length reduction at 48 weeks on RUX with AZA was 75%, which compares favorably to the 56% noted with single-agent RUX in COMFORT-II.5 Of note, 24% of the IWG-MRT responses and 28% of the spleen responses occurred after the addition of AZA.
In line with the results from the COMFORT studies, the combination was effective in controlling MF-associated symptoms (TSS improvement of ≥50% was noted in 56% of patients), but not anemia (clinical improvement in hemoglobin levels by IWG-MRT was shown in only 7% of patients). Our study confirms the limited efficacy of AZA in improving anemia in patients with MF, as was previously reported.10,27 Interestingly, some patients achieved hemoglobin stabilization over time without achieving a traditional IWG-MRT anemia response. This may be meaningful, but it needs to be followed with studies that have more patients.
Clinical responses were seen regardless of baseline mutational profile, DIPSS score, presence of anemia, or splenomegaly, consistent with what has previously been reported with RUX alone.14,28,29 Because there were only 9 patients carrying high-risk molecular mutations, we were not able to verify inferior responses in these patients as previously reported with single-agent RUX.30,31
Overall, 6 patients (13%) had disease that progressed to AML on study and/or during follow-up. We believe this is a result of patient selection. Unlike the COMFORT-I trial, which excluded patients with peripheral blood (PB) blasts ≥10%, patients with this characteristic were allowed on our study. Among the 6 patients for whom disease progressed to AML on study and/or during follow-up, 2 had PB blasts ≥10% (18% and 13%), and 1 had PB blasts of 9% at enrollment. Among these 3 patients, 2 had disease that progressed to AML on study with a median time to AML of 5 months, and only 1 was able to receive both RUX and AZA. We believe that in patients with BM and/or high PB blasts ≥5%, concomitant rather than simultaneous therapy with RUX and AZA may be the best approach, and we are applying this approach to patients prospectively enrolled on this study.
Of the 31 patients with sequential BM evaluations, 57% showed improvement in BM reticulin fibrosis at 24 months. Although these are small numbers, they compare favorably to BM reticulin fibrosis improvement rates of 15.8% at 24 months with RUX alone in a phase 1/2 study.32 Furthermore, we observed improvements in abnormal collagen or osteosclerosis in about 45% of patients, a finding rarely noted in MF trials. The improvements in BM reticulin fibrosis also manifested earlier with this combination than was seen with single-agent RUX in the COMFORT-II trial, with median time to first improvements of 12 and 24 months, respectively. Although the prognostic implications of these findings remain unclear and continued prospective evaluation of the BM morphology is ongoing, it seems that the combination may have a specific beneficial impact on fibrosis and sclerosis in patients with MF.
Although an overall discontinuation rate of 72% seems high, this includes elective discontinuation in 7 patients after completion of 15 cycles, and elective SCT in 6 patients. Treatment-related discontinuation rates for combination therapy were 8%, which is similar to treatment-related discontinuation rates of 9% to 11% with RUX alone in the COMFORT-I and COMFORT-II studies.2 The lower discontinuation rates on this study may be a result of the sequential administration strategy we adopted. This sequential strategy was developed to mitigate early and severe cytopenias, with high early discontinuation rates of 21% to 45% that were previously encountered in RUX-based combinations in which RUX and the other agent were started concomitantly.20,21 As expected, given the known myelosuppression of RUX and AZA, the most common toxicities were hematologic. New or worsening grade ≥3 anemia, thrombocytopenia, and neutropenia occurred in 35%, 26%, and 24% of patients, respectively. The majority were manageable by appropriate dose modifications and supportive measures and were not associated with treatment discontinuation. In line with previous reports from studies of RUX as a single agent, the median hemoglobin decreased during the first 12 weeks on therapy and recovered to a steady level from week 24 onwards.
Our current experience with RUX and AZA in patients with MF suggests that the combination has potential synergy for spleen length reduction and BM fibrosis improvement. This must be weighed against a higher incidence of early and late (>48 weeks) cytopenias, but with a treatment-related discontinuation rate similar to that for single-agent RUX. Longer-term follow-up and randomized trials are needed to determine the eventual impact of this combination.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was supported by Incyte Pharmaceuticals and the MD Anderson Cancer Center Leukemia Support Grant CA016672, the MD Anderson Cancer Center Leukemia Specialized Programs of Research Excellence Grant CA100632, the Charif Souki Cancer Research Fund, and generous philanthropic contributions to the MD Anderson Moon Shots Program.
Authorship
Contribution: L.M. and N.D. analyzed the data and wrote the article; N.D. and S.V. designed the study, wrote the clinical study protocol, and enrolled patients; S.P., L.Z., and L.M. compiled and summarized the data; J.N. provided statistical support for the trial; R.G. and N.D. managed the conduct of the study according to the protocol; C.E.B.-R. and J.E.H.-L. performed histomorphology review and morphological response assessment of the bone marrows; R.L. and K.P.P. performed molecular analysis; S.V., N.P., P.B., Z.E., E.J.J., F.R.-K., K.T., J.E.C., M.O., Y.A., T.M.K., C.D.D., G.B., K.B., G.G.-M., and H.M.K. enrolled patients; and all authors reviewed and approved final version of the manuscript.
Conflict-of-interest disclosure: H.M.K., J.E.C., P.B., G.G.-M., S.V., and N.D. received research funding from Incyte Corp. J.E.C., P.B., N.P., G.G.-M., C.E.B.-R., S.V., and N.D. served as consultants for Incyte Corp. The remaining authors declare no competing financial interests.
Correspondence: Naval Daver, Department of Leukemia, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 0428, Houston, TX 77030; e-mail: ndaver@mdanderson.org.