

Key Points
Primary and secondary vitreoretinal lymphomas show the same genetic profile and belong to the MCD/C5 cluster of DLBCL.
Mutational analysis of vitreous fluid is a sensitive and specific tool for diagnosis of vitreoretinal lymphoma.

Abstract
Vitreoretinal lymphoma (VRL) is a rare subtype of diffuse large B-cell lymphoma (DLBCL) considered a variant of primary central nervous system lymphoma (PCNSL). The diagnosis of VRL requires examination of vitreous fluid, but cytologic differentiation from uveitis remains difficult. Because of its rarity and the difficulty in obtaining diagnostic material, little is known about the genetic profile of VRL. The purpose of our study was to investigate the mutational profile of a large series of primary and secondary VRL. Targeted next-generation sequencing using a custom panel containing the most frequent mutations in PCNSL was performed on 34 vitrectomy samples from 31 patients with VRL and negative controls with uveitis. In a subset of cases, genome-wide copy number alterations (CNAs) were assessed using the OncoScan platform. Mutations in MYD88 (74%), PIM1 (71%), CD79B (55%), IGLL5 (52%), TBL1XR1 (48%), ETV6 (45%), and 9p21/CDKN2A deletions (75%) were the most common alterations, with similar frequencies in primary (n = 16), synchronous (n = 3), or secondary (n = 12) VRL. This mutational spectrum is similar to MYD88mut/CD79Bmut (MCD or cluster 5) DLBCL with activation of Toll-like and B-cell receptor pathways and CDKN2A loss, confirming their close relationship. OncoScan analysis demonstrated a high number of CNAs (mean 18.6 per case). Negative controls lacked mutations or CNAs. Using cell-free DNA of vitreous fluid supernatant, mutations present in cellular DNA were reliably detected in all cases examined. Mutational analysis is a highly sensitive and specific tool for the diagnosis of VRL and can also be applied successfully to cell-free DNA derived from the vitreous.
Introduction
Vitreoretinal lymphoma (VRL) is a rare malignancy, which is usually classified as a diffuse large B-cell lymphoma (DLBCL) and is considered a variant of primary central nervous system (CNS) lymphoma (PCNSL). In primary VRL, the neoplastic cells are confined to the vitreous and the retina. However, many patients develop CNS involvement, either simultaneous with or subsequent to the ocular disease. Conversely, CNS lymphoma spreads via the optic nerve to the eyes in 15% to 25% of patients, and systemic dissemination is exceptionally unusual.1-3 Given the rarity of the disease and the difficulty in obtaining diagnostic specimens, the mutational landscape of VRL remains largely unexplored. In contrast, the genetics and expression profile of PCNSL have been examined with a variety of high-throughput techniques, including whole-exome sequencing, array comparative genomic hybridization, and gene expression profiling.4-11 PCNSL usually belongs to the activated B-cell (ABC) type of DLBCL and shows a high rate of somatic hypermutation (SHM) of the rearranged immunoglobulin heavy chain (IGH) gene.12-14 Common molecular hallmarks of PCNSL include frequent coactivation of Toll-like receptor (TLR) and B-cell receptor (BCR) signaling through mutations in myeloid-derived factor 88 (MYD88) and CD79B with downstream constitutive activation of the NF-κB pathway, as well as typical homozygous losses of CDKN2A with resulting genomic instability and immune escape mechanisms, such as loss of the HLA locus on 6p21.33 and overexpression of PD-L1/2.6,15,16 With the exception of cases in immunosuppressed individuals, PCNSL lack an association with Epstein Barr-virus.12 PCNSL is set apart from conventional nodal DLBCL in the current World Health Organization lymphoma classification based on clinical features, such as confinement to the CNS throughout the disease course in most patients, as well as its distinct molecular profile.17
In contrast to PCNSL, the few studies on VRL published to date have either examined single or a few genetic alterations or investigated only a small number of cases.18-22 Based on these limited results, primary VRL seems to show a similar spectrum of mutations as PCNSL. Secondary involvement of the vitreous by systemic DLBCL is rare,23 with the notable exception of primary DLBCL of the testis, which frequently disseminates to or relapses in the CNS, including intraocular structures.24 Of interest, testicular DLBCL shares many features with PCNSL, including predominance of ABC type and frequent MYD88 and CD79B mutations, as well as losses of 6p21.33 and 9p21/CDKN2A.6,25 Because the CNS, including the vitreoretinal space and testis, is considered an immune-privileged site, immune evasion may represent a common pathogenetic mechanism for all 3 sites.26,27
The clinical presentation of VRL is variable and frequently mimics signs and symptoms of posterior uveitis and therefore is labeled a “masquerade” syndrome.1,28 This lack of specific clinical symptoms, together with the difficulty in obtaining an adequate sample, frequently results in diagnostic delay. Ultimately, the diagnosis of VRL relies on cytological and immunocytochemical or flow cytometric examination of vitrectomy specimens, supported by B-cell clonality analyses and determination of the intravitreal interleukin (IL)-10/IL-6 ratio.29,30 The diagnostic sensitivity of conventional cytology including immunocytochemistry is low, because of frequent degenerative changes of cellular constituents, especially after steroid therapy. Although molecular determination of clonality is helpful, both false-positive and false-negative results occur, related to pseudoclonal results in the presence of few B cells and SHM inhibiting primer binding, respectively.1,31,32
Mutational analysis, therefore, may represent a useful approach to increasing the diagnostic yield of vitrectomy specimens, and we and others have shown previously that detection of recurrent mutations in MYD88 and CD79B using allele-specific polymerase chain reaction (PCR) or sequencing may increase the diagnostic accuracy of VRL.18,19,21,33
The purpose of our study was to examine the mutational frequency of genes known to be commonly altered in PCNSL and to explore whether a targeted sequencing approach could be useful for diagnosing primary and secondary VRL. In addition, in a subset of cases, we explored the presence of copy number alterations (CNAs) using the OncoScan technique and investigated whether the examination of cell-free DNA (cfDNA) derived from the supernatant of vitrectomy specimens could be an adjunct for diagnosis.
Material and methods
VRL patients and clinical data
Diagnostic vitrectomy samples from patients with archival DNA obtained consecutively from 2010 through 2019 were included in the study. Twenty-nine samples were collected from the Institute of Pathology and Neuropathology (Tübingen University Hospital). Five cases were obtained from the Department of Molecular and Clinical Cancer Medicine, Institute of Systems, Molecular and Integrative Biology (University of Liverpool). Six cases had been reported as part of a previous study.18 All samples had been examined by standard cytology and immunohistochemistry for B-cell markers, as previously described,18 as well as clonality analysis for the detection of IGH rearrangements. Clinical findings were evaluated with regard to clinical presentation and relapse. Nineteen consecutive vitrectomy specimens of uveitis or infectious vitritis performed for diagnostic purposes served as negative controls. The study was approved by the Ethics Committee of the Medical Faculty of the University of Tübingen (069/2015BO2).
DNA extraction and clonality analysis
Genomic DNA was extracted from remaining vitrectomy material as previously described18 or using the Maxwell RSC DNA FFPE Kit and the Maxwell RSC Instrument (Promega, Madison, WI) according to the manufacturer’s instructions. PCR for the detection of IGH gene rearrangements (FR2 and FR3 primer sets) was performed as previously described.18,34
Targeted next-generation sequencing
Targeted mutation analysis was performed by next-generation sequencing (NGS; Ion GeneStudio S5 prime; Thermo Fisher Scientific, Waltham, MA) using an AmpliSeq Custom Panel covering the most common recurrently mutated genes in PCNSL (complete coding sequence: CD79B, PIM1, TBL1XR1, CARD11, IRF4, BTG1, BTG2, PRDM1, CREBBP, IGLL5, and ETV6; hotspot regions: MYD88 exons 2 to 5) (supplemental Table 1). Amplicon library preparation and semiconductor sequencing were performed according to the manufacturer’s manuals using the Ion AmpliSeq Library Kit version 2.0, the Ion Library TaqMan Quantitation Kit, the Ion 510 and Ion 520 and Ion 530 Kit–Chef, and the Ion 520 Chip Kit (Thermo Fisher Scientific). Five paraffin-embedded samples of PCNSL served to validate the NGS panel.
Variant calling of nonsynonymous somatic variants compared with the human reference sequence was performed with Ion Reporter Software (version 5.12.3.0; Thermo Fisher Scientific). Variants called by the Ion Reporter Software were visualized using the Integrative Genomics Viewer (IGV, version 2.5.2; Broad Institute, Cambridge, MA) to exclude panel-specific artifacts. For variant calling, standard settings were used (no allelic frequency detection limit threshold). Variants were considered at a variant allele frequency (VAF) of >2% and a coverage of at least 200×, with the exception of very few variants with <200 reads and higher VAF. The National Center for Biotechnology Information single-nucleotide polymorphism database (dbSNP; including GnomAD, ExAC, and TOPMED) was used to exclude SNPs.
Copy number analysis
DNAs were hybridized on the OncoScan FFPE array platform (Thermo Fisher Scientific). Gains and losses (copy numbers; CNs) and CN-neutral loss of heterozygosity (CNN-LOH) regions were evaluated and visually inspected using Nexus Biodiscovery version 9.0 software (Biodiscovery, Hawthorne, CA). The reference human genome was GRCh37/hg19. CNAs with a minimum size of 100 kb and CNN-LOH larger than 5 Mb were considered informative. OSCHP raw data files were evaluated using the Thermo Fisher OSCHP-TuScan algorithm, which applies the allele-specific CN analysis of tumors algorithm, in all samples except for VRL22, which was analyzed with the Affymetrix OSCHP-SNP-FASST2 algorithm, which enabled us to reset the ploidy of the sample so that only aneuploidy events would account as CNAs. In both cases, default parameters of the algorithms in Nexus Biodiscovery, version 9.0 software (Biodiscovery, Hawthorne, CA), were used.
CDKN2A copy number analysis by quantitative PCR
CDKN2A CN analysis was performed as previously described using the TaqMan DNA Copy Number Assay Hs03712084_cn (Thermo Fisher Scientific).6 RNase P served as the reference (4403326). PCR was performed with TaqMan Universal Genotyping Master Mix (Thermo Fisher Scientific), according to the manufacturer’s manual on a LightCycler480 (Roche). CN status was calculated by the 2−ΔΔCT method, normalized to the mean of 5 reactive vitreous body aspirate (VBA) samples, and centered around a CN of 1. Considering contamination of reactive cells in VRL, heterozygous or homozygous CDKN2A deletion can be higher than 0 or 0.5.
cfDNA isolation from VBA supernatant and mutation analysis
Cell-free DNA was isolated from VBA supernatants using the QIAamp Circulating Nucleid Acid Kit (Qiagen, Hilden, Germany). For patients with the MYD88 p.L265P mutation detected in the genomic DNA, MYD88 exon 5 was investigated by NGS with a single amplicon, by using the Ion Amplicon Library Preparation Fusion Method (Thermo Fisher Scientific) according to the manufacturer’s protocol (gene-specific primer sequences: forward, 5'-ACCCCTTGGCTTGCAGGT-3'; reverse, 5'-AGGATGCTGGGGAACTCTTT-3'). Amplicons were purified and quantified applying Agencourt AMPure XP (Beckman Coulter, Brea, CA) magnetic beads and the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). Amplicons were diluted to 5 pM each and pooled. Clonal amplification and semiconductor sequencing were performed as described for targeted NGS. BAM files were generated with Torrent Suite 5.10.2. Sequences were visualized and evaluated using the freely available software Integrative Genomics Viewer (IGV; Broad Institute, Cambridge, MA). For patients without MYD88 p.L265P mutations in the genomic DNA, targeted sequencing using the AmpliSeq Custom Panel was performed as described earlier.
Results
Patients and clinicopathological findings
A total of 34 samples from 31 patients with VRL were included in the study (for details, see supplemental Table 2). From 2 patients, 2 and 3 vitrectomy samples were obtained at different time points (supplemental Table 2; patients 16 and 20), and in 1 patient only a retinal biopsy specimen was available. The cohort comprised 11 men and 20 women (median age 74 years, range 34-91), who had undergone diagnostic vitrectomy for a suspected diagnosis of VRL or verification of relapse. Nineteen patients presented with primary VRL (PVRL), of which 3 had synchronous manifestations in the CNS and 6 developed CNS involvement during the disease course (supplemental Table 2). In 12 patients, secondary VRL developed after PCNSL (6 patients), or after extracranial DLBCL (6 patients), respectively, including 3 with primary testicular lymphoma. Three available extracranial DLBCL samples (including 2 testicular and 1 cutaneous DLBCL of the “leg type”) were analyzed for the investigation of shared mutations and clonal evolution.
Cytological findings were either suspect or, less frequently, clearly positive for VRL in 25 of 33 (76%) vitreous samples, whereas the remaining specimens showed either a predominance of small T cells or severe degenerative changes precluding a cytological diagnosis (Figure 1; supplemental Table 2).
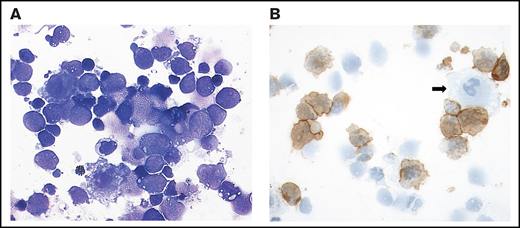
Cytological features of VRL: case 3. (A) The cytospin shows a cellular vitreous aspirate with large, atypical cells with irregular nuclei, frequent prominent nucleoli, and basophilic cytoplasm containing vacuoles admixed with small lymphocytes. Degenerative changes with frequent cell shadows are evident. May Grunwald-Giemsa stain, original magnification ×400. (B) CD20 staining of many atypical cells. The negative cells are likely to be T cells, macrophages (arrow), or degenerated tumor cells lacking preserved membrane staining. Immunoperoxidase; original magnification ×400.
Cytological features of VRL: case 3. (A) The cytospin shows a cellular vitreous aspirate with large, atypical cells with irregular nuclei, frequent prominent nucleoli, and basophilic cytoplasm containing vacuoles admixed with small lymphocytes. Degenerative changes with frequent cell shadows are evident. May Grunwald-Giemsa stain, original magnification ×400. (B) CD20 staining of many atypical cells. The negative cells are likely to be T cells, macrophages (arrow), or degenerated tumor cells lacking preserved membrane staining. Immunoperoxidase; original magnification ×400.
Clonality and identification of recurrent mutations by targeted NGS
Clonal IGH rearrangements were detected in 21 of 32 (66%) examined samples. All 34 samples were analyzed by NGS. The mean average read depth of the NGS sequence analysis was 2934 (range, 30-27028). In the 34 samples, 291 mutations were identified (supplemental Table 3), with a mean of 8.6 mutations per case. The most frequently mutated gene was MYD88 with 23 mutations in 31 patients (74%) and a median VAF of 52.2% (range, 9% to 94.9%; supplemental Table 3). The next most frequently mutated genes were PIM1 in 22 of 31 (71%) patients with a median VAF of 36.3% (range, 2.9% to 96.4%), CD79B in 17 of 31 (55%) with median VAF of 43.6% (range, 8% to 79.4%) and IGLL5 in 16 of 31 (52%) with median VAF of 30.5% (range, 2.4% to 86.4%; Figure 2A; supplemental Figure 1). Other mutated genes were TBL1XR1 (15 of 31; 48%), ETV6 (14 of 31; 45%); BTG2 (12 of 31; 39%), BTG1 (12 of 31; 39%), PRDM1 (6 of 31; 19%), IRF4 (5 of 31; 16%), CARD11 (4 of 31; 13%), and CREBBP (2 of 31; 7%). The highest mean VAFs were detected in MYD88, PRDM1, TBL1XR1, and CD79B (52.2%, 46.6%, 45.5%, and 43.6%; supplemental Figure 1). TBLXR1 mutations always cooccurred with MYD88 mutations and CD79B with MYD88 mutations in all but 1 case.
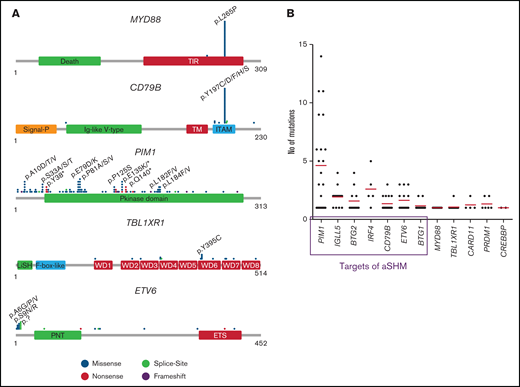
Mutation distribution and number in mutated genes. (A) Schematic presentation of the most important mutated genes in VRL. Relative positions of mutations are shown for CD79B, ETV6, MYD88, PIM1, and TBL1XR1. The approximate location of somatic mutations identified in each gene is indicated. Domains of the protein are represented according to the Uniprot database (www.uniprot.org). (B) Analysis of mutation numbers per gene per sample. Red lines indicate the median number of mutations per gene. Genes in the frame are known targets of aSHM.
Mutation distribution and number in mutated genes. (A) Schematic presentation of the most important mutated genes in VRL. Relative positions of mutations are shown for CD79B, ETV6, MYD88, PIM1, and TBL1XR1. The approximate location of somatic mutations identified in each gene is indicated. Domains of the protein are represented according to the Uniprot database (www.uniprot.org). (B) Analysis of mutation numbers per gene per sample. Red lines indicate the median number of mutations per gene. Genes in the frame are known targets of aSHM.
With the exception of 1 case (p.S243N), the MYD88 mutations were in hotspot p.L265P and located in the TIR domain. CD79B mutations (92% missense, 8% splice site) were mostly (92%) identified in the ITAM domain, of which 72% occurred in the hotspot codon 197. ETV6 mutations included 13 missense (43%), 2 nonsense, and 7 splice site mutations and 1 frameshift deletion. Sixty-one percent of alterations were found in exon 1, which does not code for a special domain. TBL1XR1 mutations were distributed across the WD domains with 5 mutations (31%) concentrating in a minor hotspot in codons 394 of 395 (mostly p.Y395C). As a target of SHM, PIM1 showed a large variety of alterations (n = 104) including 81 missense, 13 nonsense, and 3 splice site mutations and 1 in-frame deletion and 5 frameshift alterations distributed across the complete coding region with some minor hotspots (Figure 2A).
In 7 of 12 genes investigated, 19 splice site mutations (mostly substitutions of the first base 5' and 3' in the intron [guanine] or deletions up to 3 or 4 bases in the intron boundary) were found in addition to mutations within the exonic regions (supplemental Table 3). ETV6 showed splice site mutations in the highest percentage of cases (5 of 14; 36%).
Sequential samples from different time points were analyzed from 2 patients (supplemental Table 2). Case 16 lacked mutations in both samples, but displayed homozygous deletion of 9p21/CDKN2A in both samples (described in “CN and CNN-LOH alterations in VRL”). Case 20, in contrast, revealed shared mutations in MYD88, PIM1, CD79B, and BTG2 in all 3 samples and displayed an additional BTG1 mutation with a VAF of 19.7% in the second biopsy specimen.
More than 2 mutations per gene in individual cases were observed in several genes, which are known targets of aberrant SHM (aSHM). In PIM1, 4.6 mutations; IGLL5, 1.9; BTG2, 1.6; IRF4, 2.6; and CD79B, 1.4 were detected as the average per patient (Figure 2B; supplemental Table 3). The range of VAFs was broader in SHM target genes (supplemental Figure 2).
All 3 investigated systemic DLBCLs were clonally related to the respective VRLs, as evidenced by shared mutations in MYD88, CD79B, and TBL1XR1, but additionally exhibited private mutations in both samples in genes targeted by SHM, suggesting clonal evolution (supplemental Table 4).
As a control group, VBAs from 19 cases of inflammatory ocular diseases, including uveitis and infectious vitritis were analyzed. None of the control patients showed evidence of VRL or PCNSL during follow-up. No mutations were detected in any of the cases (data not shown).
CN and CNN-LOH alterations in VRL
CN analysis of 7 samples of 6 VRL patients and 3 uveitis samples identified CNAs in all VRL cases, but in none of the 3 controls. A total of 112 CNAs with a mean of 18.6 imbalances per case (range, 4-38) were identified (Figure 3A; supplemental Table 5). Specifically, 40 gains, 61 CN losses, 15× CNN-LOH, and 8 homozygous deletions were detected. Recurrent gains were observed in 2q35-36.3 (4 of 6), 12q12-14.2 (4 of 6), and 18q (3 of 6). The most frequent region of loss in 5 of 6 cases (83%) was at 9p21/CDKN2A. Loss of 17p13.1/TP53 was found in 2 of 6 cases (33%). All cases showed CNN-LOH (73% potentially acquired and 27% germ line). In 3 of 6 (50%) patients CNN-LOH in 6p (HLA) was detected. As mentioned earlier, the homozygous deletion of 9p21/CDKN2A was the only alteration found in both samples of patient 16 (supplemental Figure 2). When OncoScan and quantitative PCR (qPCR) results were combined, homozygous (4 of 11) or heterozygous deletions of 9p21/CDKN2A were identified in 9 of 12 (75%) patients (Figure 3B).
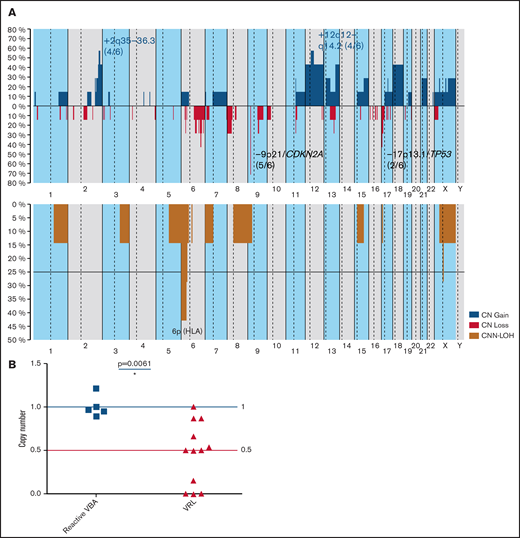
CN profile of VRL. (A) Global CN (top) and CNN-LOH (bottom) profile of 6 VRL cases. The x-axis depicts chromosome positions with dotted lines indicating centromeres. The y-axis indicates the frequency of the genomic aberration among the analyzed cases. Each probe is aligned from chromosomes 1 to 22 and p to q. Gains are depicted in blue; losses are depicted in red, and regions of CNN-LOH are represented in mustard. Recurrent CN and CNN-LOH regions are indicated. (B) CDKN2A CN analysis of 12 VRL cases and reactive control samples. A CDKN2A CN assay was performed on 12 VRL cases (including TaqMan and OncoScan data) and 5 samples from reactive VBA cases in comparison. Data were analyzed according to the 2-ΔΔCp method. Results are depicted as CNs relative to mean levels of reactive VBA (CN status, 1). Blue and red lines indicate wild-type CN status (1) and heterozygous deletion (0.5), respectively. A CN status of 0 indicates homozygous deletion. Statistical analysis was performed with the Mann-Whitney test. *P > .05.
CN profile of VRL. (A) Global CN (top) and CNN-LOH (bottom) profile of 6 VRL cases. The x-axis depicts chromosome positions with dotted lines indicating centromeres. The y-axis indicates the frequency of the genomic aberration among the analyzed cases. Each probe is aligned from chromosomes 1 to 22 and p to q. Gains are depicted in blue; losses are depicted in red, and regions of CNN-LOH are represented in mustard. Recurrent CN and CNN-LOH regions are indicated. (B) CDKN2A CN analysis of 12 VRL cases and reactive control samples. A CDKN2A CN assay was performed on 12 VRL cases (including TaqMan and OncoScan data) and 5 samples from reactive VBA cases in comparison. Data were analyzed according to the 2-ΔΔCp method. Results are depicted as CNs relative to mean levels of reactive VBA (CN status, 1). Blue and red lines indicate wild-type CN status (1) and heterozygous deletion (0.5), respectively. A CN status of 0 indicates homozygous deletion. Statistical analysis was performed with the Mann-Whitney test. *P > .05.
Genetic comparison of primary and secondary VRL
Mutational frequencies were compared for primary (19 cases) and secondary (12 cases) VRL (Figure 4). Overall, few differences were observed, with lower frequencies of CD79B (42% vs 75%; P = .14), BTG1 (21% vs 67%; P = .024, Fisher’s exact test), and BTG2 (31.5% vs 50%) mutations in primary cases. Secondary VRL after PCNSL (6 cases) and extracranial DLBCL, including testicular lymphoma, showed very similar mutational profiles with identical frequencies for MYD88, PIM1 (5 of 6 cases each), and CD79B (4 of 6 cases each).
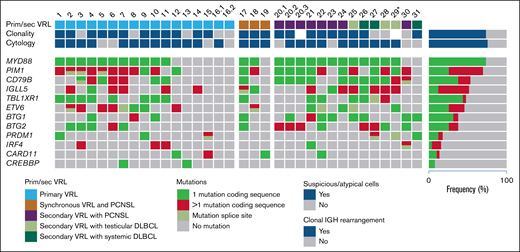
Mutational landscape of VRL. Each column represents a sample and each row a specific analysis. Genes are ordered from top to bottom according to the mutational frequency across all samples, with the percentages shown on the right side. In cases 16 and 20, 2 and 3 vitreous samples, respectively, from different time points were analyzed. *For case 29, only a retinal biopsy specimen was available.
Mutational landscape of VRL. Each column represents a sample and each row a specific analysis. Genes are ordered from top to bottom according to the mutational frequency across all samples, with the percentages shown on the right side. In cases 16 and 20, 2 and 3 vitreous samples, respectively, from different time points were analyzed. *For case 29, only a retinal biopsy specimen was available.
VBA supernatant as a liquid biopsy sample
In 8 cases, cfDNA was isolated from VBA supernatants and analyzed for the presence of mutations found in the cell-derived DNA. In all cases analyzed, the known mutations, including MYD88 L265P in 6 cases studied by single-amplicon testing, and a CARD11 (patient 19) and both a CARD11 and a PRDM1 mutation (patient 15) were detected using the panel in the cfDNA, with a higher VAF than in genomic DNA (Figure 5).
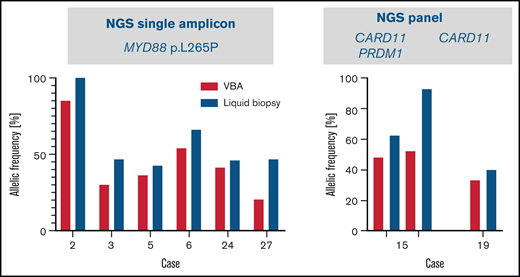
Results of liquid biopsy of vitreous body fluid from 8 cases of VRL. Allelic frequencies of mutations analyzed by targeted NGS of cell-free DNA isolated from vitreous body fluids (blue) are represented in comparison with allelic frequencies obtained from the cellular samples (red; Figure 1). In 6 cases MYD88 hotspot codon 265 was analyzed using single amplicon sequencing (left). In cases 19 and 15, mutations in CARD11 and/or PRDM1 were analyzed by using panel sequencing (case 19: CARD11 c.1871A>C/p.Q624P; case 15: CARD11 c.1078A>G/p.M360V, c.1034A>G/p.E345G, both with similar allelic frequencies, PRDM1: c.1801C>T/p.R601W) (right).
Results of liquid biopsy of vitreous body fluid from 8 cases of VRL. Allelic frequencies of mutations analyzed by targeted NGS of cell-free DNA isolated from vitreous body fluids (blue) are represented in comparison with allelic frequencies obtained from the cellular samples (red; Figure 1). In 6 cases MYD88 hotspot codon 265 was analyzed using single amplicon sequencing (left). In cases 19 and 15, mutations in CARD11 and/or PRDM1 were analyzed by using panel sequencing (case 19: CARD11 c.1871A>C/p.Q624P; case 15: CARD11 c.1078A>G/p.M360V, c.1034A>G/p.E345G, both with similar allelic frequencies, PRDM1: c.1801C>T/p.R601W) (right).
Discussion
In this study, the genetic profile of a large series of VRLs, focusing on genes frequently mutated in PCNSL was identified, and its close relationship with PCNSL was supported. Furthermore, we demonstrated that NGS-based mutational analysis of vitreous aspirate is a valuable diagnostic tool of high sensitivity and specificity.
Primary VRL, a very rare disease with an estimated incidence below 0.05 per 100000 per year, is considered a variant of PCNSL based on biological features and the high frequency of simultaneous or metachronous lymphoma manifestations at both sites. Systemic dissemination of both PVRL and PCNSL is rare, and most cases of secondary VRL are associated with PCNSL.
Our series of VRL shows a high incidence of MYD88 and CD79B mutations and frequent homozygous deletions of 9p21/CDKN2A, regardless of primary or secondary ocular involvement. Furthermore, the cases studied by OncoScan array exhibited a high frequency of CNAs (18.6 alterations per case), which could reflect genomic instability. The only previous study addressing CNA in VRL using SNP array karyotyping also identified CN gains in 12q and 18q and loss in 9p21/CDKN2A, similar to our data.35 In addition to the high frequency of MYD88 mutations18-22 found in 74% of our series, commonly affected genes included PIM1, CD79B, IGLL5, TBL1XR1, ETV6, and BTG1/2, with mutational frequencies ranging from 39% to 71% of cases. Despite the limitations of our targeted approach, the mutational pattern observed is similar to that of PCNSL4-7,11,15,16 and characterizes a specific subgroup of DLBCL of ABC subtype with poor prognosis and common extranodal manifestation identified in 2 recent, large genomic-profiling studies and designated as MCD or cluster 5 (C5).36,37 MCD/C5 DLBCLs show the highest imprint of aSHM, as well as signs of immune evasion, including MHC antigen loss and expression of PD-L1/2.14,36,37 DLBCL of ABC subtype including MCD/C5 was previously thought to arise from plasmablasts; however, recent studies suggest that aberrant memory B cells, rather than plasmablasts, are the true cell of origin of this DLBCL subtype.26,38 This aberrant memory B-cell phenotype drives the cells to repeated rounds of reentry into the germinal center reaction with consecutive cycles of mutagenesis. Consistent with this finding, several of the most commonly mutated genes in this VRL series are typical targets of aSHM and frequently displayed multiple mutations per patient.14
TBL1XR1, which was mutated in half of our VRL cases, forms part of the SMRT/NCOR1 transcriptional corepressor, and its dysfunction caused by mutations is thought to be a key mechanism for the skewing of germinal center B-cell maturation toward memory B cells mentioned in “Clonality and identification of recurrent mutations by targeted NGS”.9 In line with this hypothesis, TBL1XR1-deficient mice develop MCD/C5-like aggressive lymphomas with very high aSHM signatures.38 In addition, TBL1XR1 modulates Toll-like/MYD88 signaling, which is constitutively active in most MCD/C5 DLBCLs, including PCNSL and VRL.39
The transcriptional regulators BTG1 and BTG2, each mutated in 39% of VRL cases, are considered tumor suppressors because of their frequent mutation or deletion in B-cell malignancies.40 Both BTG1 and TBL1XR1 are transcriptional cofactors of ETV6, another commonly affected gene in our VRL series.41 ETV6 is a strong transcriptional repressor playing key roles in hematopoiesis and is involved in a variety of hematological neoplasms. In PCNSL, in addition to common mutations, ETV6 has been found to be involved in recurrent translocations with the IGH locus in 18% of cases, probably resulting in ETV6 haploinsufficiency.5,6,42 ETV6 exon deletions in PCNSL discovered on the mRNA level in the study by Chapuy et al are probably consistent with ETV6 splice site mutations found in this study.6 Remarkably, the single study looking at VRL by whole-exome sequencing in a few cases identified a similar mutation spectrum compared with our limited gene panel (MYD88, PIM1, IGLL5, BTG2, and BTG1)22 ; however, neither ETV6 nor TBL1XR1 mutations, each present in nearly half of our cases and in 20% to 30% of published PCNSL series, were observed.4-7 In concordance with data on PCNSL, TBL1XR1 and CD79B mutations were found almost exclusively in MYD88mut cases. Several of the recurrently mutated genes in our series, most prominently MYD88, showed high allele frequencies indicative of homozygosity. The presence of homozygous MYD88 mutations has already been documented in Waldenström’s macroglobulinemia and has recently been found in VRL by using a single-cell assay.43,44
Because both primary and secondary VRL were investigated in our study, mutational profiles were compared for both groups. Overall, no significant differences were identified. Similar to our results, a lower rate of CD79B mutations in PVRL vs PCNSL was identified previously in 2 studies (22% to 35% vs 45% to 83%), with 1 of them showing a significantly longer time to CNS involvement for CD79B wild-type cases.21,22 In contrast, in our series the incidence of CD79B mutations in PVRL without CNS involvement 5 of 10 (50%) was higher than in cases with CNS involvement 3 of 9 (33%), although the difference was not significant (P = .6). Of note, half of the cases of secondary VRL in this series arose from systemic DLBCL, rather than PCNSL. Nevertheless, they showed a similar genetic profile with high frequencies of MYD88 (5 of 6), CD79B (5 of 6), IGLL5 (5 of 6), ETV6 (4 of 6), and TBL1XR1 (4 of 6) mutations. A clonal relationship was confirmed by the presence of shared mutations in all 3 examined cases. This further supports the existence of a specific DLBCL subtype, MCD/C5, sharing a common pathogenesis and molecular profile, irrespective of primary location,36,37 and indicates that secondary involvement of the vitreoretinal space and the CNS is a result of the specific properties of this DLBCL subtype, rather than a random occurrence.
We also analyzed the potential of cfDNA analysis from vitreous aspirates, because this might open an avenue for molecular diagnosis of VRL using microaspirates rather than vitrectomy specimens.45 Using cell-free supernatant, mutations present in matched, cell-derived DNA were reliably detected, and surprisingly, the allelic frequency was universally higher in the cfDNA in all cases investigated, most likely because of the higher vulnerability of lymphoma cells with increased rates of apoptosis relative to the reactive cell component, mostly T cells and macrophages, with consecutive high cell-free tumor DNA concentrations in the vitreous fluid. The detection of MYD88 mutations in cfDNA has also been reported in cerebrospinal fluid samples of patients with PCNSL.46 The median VAF of 7% in this study, however, was significantly lower than in our cfDNA samples, probably because of the confined space of the vitreous fluid.
In summary, frequently concurrent constitutive activation of TLR and BCR signaling and deletions of CDKN2A are molecular hallmarks of VRL. Its mutational profile is similar to PCNSL and specifically to the molecular MCD/C5 cluster, confirming the existence of a subset of DLBCL with specific clinical, biological and genetic features and frequent involvement of immune-privileged extranodal sites. In light of the frequent difficulties in reaching a diagnosis of VRL with conventional methods, targeted mutational analysis of vitreoretinal aspirates is a useful addition to the diagnostic repertoire, with high sensitivity and specificity. Analysis of cfDNA derived from small-volume aspirates holds promise for minimally invasive diagnosis and follow-up, given the significant shedding of tumor DNA fragments into the vitreous fluid.
Acknowledgments
The authors thank Sema Colak for excellent technical assistance.
E.C. is supported by Spanish Ministerio de Economía y Competitividad (RTI2018-094274-B-I00) and Generalitat de Catalunya Suport Grups de Recerca (2017-SGR-1142) and is also an Academia Researcher of the “Institució Catalana de Recerca i Estudis Avançats” (ICREA) of the Generalitat de Catalunya.
Authorship
Contributions: I.B. and F.F. conceived and designed the study, supervised the experimental work, analyzed the data and wrote the manuscript; P. Sander, M.B., J. Steinhilber, and J.S-V. analyzed and interpreted the data; P. Sander and J.S-V. helped to write the manuscript; J.S-V. and E.K. performed genetic analysis; I.S. supervised the CNA analyses and helped interpret the data; D.S., S.E.C., P. Szurman, F.G., M.S.S., and J. Schittenhelm contributed samples, evaluated the data, and performed the clinicopathologic correlations; and E.C., S.E.C., and L.Q-M. performed pathological review and helped in data interpretation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Irina Bonzheim, Institute of Pathology and Neuropathology, University Hospital Tübingen, Liebermeisterstraße 8,72076 Tübingen, Germany; e-mail: irina.bonzheim@med.uni-tuebingen.de; and Falko Fend, Institute of Pathology and Neuropathology, Tübingen University Hospital, Liebermeisterstraße 8, 72076 Tübingen, Germany; e-mail: falko.fend@med.uni-tuebingen.de.

