

Key Points
Treatment with IFNα was associated with distinct molecular responses in patients with JAK2-mutated MPN compared with CALR-mutated MPN.
Among patients treated with IFNα who did not achieve CHR, DNMT3A mutations emerged more frequently than non-DNMT3A mutations.

Abstract
Although somatic mutations influence the pathogenesis, phenotype, and outcome of myeloproliferative neoplasms (MPNs), little is known about their impact on molecular response to cytoreductive treatment. We performed targeted next-generation sequencing (NGS) on 202 pretreatment samples obtained from patients with MPN enrolled in the DALIAH trial (A Study of Low Dose Interferon Alpha Versus Hydroxyurea in Treatment of Chronic Myeloid Neoplasms; #NCT01387763), a randomized controlled phase 3 clinical trial, and 135 samples obtained after 24 months of therapy with recombinant interferon-alpha (IFNα) or hydroxyurea. The primary aim was to evaluate the association between complete clinicohematologic response (CHR) at 24 months and molecular response through sequential assessment of 120 genes using NGS. Among JAK2-mutated patients treated with IFNα, those with CHR had a greater reduction in the JAK2 variant allele frequency (median, 0.29 to 0.07; P < .0001) compared with those not achieving CHR (median, 0.27 to 0.14; P < .0001). In contrast, the CALR variant allele frequency did not significantly decline in those achieving CHR or in those not achieving CHR. Treatment-emergent mutations in DNMT3A were observed more commonly in patients treated with IFNα compared with hydroxyurea (P = .04). Furthermore, treatment-emergent DNMT3A mutations were significantly enriched in IFNα–treated patients not attaining CHR (P = .02). A mutation in TET2, DNMT3A, or ASXL1 was significantly associated with prior stroke (age-adjusted odds ratio, 5.29; 95% confidence interval, 1.59-17.54; P = .007), as was a mutation in TET2 alone (age-adjusted odds ratio, 3.03; 95% confidence interval, 1.03-9.01; P = .044). At 24 months, we found mutation-specific response patterns to IFNα: (1) JAK2- and CALR-mutated MPN exhibited distinct molecular responses; and (2) DNMT3A-mutated clones/subclones emerged on treatment.
Introduction
Philadelphia chromosome–negative chronic myeloproliferative neoplasms (MPNs) comprise essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF), including prefibrotic myelofibrosis (Pre-MF). MPNs are clonal hematopoietic neoplasms characterized by excessive proliferation of mature hematopoietic cells from one or more of the myeloid lineages.1,2 The diseases are associated with an increased risk of thrombohemorrhagic events and reduced life expectancy compared with the general population.3,4 ET and PV may progress into post-ET and post-PV myelofibrosis, and all disease entities may transform into secondary acute myeloid leukemia, which has a dismal prognosis.5
The majority of MPNs are driven by somatic mutations in JAK2, CALR, or MPL that arise in the hematopoietic stem cell compartment (ie, MPN phenotypic driver mutations).6 All 3 MPN phenotypic driver mutations lead to uncontrolled myeloproliferation by constitutive activation of the JAK-STAT signal transduction pathway through ligand-independent activation and hypersensitivity of type I cytokine receptors.7 Approximately 95% to 97% of patients with PV and 50% to 60% of patients with ET or PMF harbor a point mutation in exon 14 of the JAK2 gene.8-11 The remaining 2% to 3% of patients with PV carry mutations in JAK2 exon 12.12 CALR or MPL mutations are present in the majority of patients with JAK2-negative ET and PMF. Approximately 10% of patients with MPN carry none of the 3 phenotypic driver mutations and are referred to as “triple-negative.”13,14
The emergence of next-generation sequencing (NGS) has expanded insights into the molecular complexity of MPN, and >50 genes have been reported to be recurrently mutated.15 Mutations outside of JAK2, CALR, and MPL (ie, concomitant somatic mutations) are observed in >50% of patients with MPN, and increasing numbers are observed with disease progression.16,17 The most common classes of concomitant mutations consist of genes involved in DNA methylation (TET2, DNMT3A, IDH1, and IDH2), chromatin modification (ASXL1 and EZH2), RNA splicing (SRSF2, U2AF1, SF3B1, and ZRSR2), signaling pathways (LNK/SH2B3, CBL, NRAS, KRAS, and PTPN1), transcription factors (RUNX1 and NFE2), and DNA damage response/stress signaling (TP53 and PPM1D).7 These mutations may precede the acquisition of the phenotypic driver mutation or occur subsequently in the same or a different clone.16 Concomitant mutations may contribute to phenotype and are often associated with disease progression and inferior survival.15,18-20 Furthermore, the presence of specific concomitant mutations,18 as well as the total number19 and order of acquisition, influences prognosis.21
Internationally, the most widely used first-line cytoreductive therapy in patients with high-risk ET or PV is hydroxyurea (HU). HU effectively reduces elevated peripheral blood counts and the risk of thrombosis.22-24 However, there is conflicting evidence regarding the potential of HU to induce a continuous reduction of the JAK2V7617F-mutated clone.25-27 In contrast, recombinant interferon-α (IFNα), which has been used off-label for the treatment of MPN for >3 decades, has been associated with molecular responses in JAK2V617F-mutated MPN.28-36 A subset of patients achieve molecular remissions and normalization of the bone marrow after long-term treatment, which may be sustained in a minority of patients even after treatment discontinuation, an effect never observed for HU.36-39
Increasing knowledge regarding the complex molecular landscape of MPN has enabled more accurate personalized prediction of outcomes and improved clinical decision-making, particularly in myelofibrosis. However, the predictive role of somatic mutations regarding response and resistance to cytoreductive therapy remains unclear. To address this question, we performed serial genomic profiling on patients enrolled in the DALIAH trial (A Study of Low Dose Interferon Alpha Versus Hydroxyurea in Treatment of Chronic Myeloid Neoplasms), which to our knowledge is the largest randomized controlled phase 3 trial of IFNα vs HU in patients with newly diagnosed MPN.
Methods
Trial design
Genomic profiling by NGS was performed in 202 pretreatment samples and 135 samples obtained after 24 months from patients enrolled in the DALIAH trial. This study was an investigator-initiated, open-label, randomized controlled, parallel-design, clinical phase 3 trial (ClinicalTrials.gov identifier: #NCT01387763). The study was approved by the Danish Regional Science Ethics Committee and the Danish Medicines Agency and was conducted in compliance with the Declaration of Helsinki and Good Clinical Practice. All study participants provided written informed consent before entering the trial.
Patients aged ≥18 years with a diagnosis of ET, PV, Pre-MF, or PMF according to the World Health Organization 2008 criteria40 and evidence of active disease regardless of risk group were eligible to be enrolled. Detailed inclusion and exclusion criteria are provided in the supplemental Methods. Patients aged >60 years were randomly allocated (1:1:1) to receive HU, IFNα-2a, or IFNα-2b; patients aged ≤60 years were randomly allocated (1:1) to receive IFNα-2a or IFNα-2b. Treatment dose was modified based on efficacy and toxicity according to predefined dose levels (supplemental Tables 1 and 2). Clinicohematologic response (CHR) assessment was performed by central review according to the modified 2009 European LeukemiaNet (ET, PV, and Pre-MF)41 and the 2005 European Myelofibrosis Network criteria (PMF).42
NGS analysis
Genomic profiling comprised targeted NGS of 120 myeloid malignancy–associated genes and 1609 informative single nucleotide polymorphisms on chromosome 9p. Detailed information on the sequencing, including a list of sequenced genes and genomic coordinates of all target regions, is provided in the supplemental Methods and supplemental Tables 6 to 8.
Statistical methods
Statistical methods are presented in the supplemental Methods.
Results
Clinical characteristics at baseline
NGS was performed on 202 pretreatment samples from patients randomly allocated to treatment with HU (n = 38) or IFNα-2a (n = 164), and 135 samples were obtained 24 months after initiation of therapy (HU, n = 34; IFNα, n = 101) (Figure 1). Seventy-two patients (36%) had ET, 89 (44%) had PV, 16 (8%) had Pre-MF, and 25 (12%) had PMF (Table 1). The median age was 62 years (range, 20-88 years), and 112 (55%) were male. Thirty-nine (19%) patients had experienced previous major thrombosis, including 17 (8%) with prior stroke (ET, 4 of 72 [6%]; PV, 10 of 89 [11%]; Pre-MF, 1 of 16 [6%]; PMF, 2 of 25 [8%]). Twenty-one (10%) patients received HU from screening and until random allocation in the study due to major thrombosis at diagnosis or platelet count >1000 × 109/L at screening. The median time from screening to randomization in these patients was 21 days (range, 3-45 days). Prior phlebotomy was performed in 90 (45%) patients, with a median number of 3 phlebotomies in each patient (range, 1-29). Due to the design of the study, median age was higher in patients allocated to receive HU (68 years; interquartile range [IQR], 64-71 years) compared with IFNα (59 years; IQR, 46-67 years). Baseline demographic and clinical characteristics are presented in Table 1 and supplemental Tables 9 and 10.
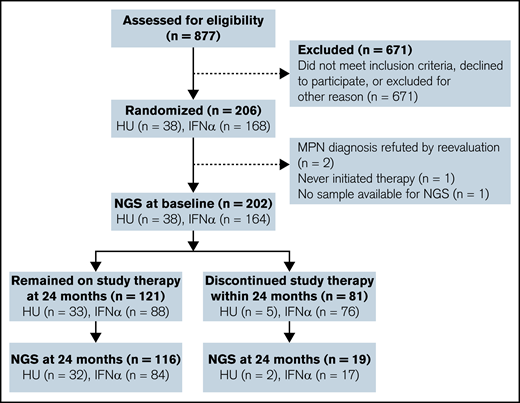
Trial flowchart. NGS was performed on 202 primary MPN samples and 135 samples obtained 24 months after initiation of therapy with either HU or IFNα (IFNα-2a or IFNα-2b). One patient allocated to IFN-α died within 24 months.
Trial flowchart. NGS was performed on 202 primary MPN samples and 135 samples obtained 24 months after initiation of therapy with either HU or IFNα (IFNα-2a or IFNα-2b). One patient allocated to IFN-α died within 24 months.
Baseline demographic and clinical characteristics according to treatment group
| HU (n = 38) | IFNα-2a (n = 82) | IFNα-2b (n = 82) | Total (n = 202) | |
|---|---|---|---|---|
| Patient-related variable | ||||
| MPN subtype | ||||
| ET | 9 (24) | 30 (37) | 33 (40) | 72 (36) |
| PV | 21 (55) | 34 (41) | 34 (41) | 89 (44) |
| Pre-MF | 1 (3) | 9 (11) | 6 (7) | 16 (8) |
| PMF | 7 (18) | 9 (11) | 9 (11) | 25 (12) |
| Age, median (range), y | 68 (60-80) | 60 (21-88) | 58 (20-81) | 62 (20-88) |
| Age group | ||||
| ≤60 y | 0 (0) | 45 (55) | 45 (55) | 90 (45) |
| >60 y | 38 (100) | 37 (45) | 37 (45) | 112 (55) |
| Biological sex | ||||
| Female | 14 (37) | 37 (45) | 39 (48) | 90 (45) |
| Male | 24 (63) | 45 (55) | 43 (52) | 112 (55) |
| History of major thrombosis | 6 (16) | 21 (25) | 12 (15) | 39 (19) |
| History of prior stroke | 3 (8) | 10 (12) | 4 (5) | 17 (8) |
| Phenotypic driver mutation | ||||
| JAK2* | 31 (84) | 62 (80) | 57 (80) | 150 (74) |
| CALR | 6 (16) | 10 (14) | 13 (17) | 29 (14) |
| MPL† | 1 (3) | 4 (6) | 5 (6) | 10 (5) |
| Triple-negative | 1 (3) | 4 (5) | 11 (12) | 16 (8) |
| Disease-related variable | ||||
| Hemoglobin, mmol/L | 9.3 (7.9-10.2) | 9.0 (8.3-9.9) | 8.9 (8.1-9.5) | 9.0 (8.2-9.8) |
| Hematocrit, vol % | 45 (41-52) | 45 (42-47) | 43 (40-47) | 44 (41-49) |
| WBC, ×109/L | 9.9 (8.1-11.5) | 8.9 (7.6-11.6) | 9.5 (7.8-12.7) | 9.4 (7.7-11.7) |
| Platelets, ×109/L | 664 (552-895) | 712 (480-930) | 615 (484-852) | 667 (502-904) |
| Lactate dehydrogenase, U/L | 242 (216-288) | 232 (180-296) | 224 (177-294) | 229 (184-294) |
| Splenomegaly on imaging, ≥13 cm | 15/30 (50) | 21/50 (42) | 31/60 (52) | 67/140 (48) |
| Disease-related symptoms‡ | 19 (50) | 51 (62) | 40 (49) | 110 (54) |
| Pretreatment | ||||
| HU | 4 (11) | 10 (12) | 7 (9) | 21 (10) |
| Phlebotomy | 17 (45) | 34 (41) | 39 (48) | 90 (45) |
| HU (n = 38) | IFNα-2a (n = 82) | IFNα-2b (n = 82) | Total (n = 202) | |
|---|---|---|---|---|
| Patient-related variable | ||||
| MPN subtype | ||||
| ET | 9 (24) | 30 (37) | 33 (40) | 72 (36) |
| PV | 21 (55) | 34 (41) | 34 (41) | 89 (44) |
| Pre-MF | 1 (3) | 9 (11) | 6 (7) | 16 (8) |
| PMF | 7 (18) | 9 (11) | 9 (11) | 25 (12) |
| Age, median (range), y | 68 (60-80) | 60 (21-88) | 58 (20-81) | 62 (20-88) |
| Age group | ||||
| ≤60 y | 0 (0) | 45 (55) | 45 (55) | 90 (45) |
| >60 y | 38 (100) | 37 (45) | 37 (45) | 112 (55) |
| Biological sex | ||||
| Female | 14 (37) | 37 (45) | 39 (48) | 90 (45) |
| Male | 24 (63) | 45 (55) | 43 (52) | 112 (55) |
| History of major thrombosis | 6 (16) | 21 (25) | 12 (15) | 39 (19) |
| History of prior stroke | 3 (8) | 10 (12) | 4 (5) | 17 (8) |
| Phenotypic driver mutation | ||||
| JAK2* | 31 (84) | 62 (80) | 57 (80) | 150 (74) |
| CALR | 6 (16) | 10 (14) | 13 (17) | 29 (14) |
| MPL† | 1 (3) | 4 (6) | 5 (6) | 10 (5) |
| Triple-negative | 1 (3) | 4 (5) | 11 (12) | 16 (8) |
| Disease-related variable | ||||
| Hemoglobin, mmol/L | 9.3 (7.9-10.2) | 9.0 (8.3-9.9) | 8.9 (8.1-9.5) | 9.0 (8.2-9.8) |
| Hematocrit, vol % | 45 (41-52) | 45 (42-47) | 43 (40-47) | 44 (41-49) |
| WBC, ×109/L | 9.9 (8.1-11.5) | 8.9 (7.6-11.6) | 9.5 (7.8-12.7) | 9.4 (7.7-11.7) |
| Platelets, ×109/L | 664 (552-895) | 712 (480-930) | 615 (484-852) | 667 (502-904) |
| Lactate dehydrogenase, U/L | 242 (216-288) | 232 (180-296) | 224 (177-294) | 229 (184-294) |
| Splenomegaly on imaging, ≥13 cm | 15/30 (50) | 21/50 (42) | 31/60 (52) | 67/140 (48) |
| Disease-related symptoms‡ | 19 (50) | 51 (62) | 40 (49) | 110 (54) |
| Pretreatment | ||||
| HU | 4 (11) | 10 (12) | 7 (9) | 21 (10) |
| Phlebotomy | 17 (45) | 34 (41) | 39 (48) | 90 (45) |
Data are presented as no. (%) or median (IQR) unless otherwise indicated.
Mutated JAK2V617F or JAK2 exon 12 mutation.
Coexistence of mutated MPL and JAK2V617F was detected in 3 patients.
Constitutional symptoms, microcirculatory disturbances, or pruritus.
Somatic mutations at baseline
Somatic mutations in 34 genes were detected by NGS in 191 (95%) patients at baseline. MPN phenotypic driver mutations were present in 92% of the patients: JAK2 (74%; JAK2V617F, 73%; JAK2 exon 12, 1%), CALR (14%; type 1, 11%; type 2, 3%), or MPL (5%) (Figure 2; supplemental Table 11). No somatic mutations were detected in 11 patients (5%), 1 mutation was detected in 88 patients (44%), 2 mutations in 55 patients (27%), and ≥3 mutations in 48 patients (24%). The number of mutations was significantly different based on diagnosis (mean number ± standard deviation of mutations in ET, 1.6 ± 1.5; PV, 2.0 ± 1.3; Pre-MF, 2.1 ± 1.2; PMF, 1.9 ± 1.9). Although the presence of phenotypic driver mutations is usually considered mutually exclusive, we found coexistence of JAK2V617F and MPL mutations in 3 patients (1%). Sixteen (8%) patients (ET, n = 12; PV, n = 2; Pre-MF, n = 1; PMF, n = 1) were triple-negative for JAK2, CALR, and MPL mutations. The median JAK2 variant allele frequency (VAF) at baseline was 0.25 (range, 0.01-0.94), and JAK2 uniparental disomy (JAK2-UPD) was observed in 28%. The median JAK2 VAF was significantly higher among patients with JAK2-UPD (0.48; IQR, 0.35-0.68) compared with those without JAK2-UPD (0.15; IQR, 0.09-0.26; P < .0001). The most frequent concomitant mutations at baseline affected 3 genes: TET2 (24%), DNMT3A (16%), and ASXL1 (10%). Spliceosome gene mutations were found in 4% (SF3B1, n = 6; SRSF2, n = 2; U2AF1, n = 1; ZRSR2, n = 1), and mutations involving RAS/MAPK signaling, including CBL, KRAS, NRAS, NF1, PTPN11, and RIT1, were detected in 6%.
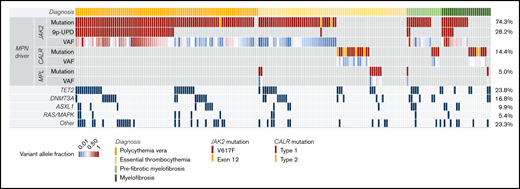
Genomic profiling of somatic mutations in baseline samples by NGS (comutation plot). Each column represents 1 patient (n = 202), and the rows represent different somatic mutations. The VAF for each phenotypic driver mutation is color coded. The frequency of specific somatic mutations is listed on the right border of the figure. Somatic mutations in 34 different genes were detected in 191 (95%) patients, including 92% with MPN phenotypic driver mutations: JAK2, 74%; CALR, 14%; or MPL, 5%. JAK2-UPD was observed in 28% and was significantly associated with PV (Kruskal-Wallis test, P < .0001). The most frequent concomitant mutations affected 3 genes: TET2 (24%), DNMT3A (16%), and ASXL1 (10%). 9p-UPD, uniparental disomy of chromosome 9p.
Genomic profiling of somatic mutations in baseline samples by NGS (comutation plot). Each column represents 1 patient (n = 202), and the rows represent different somatic mutations. The VAF for each phenotypic driver mutation is color coded. The frequency of specific somatic mutations is listed on the right border of the figure. Somatic mutations in 34 different genes were detected in 191 (95%) patients, including 92% with MPN phenotypic driver mutations: JAK2, 74%; CALR, 14%; or MPL, 5%. JAK2-UPD was observed in 28% and was significantly associated with PV (Kruskal-Wallis test, P < .0001). The most frequent concomitant mutations affected 3 genes: TET2 (24%), DNMT3A (16%), and ASXL1 (10%). 9p-UPD, uniparental disomy of chromosome 9p.
Association between somatic mutations and clinical characteristics at baseline
At baseline, mutations in JAK2 were detected in 98% of patients with PV and 53%, 69%, and 56% of patients with ET, Pre-MF, and PMF, respectively. JAK2-UPD was most commonly found in patients with PV (54%) (Figure 2), where it was significantly associated with higher hemoglobin level (P = .0003), higher hematocrit level (P < .0001), higher neutrophil count (P = .039), and lower platelet count (P < .0001) compared with PV patients without JAK2-UPD (supplemental Table 12). JAK2-UPD was not detected in any patients with ET. Among patients with ET, Pre-MF, and PMF, patients with ET were more likely to present with triple-negative disease (ET, 18%; PV, 2%; Pre-MF, 6%; PMF, 4%; P = .007), which was significantly associated with younger age compared with patients harboring 1 of the 3 phenotypic driver mutations (median, 44 years vs 64 years; P = .006). Among patients with ET, Pre-MF, and PMF, mutated CALR was significantly associated with higher platelet count (P = .004) or elevated lactate dehydrogenase levels (P = .0008) compared with patients with JAK2 (+/−MPL)-mutated MPN or patients with triple-negative MPN (supplemental Table 13).
The most frequent concomitant mutations (ie, in TET2, DNMT3A, ASXL1, RAS/MAPK signaling, and RNA splicing genes) among all MPN subtypes were detected in both JAK2-mutated and JAK2 wild-type (WT) patients. However, coexistence of ASXL1 was significantly associated with JAK2, as it was present in 13% of JAK2-mutated patients compared with 2% of JAK2 WT patients (P = .029) (supplemental Table 14). Mutations in TET2, DNMT3A, or ASXL1 were significantly associated with older age (≥60 years) (54% vs 26%; P < .0001), as well as with a history of major thrombosis (odds ratio [OR], 2.11 [95% confidence interval (CI), 1.04-4.37; P = .038]; age-adjusted OR, 1.96 [95% CI, 0.94-4.12; P = .073]) and in particular prior stroke (OR, 5.21 [95% CI, 1.64-16.67; P = .005]; age-adjusted OR, 5.29 [95% CI, 1.59-17.54; P = .007]) compared with patients without these mutations. Also, TET2 alone was significantly associated with prior stroke (age-adjusted OR, 3.03; 95% CI, 1.03-9.01; P = .044). No other significant baseline associations were detected between clinical characteristics and baseline mutational status.
Treatment discontinuation within 24 months
At 24 months, 40% of all patients had discontinued study medication (supplemental Table 15). The most frequent reason for treatment discontinuation across all treatment groups was treatment-related toxicity: HU, 8%; IFNα-2a, 30%; and IFNα-2b, 38%. One patient with CALR-positive PMF and a history of chronic obstructive pulmonary disease died of pneumonia after ∼17 months of treatment with IFNα-2b. None of the patients transformed to post-ET/PV myelofibrosis or secondary acute myeloid leukemia.
CHR at 24 months
At 24 months, 121 patients were on study medication and eligible for CHR assessment. Missing data made response evaluation impossible in 3 of these patients. CHR was achieved in 21% (95% CI, 10-37) treated with HU and 26% (95% CI, 19-33) treated with IFNα (IFNα-2a, 30%; IFNα-2b, 21%) (P = .68) (Figure 3A-B; supplemental Table 16). Median time to CHR was 5.7 months (IQR, 1.8-10.5 months) for HU, 4.9 months (IQR, 2.1-8.9 months) for IFNα-2a, and 6.0 months (IQR, 1.8-10.1 months) for IFNα-2b. Of note, 31 (19%) patients allocated to receive IFNα received either pretreatment with HU (n = 17) and/or combination treatment with IFNα and HU (n = 28) within 24 months after treatment allocation (supplemental Table 17). At CHR assessment at 24 months, 7 patients (HU, n = 1; IFNα, n = 6) received combination treatment. The median duration of combination treatment among these patients was 14.3 months (range, 6.2-18.4 months). Two were in CHR, 1 was not evaluable due to missing data, and 4 were nonresponders.
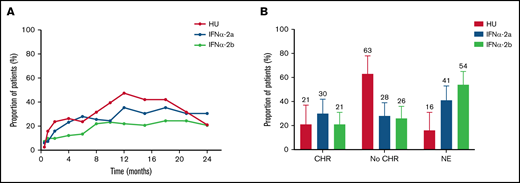
Complete CHR response at 24 months. (A) Proportion of patients with complete CHR over time according to treatment group. Median time to CHR was 5.7 months (IQR, 1.8-10.5 months) for HU, and 4.9 months (IQR, 2.1-8.9 months) and 6.0 months (IQR, 1.8-10.1 months) for patients treated with IFNα-2a or IFNα-2b, respectively. The CHR rate reached a maximum after 12 months among patients treated with HU (47%), whereas the CHR rate increased almost gradually over time among patients treated with IFNα-2a or IFNα-2b. (B) Proportion of patients with CHR at 24 months according to treatment group. CHR was achieved in 8 of 38 (21%; 95% CI, 10-37) patients treated with HU, 25 of 82 (30%; 95% CI, 21-42) patients treated with IFNα-2a, and in 17 of 82 (17%; 95% CI, 13-31) patients treated with IFNα-2b. No significant difference in the CHR rate was detected between HU and the two IFNα groups combined. Patients considered nonevaluable (NE) at 24 months had all discontinued the study therapy to which they were allocated, except 3 patients in whom complete diagnostic workup was not available at 24 months (HU, n = 1; IFNα-2a, n = 2). Error bars are 95% CI upper limits.
Complete CHR response at 24 months. (A) Proportion of patients with complete CHR over time according to treatment group. Median time to CHR was 5.7 months (IQR, 1.8-10.5 months) for HU, and 4.9 months (IQR, 2.1-8.9 months) and 6.0 months (IQR, 1.8-10.1 months) for patients treated with IFNα-2a or IFNα-2b, respectively. The CHR rate reached a maximum after 12 months among patients treated with HU (47%), whereas the CHR rate increased almost gradually over time among patients treated with IFNα-2a or IFNα-2b. (B) Proportion of patients with CHR at 24 months according to treatment group. CHR was achieved in 8 of 38 (21%; 95% CI, 10-37) patients treated with HU, 25 of 82 (30%; 95% CI, 21-42) patients treated with IFNα-2a, and in 17 of 82 (17%; 95% CI, 13-31) patients treated with IFNα-2b. No significant difference in the CHR rate was detected between HU and the two IFNα groups combined. Patients considered nonevaluable (NE) at 24 months had all discontinued the study therapy to which they were allocated, except 3 patients in whom complete diagnostic workup was not available at 24 months (HU, n = 1; IFNα-2a, n = 2). Error bars are 95% CI upper limits.
Somatic mutations on serial sampling
NGS was performed in 135 patients at 24 months, including 113 of 121 patients eligible for CHR assessment (HU, n = 32; IFNα, n = 84) and in 19 who had discontinued study treatment (HU, n = 2; IFNα, n = 17) (supplemental Figure 2). Phenotypic driver mutations remained detectable by NGS at 24 months in all patients. JAK2 VAF decreased in 94% of the patients treated with IFNα and in 75% treated with HU (P = .01). The median absolute JAK2 VAF reduction (baseline to 24 months) was significantly greater in patients treated with IFNα (HU 0.05 vs IFNα 0.11; P = .005). The change in CALR VAF with treatment was more heterogeneous. The CALR VAF decreased in 80% of patients allocated to HU and 78% allocated to IFNα (P = .99) (median VAF reduction, HU 0.02 vs IFNα 0.04; P = .63) (supplemental Table 16). Among patients treated with IFNα, those with JAK2-UPD had a greater absolute JAK2 VAF reduction (median, 0.49 to 0.17) compared with those without JAK2-UPD (median, 0.15 to 0.08) (P < .0001). No significant reduction in JAK2 VAF was observed among patients with JAK2-UPD treated with HU (median, 0.44 to 0.30) than those without JAK2-UPD (median, 0.22 to 0.08) (P = .76).
Mutations were detected in 30 genes at 24 months, including 3 not observed at baseline (EP300, IDH2, and PHF6) (supplemental Figure 2). Thirty-eight treatment-emergent mutations were detected in 32 patients (HU, n = 14; IFNα, n = 18), of whom 4 patients had discontinued treatment.
DNMT3A was the most frequent treatment-emergent mutation (n = 15 [39%]), followed by TET2 (n = 4 [11%]), ASXL1 (n = 3 [8%]), PPM1D (n = 3 [8%]), and TP53 (n = 3 [8%]) (Figure 4A; supplemental Table 18). The VAF of treatment-emergent mutations was low (median, 1.5%), and they primarily occurred in JAK2-positive patients (97%) (Figure 4B). The NGS platform enabled simultaneous evaluation of the following: (1) the molecular response of MPN phenotypic driver mutations; (2) JAK2-UPD at 9p; and (3) detection of treatment-emergent mutations in any of the >100 genes assessed, allowing us to uncover the complexity of molecular responses (Figure 4C-D). Treatment-emergent mutations in DNMT3A were more commonly observed in patients treated with IFNα (11 of 18 [61%]) than HU (3 of 14 [21%]) (P = .046). In contrast, treatment-emergent mutations in PPM1D or TP53 were more common in patients who received HU (5 of 14 [36%]) compared with IFNα (1 of 18 [6%]) (P = .06) (supplemental Figure 3).
![Treatment-emergent mutations at 24 months. (A) Number of treatment-emergent mutations at 24 months. Thirty-eight treatment-emergent mutations were detected in 32 patients, 4 of whom had discontinued treatment. Mutations were defined as treatment-emergent if: (1) the VAF was <0.01 in the baseline sample and ≥0.01 in the 24-month sample (n = 36); or (2) if the VAF was ≥0.01 in the baseline sample and had a more than fourfold increase in the 24-month sample (n = 2). The most frequent treatment-emergent mutations were detected in DNMT3A (n = 15 [39%]), followed by TET2 (n = 4 [11%]), ASXL1 (n = 3 [8%]), PPM1D (n = 3 [8%]), and TP53 (n = 3 [8%]). (B) VAF of treatment-emergent mutations at baseline and posttreatment at 24 months. The median VAF of treatment-emergent mutations was low (median, 1.5%) and primarily occurred in JAK2-mutated patients (97%). Representative examples of treatment-emergent mutations detected in patients treated with HU (C) and IFNα (D). MPN phenotypic driver mutations are depicted with blue lines, treatment-emergent concomitant mutations with red lines, and other concomitant mutations with black lines. The upper rows of panels C and D also show uniparental disomy of chromosome 9p (9p-UPD) analysis. In all examples, 9p-UPD is no longer detectable posttreatment at 24 months, which is concordant with the decrease in mutant JAK2 VAF.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/7/10.1182_bloodadvances.2021004856/3/m_advancesadv2021004856f4.png?Expires=1765922775&Signature=5GMG5PKAjq7iH5NJ4JF0SD-q~cR~74YqW0bHslPDdt7DMo0BtrhxY44CKAn0USi1gHYqWfLw0bsmCSaDISKs~oFBYUCgzgrbp9d6td3sDa9wsoZuDVhz-y1AuoSfQTCJJgqMblJqApZhVAPwb1zJ4p006cMtUMw8lxBkdZxAVfwlH3vFQZZAAsYeqyglJP8NSVjDmY55l8-~Uc7~Nb05RdzX6m4MjedIzxyNsZMMJJuKcv-OFpX3PXbYRd4gmSs10-4wu0POSYssH7m-puHD9ZBGLTY5aGNi7jrtOw8m8qDcLptKAoG5wbgbKwqiBQwdWlldjXyXjhIZomWQzM8RkA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Treatment-emergent mutations at 24 months. (A) Number of treatment-emergent mutations at 24 months. Thirty-eight treatment-emergent mutations were detected in 32 patients, 4 of whom had discontinued treatment. Mutations were defined as treatment-emergent if: (1) the VAF was <0.01 in the baseline sample and ≥0.01 in the 24-month sample (n = 36); or (2) if the VAF was ≥0.01 in the baseline sample and had a more than fourfold increase in the 24-month sample (n = 2). The most frequent treatment-emergent mutations were detected in DNMT3A (n = 15 [39%]), followed by TET2 (n = 4 [11%]), ASXL1 (n = 3 [8%]), PPM1D (n = 3 [8%]), and TP53 (n = 3 [8%]). (B) VAF of treatment-emergent mutations at baseline and posttreatment at 24 months. The median VAF of treatment-emergent mutations was low (median, 1.5%) and primarily occurred in JAK2-mutated patients (97%). Representative examples of treatment-emergent mutations detected in patients treated with HU (C) and IFNα (D). MPN phenotypic driver mutations are depicted with blue lines, treatment-emergent concomitant mutations with red lines, and other concomitant mutations with black lines. The upper rows of panels C and D also show uniparental disomy of chromosome 9p (9p-UPD) analysis. In all examples, 9p-UPD is no longer detectable posttreatment at 24 months, which is concordant with the decrease in mutant JAK2 VAF.
Treatment-emergent mutations at 24 months. (A) Number of treatment-emergent mutations at 24 months. Thirty-eight treatment-emergent mutations were detected in 32 patients, 4 of whom had discontinued treatment. Mutations were defined as treatment-emergent if: (1) the VAF was <0.01 in the baseline sample and ≥0.01 in the 24-month sample (n = 36); or (2) if the VAF was ≥0.01 in the baseline sample and had a more than fourfold increase in the 24-month sample (n = 2). The most frequent treatment-emergent mutations were detected in DNMT3A (n = 15 [39%]), followed by TET2 (n = 4 [11%]), ASXL1 (n = 3 [8%]), PPM1D (n = 3 [8%]), and TP53 (n = 3 [8%]). (B) VAF of treatment-emergent mutations at baseline and posttreatment at 24 months. The median VAF of treatment-emergent mutations was low (median, 1.5%) and primarily occurred in JAK2-mutated patients (97%). Representative examples of treatment-emergent mutations detected in patients treated with HU (C) and IFNα (D). MPN phenotypic driver mutations are depicted with blue lines, treatment-emergent concomitant mutations with red lines, and other concomitant mutations with black lines. The upper rows of panels C and D also show uniparental disomy of chromosome 9p (9p-UPD) analysis. In all examples, 9p-UPD is no longer detectable posttreatment at 24 months, which is concordant with the decrease in mutant JAK2 VAF.
Association between somatic mutations and complete CHR on serial sampling
The probability of CHR at 24 months was not associated with JAK2 (P = .27), JAK2-UPD (P = .35), or CALR (P = .10) baseline mutational status or concomitant mutations in DNMT3A, TET2, or ASXL1 (P = .40) in the entire cohort or when stratifying according to treatment group (HU vs IFNα). Analysis for associations with other concomitant mutations was not feasible due to their low frequency in the cohort. CHR at 24 months was obtained in 34 (23%) of 150 patients with JAK2 mutations, 11 (37%) of 29 patients with CALR mutations, and in 18 (21%) of 84 patients with DNMT3A, TET2, or ASXL1 mutations. The JAK2 VAF declined significantly in patients randomized to receive HU achieving CHR (median, 0.25 to 0.08; P = .03) but not in those not achieving CHR (median, 0.30 to 0.26; P = .10). Among JAK2-positive patients randomized to receive IFNα, those attaining CHR had a greater reduction in the JAK2 VAF (median, 0.29 to 0.07; P < .0001) compared with patients who did not achieve CHR (median, 0.27 to 0.14; P < .0001) (Figure 5A). In contrast, the mutant CALR VAF did not significantly decline in either those achieving CHR during treatment with IFNα (median, 0.17 to 0.13; P = .078) or in those not achieving CHR (median, 0.21 to 0.17; P = .066) (Figure 5B). Of note, only 18 CALR-positive patients allocated to receive IFNα were evaluable for response at 24 months. None of the 5 CALR-positive patients allocated to receive HU achieved CHR.
![Association between somatic mutations and complete CHR on serial sampling. (A-B) Molecular response among patients allocated to IFNα at baseline (Pre) and at 24 months (Post) of treatment by complete CHR. (A) Among JAK2-mutated patients, those attaining CHR at 24 months had a greater reduction in the JAK2 VAF (median, 0.29 to 0.07; P < .0001) compared with JAK2-mutated patients who did not achieve CHR (median, 0.27 to 0.14; P < .0001). (B) The CALR VAF did not significantly decline among patients achieving CHR nor among those not achieving CHR at 24 months. The middle horizontal lines indicate the median value; box limits indicate the 5th and 95th percentiles, and whiskers indicate the range. All observations are represented by a dot. (C) Number of patients with no treatment-emergent mutations. Significantly more patients with no treatment-emergent mutations treated with IFNα achieved CHR (35 of 68 [51%]) compared with patients treated with HU (4 of 18 [22%]) (P = .03). (D) Number of patients with treatment-emergent mutations. No difference in the number of patients failing to achieve CHR was observed between patients treated with HU (10 of 13 [77%]) or IFNα (9 of 14 [64%]) (P = .68). (E) Number of patients with treatment-emergent DNMT3A mutations. (F) Number of patients with treatment-emergent non-DNMT3A mutations. Treatment-emergent DNMT3A mutations were significantly enriched in patients treated with IFNα failing to achieve CHR (8 of 9 [89%]) compared with treatment-emergent non-DNMT3A mutations (1 of 5 [20%]) (P = .02).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/7/10.1182_bloodadvances.2021004856/3/m_advancesadv2021004856f5.png?Expires=1765922775&Signature=bFbmMS~HawNdkcfqFOERYUj9ltts4GeyIDijKbPoosChO9-IQRqa7BR-essvLkeT3jPocjz09fLuNQVjlsGNKC50x1u3DNrhoobde~QuBowD59pywNXcWB58mmx7P6gIukzibriSM9579QsyA~yEuwvEk1K8EO7UkUh0m8eeHjSxK9ssnhsfKgpRVkzEwq4JMXIQhce6IwfULUpdhVJAsjT85m5aLohr1fStLt6wW9T2o2xPpmwKc3VFJDzTjz4hWgKGywY8HjDKbZ34bjpEV1wmU5JttY5wjIbi8ie58FtMQk~tIf2V-7mbEFZ4P~-RXxoNghVHlf1iQvpkqvKJpQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Association between somatic mutations and complete CHR on serial sampling. (A-B) Molecular response among patients allocated to IFNα at baseline (Pre) and at 24 months (Post) of treatment by complete CHR. (A) Among JAK2-mutated patients, those attaining CHR at 24 months had a greater reduction in the JAK2 VAF (median, 0.29 to 0.07; P < .0001) compared with JAK2-mutated patients who did not achieve CHR (median, 0.27 to 0.14; P < .0001). (B) The CALR VAF did not significantly decline among patients achieving CHR nor among those not achieving CHR at 24 months. The middle horizontal lines indicate the median value; box limits indicate the 5th and 95th percentiles, and whiskers indicate the range. All observations are represented by a dot. (C) Number of patients with no treatment-emergent mutations. Significantly more patients with no treatment-emergent mutations treated with IFNα achieved CHR (35 of 68 [51%]) compared with patients treated with HU (4 of 18 [22%]) (P = .03). (D) Number of patients with treatment-emergent mutations. No difference in the number of patients failing to achieve CHR was observed between patients treated with HU (10 of 13 [77%]) or IFNα (9 of 14 [64%]) (P = .68). (E) Number of patients with treatment-emergent DNMT3A mutations. (F) Number of patients with treatment-emergent non-DNMT3A mutations. Treatment-emergent DNMT3A mutations were significantly enriched in patients treated with IFNα failing to achieve CHR (8 of 9 [89%]) compared with treatment-emergent non-DNMT3A mutations (1 of 5 [20%]) (P = .02).
Association between somatic mutations and complete CHR on serial sampling. (A-B) Molecular response among patients allocated to IFNα at baseline (Pre) and at 24 months (Post) of treatment by complete CHR. (A) Among JAK2-mutated patients, those attaining CHR at 24 months had a greater reduction in the JAK2 VAF (median, 0.29 to 0.07; P < .0001) compared with JAK2-mutated patients who did not achieve CHR (median, 0.27 to 0.14; P < .0001). (B) The CALR VAF did not significantly decline among patients achieving CHR nor among those not achieving CHR at 24 months. The middle horizontal lines indicate the median value; box limits indicate the 5th and 95th percentiles, and whiskers indicate the range. All observations are represented by a dot. (C) Number of patients with no treatment-emergent mutations. Significantly more patients with no treatment-emergent mutations treated with IFNα achieved CHR (35 of 68 [51%]) compared with patients treated with HU (4 of 18 [22%]) (P = .03). (D) Number of patients with treatment-emergent mutations. No difference in the number of patients failing to achieve CHR was observed between patients treated with HU (10 of 13 [77%]) or IFNα (9 of 14 [64%]) (P = .68). (E) Number of patients with treatment-emergent DNMT3A mutations. (F) Number of patients with treatment-emergent non-DNMT3A mutations. Treatment-emergent DNMT3A mutations were significantly enriched in patients treated with IFNα failing to achieve CHR (8 of 9 [89%]) compared with treatment-emergent non-DNMT3A mutations (1 of 5 [20%]) (P = .02).
We divided the patients available for CHR assessment and serial sampling (n = 113) into 2 groups: (1) those in whom no treatment-emergent mutations were detected (HU, n = 18; IFNα, n = 68) (Figure 5C); and (2) those in whom treatment-emergent mutations were detected (HU, n = 13; IFNα, n = 14) (Figure 5D). We further divided the latter group into those in whom DNMT3A mutations were detected (HU, n = 2; IFNα, n = 9) (Figure 5E) and those in whom non-DNMT3A treatment-emergent mutations were detected (HU, n = 11; IFNα, n = 5) (Figure 5F). Within the group in whom no treatment-emergent mutations were detected, significantly more patients treated with IFNα achieved CHR (35 of 68 [51%]) compared with patients treated with HU (4 of 18 [22%]) (P = .034). Of 27 patients with treatment-emergent mutations at 24 months and available for response assessment, 19 (70%) failed to achieve CHR (HU, 10 of 13 [77%]; IFNα, 9 of 14 [64%]) (P = .68). We found that treatment-emergent DNMT3A mutations were significantly enriched among patients treated with IFNα failing to achieve CHR (8 of 9 [89%]) compared with treatment-emergent non-DNMT3A mutations (1 of 5 [20%]) (P = .02). Among patients randomized to receive HU, the 2 patients with treatment-emergent DNMT3A mutations did not obtain CHR compared with CHR in 3 (27%) of 11 patients with treatment-emergent non-DNMT3A mutations.
Discussion
To determine the impact of molecular genetics on response to front-line cytoreductive therapy in MPN, we performed sequential molecular profiling on samples obtained from patients enrolled in the DALIAH trial, a randomized controlled, phase 3 clinical trial of IFNα vs HU in patients newly diagnosed with MPN.
To enable detailed molecular profiling, we first developed a custom targeted NGS assay encompassing 120 myeloid malignancy–associated genes. A significant age-independent association was found between the presence of a mutation in TET2, DNMT3A, or ASXL1 at baseline and a history of stroke, which remained significant for mutated TET2 alone. We also found an association between the presence of a TET2, DNMT3A, or ASXL1 mutation and a history of major thrombosis. However, this association did not retain significance when adjusted for age. Previous studies have found an age-independent association between the presence of one or more mutations in TET2, DNMT3A, or ASXL1 and thrombotic events in PV, which was retained for the presence of a TET2 mutation alone.43 However, an association between TET2, DNMT3A, or ASXL1 mutations and thrombosis in PV was not found in earlier studies.44,45 A novel feature of the NGS assay was the ability to determine the presence of JAK2-UPD on 9p, allowing us to distinguish patients who were heterozygous for the JAK2 mutation from those who were homozygous. This is particularly informative in patients with a JAK2 VAF <50%. We incorporated JAK2-UPD into our analysis of the molecular response, and by combining sequential mutational and JAK2-UPD analyses, we were able to uncover distinct treatment responses in independent clones/subclones in individual patients (discussed later).
In terms of treatment response, we first focused our attention on the 2 most common MPN phenotypic driver mutations, JAK2 and CALR. We found that more patients treated with IFNα had a decrease in the mutant JAK2 VAF than patients treated with HU. Furthermore, the median mutant JAK2 VAF reduction was significantly greater among patients treated with IFNα than with HU. In contrast, there was no difference in the magnitude of decrease in mutant CALR VAF in patients treated with HU than with IFNα, and the median reduction in mutant CALR VAF was <5% for both HU and IFNα. We found that patients with JAK2-UPD treated with IFNα had a greater decrease in JAK2 VAF than JAK2-mutated patients without JAK2-UPD (not seen with HU). This finding is consistent with a small prospective study of MPN (n = 33 patients) by Mosca et al,46 who reported (in abstract form) that hematopoietic stem cells homozygous for mutated JAK2V167F were more effectively targeted by IFNα than heterozygous cells.
We next evaluated the association between CHR at 24 months and molecular response. CHR rates were similar at 24 months between HU and IFNα, which is in accordance with data (presented in abstract form) from the randomized Myeloproliferative Disorders–Research Consortium (MPD-RC) 112 study of high-risk ET or PV comparing HU with IFNα-2a.47 Interestingly, we found that CHR at 24 months was associated with a significant VAF reduction in JAK2-mutated patients but not in CALR-mutated patients treated with IFNα. Although reductions in mutant CALR VAF in response to IFNα treatment have been reported in MPN,48,49 previous smaller studies, including a recent retrospective study (n = 38 patients) reported by Czech et al,50 have suggested that CALR-mutant MPN cells are less sensitive to IFNα than JAK2-mutated cells.51 Strengths of our findings on this point include that patients were treated in a large prospective randomized trial (n = 202 patients) and that JAK2 and CALR VAF were assessed simultaneously using the same NGS platform. Limitations include the fact that almost one-third of the CALR mutant group were patients with PMF (31%), in addition to patients with ET (55%) and Pre-MF (14%), whereas the JAK2 mutant group was composed primarily of patients with PV (59%) and ET (24%), in addition to patients with Pre-MF (7%) and PMF (10%).
We next turned our attention to treatment-emergent mutations. By serial sampling at 24 months, 38 treatment-emergent mutations were found in 32 patients. Notably, approximately one-half the time a treatment-emergent mutation was detected on serial sampling, the JAK2 VAF was found to have declined by >50%, suggesting that the treatment-emergent mutation had arisen independently or was subclonal to the JAK2 mutant clone. This finding highlights the importance of not restricting molecular analysis in clinical trials to MPN phenotypic driver genes only.
The gene in which we most commonly identified treatment-emergent mutations was DNMT3A (39%), and we found that treatment-emergent DNMT3A mutations were significantly more prevalent in patients treated with IFNα failing to achieve CHR. DNMT3A mutations have been reported to both precede and follow JAK2V617F acquisition, in addition to arising in independent clones in MPN.52,53 As such, these DNMT3A mutations could reflect either treatment-resistant subclones or genetically unrelated clones that develop in parallel to the phenotypic driver clone. The methodology we used in this study did not allow us to distinguish whether preexisting DNMT3A-mutated clones expanded during treatment with IFNα or de novo DNMT3A mutations were induced by IFNα. However, we believe it is highly likely that treatment-emergent DNMT3A mutations were preexisting at baseline and selected for with IFNα therapy. In agreement with this model, studies using ultrasensitive error-corrected sequencing have found that most adults aged >50 years have evidence of clonal hematopoiesis, most commonly involving mutations in DNMT3A.54 In accordance with our finding, Quintás-Cardama et al33 found that the acquisition of a DNMT3A mutation was associated with failure to achieve complete molecular remission in patients with PV and ET treated with IFNα (n = 83). More recently, Stetka et al55 reported (in abstract form) that genetic loss of Dnmt3a confers resistance to treatment with IFN in a JAK2V617F-driven MPN mouse model. Clues to the mechanism by which Dnmt3a loss could render hematopoietic stem and progenitor cells resistant to IFNα are suggested by Jacquelin et al, who found that Dnmt3a loss induced aberrant self-renewal of Jak2-mutant hematopoietic stem and progenitor cells and augmented pro-inflammatory signaling due to increased chromatin accessibility.56,57 It is important to note that the majority of DNMT3A mutations found in MPN (and in this study) are heterozygous missense mutations that do not result in complete loss of DNMT3A function. Mutations in PPM1D or TP53 were found more frequently in patients treated with HU, a finding consistent with several earlier reports linking mutations in these genes to chemotherapy exposure in other contexts.58-61 However, it is important to note that low allele burden TP53 mutations have been associated with older age in chronic-phase MPN, and randomization to HU was restricted to patients aged >60 years in our study.62
In the current study, molecular response was assessed at 24 months. In previous studies, the JAK2 molecular response has been shown to increase gradually with time upon treatment with IFNα,28-36 whereas the molecular response is often transient in patients treated with HU.25-27,30 In the recently reported randomized CONTINUATION-PV trial of 171 patients with PV allocated to receive ropeginterferon α-2b (ropeg) or best available therapy (mainly HU), significantly higher JAK2 molecular responses were observed among patients treated with ropeg after 24 and 36 months of treatment.30 Furthermore, the higher JAK2 molecular response rate in the ropeg arm was even more striking after 48 months and was sustained at 60 months.63 This is consistent with the reported durable JAK2 molecular responses beyond 5 years in patients with ET and PV treated with IFNα-2a.32 Notably, ropeg, which is dosed every 2 weeks, seemed to be well tolerated in the CONTINUATION-PV trial,64 in contrast to our study, in which 34% of the IFNα–treated patients discontinued study medication for toxicity within 24 months, despite a low-dose regimen.
Although NGS technologies are increasingly used in clinical practice to provide prognostic information and guide treatment decisions in MPN, sequential genomic profiling is not usually performed outside of clinical trials. In terms of counseling patients on the possible molecular consequences of cytoreductive therapy, our findings can be summarized as follows: (1) coexisting mutations are present at diagnosis in ∼50% of patients with ET and PV; (2) concomitant mutations may be present in the same cell or a different cell than the MPN disease-initiating mutation (ie, JAK2, CALR, MPL); and (3) not all mutations respond in the same way to IFNα or HU treatment. It is important to acknowledge that we currently have an incomplete understanding of the clinical significance of concomitant mutations in ET and PV, particularly with respect to treatment. Therefore, additional studies with long follow-up are required to understand the clinical significance of an IFNα–induced reduction in JAK2V617F allele burden and mutations, such as DNMT3A, expanding during IFNα treatment.
Because the primary goal of cytoreductive therapy in MPN is to reduce the risk of thrombotic and vascular events, we are not suggesting any immediate change in clinical practice based on our results. However, we suggest several next steps to further advance the understanding of the differential effects of cytoreductive therapy on clonal MPN cells: (1) aggregate currently available molecular genetic data on patients treated with IFNα to increase statistical power and further validate key findings; (2) perform sequential NGS analysis in prospective clinical trials of cytoreductive therapy in PV and ET, and correlate early molecular findings with long-term clinical outcomes (ie, identify molecular genetic biomarkers that predict clinical outcome); and (3) develop low-cost methodologies to enable sequential molecular genetic analysis as a routine component of MPN clinical care.
Finally, newly emerging data have shown that acquisition of the JAK2V617F mutation may occur decades before the development of MPN,65,66 consistent with a long preclinical phase termed JAK2-mutant clonal hematopoiesis (CH). Although not all patients with JAK2-mutant CH develop MPN, it is a clinically relevant entity associated with an increased risk of cardiovascular disease67 and venous thrombosis.68,69 Due to the ability of IFNα to reduce the JAK2-mutated clone,70 early identification and upfront treatment of individuals with JAK2-mutant CH raise the possibility that IFNα could have the potential to prevent the development of MPN and/or decrease JAK2-mutant CH-associated morbidity and mortality. The development of specialized CH clinics to identify such individuals and offer them clinical trials (eg, with IFNα) is a recent development in this regard.71
In conclusion, we performed comprehensive molecular profiling of patients with newly diagnosed MPN treated with front-line cytoreductive therapy (IFNα vs HU) and identified treatment- and mutation-specific patterns of response that have clinical implications.
Acknowledgments
The authors thank all the participating patients and their families, all investigators, research coordinators, and site staff in Denmark.
The sponsors of this study are nonprofit organizations that support science in general. They had no role in gathering, analyzing, or interpreting the data.
This research was supported by grants from OUH Frie Forskningsmidler, OUH-Region Sjaelland Faelles Forskningspulje, Fonden til Lægevidenskabens Fremme, Ellen og Aage Fausboells Helsefond af 1975, Swedish Orphan, Region Sjaellands Sundhedsvidenskabelige Forskningsfond (Ordinaer), Region Sjællands Sundhedsvidenskabelige Forskningsfond (RSSF) 2018, Gangstefonden, and the National Institutes of Health, National Cancer Institute (K08CA204734, R.C.L.). The NGS studies were funded by an Interferon Initiative grant (A.M.) from the MPN Research Foundation. A.M. acknowledges funding from the National Institutes of Health, National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL131835). A.M. is a Scholar of The Leukemia & Lymphoma Society.
Authorship
Contribution: A.M., H.C.H., and R.C.L designed the study, analyzed and interpreted data, edited the manuscript, and oversaw the study; R.C.L. designed and performed genomic analyses and created figures; T.A. Knudsen collected clinical data, analyzed and interpreted data, created figures, and wrote the first version of the manuscript; L.F.O. collected clinical data; D.L.H. constructed the clinical database; C.E., D.L.H., L.K., T.S.L., and V.S. analyzed and interpreted data and edited the manuscript; W.D. processed samples and analyzed data; C.L. analyzed data; D.S.N., L.W., K.S., and T.A. Knudsen performed statistical analysis; D.E.F., D.L.H., J. Starklint, J. Stentoft, K.d.S., M.F., M.B., M.T., M.T.S., O.W.B., T.A. Kruse, T.M.-A., T.K.K., T.S.L., and U.M.O. performed research; C.J.G. analyzed NGS data; A.N., A.R.T., and B.W. oversaw the generation of NGS data; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: D.L.H. received research funding from Alexion. D.S.N. received research funding from Celgene and Pharmacyclics; and has equity ownership in Madrigal Pharmaceuticals. H.C.H. has received research funding from Novartis; and is on the data monitoring board for AOP Orphan. R.C.L. has received research funding from Jazz Pharmaceuticals and MedImmune; and consultancy for Takeda Pharmaceuticals and Bluebird Bio. A.M. reports research funding and consulting for Janssen; research funding from Actuate Therapeutics; advisory board membership for Constellation; steering committee membership for PharmaEssentia; and consulting for Relay Therapeutics.
Correspondence: Ann Mullally, Harvard Institutes of Medicine Building, Room 738, 77 Ave Louis Pasteur, Boston, MA 02115; e-mail: ann_mullally@dfci.harvard.edu.

