

Key Points
Pre- and posttreatment anti-mCAR19 antibodies did not alter tisagenlecleucel cellular kinetics, efficacy, or safety in r/r B-ALL or r/r DLBCL.
T-cell responses to mCAR19 peptides did not influence patient outcomes or cellular expansion in r/r B-ALL or r/r DLBCL.

Abstract
Tisagenlecleucel is indicated for pediatric and young adult patients with relapsed/refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) and adult patients with r/r diffuse large B-cell lymphoma (DLBCL). The tisagenlecleucel chimeric antigen receptor (CAR) contains a murine single-chain variable fragment domain; we examined the effects of humoral and cellular immune responses to tisagenlecleucel on clinical outcomes using 2 validated assays. Data were pooled from the ELIANA (registered at www.clinicaltrials.gov as #NCT02435849) and ENSIGN (#NCT02228096) trials in r/r B-ALL (N = 143) and the JULIET trial (#NCT02445248) in r/r DLBCL (N = 115). Humoral responses were determined by flow cytometric measurement of anti-murine CAR19 (mCAR19) antibodies in serum. Cellular responses were determined using T-cell production of interferon-γ in response to 2 different pools of mCAR19 peptides. Pretreatment anti-mCAR19 antibodies were detected in 81% of patients with r/r B-ALL and 94% of patients with r/r DLBCL. Posttreatment anti-mCAR19 antibodies were higher than patient-specific baseline in 42% of r/r B-ALL and 9% of r/r DLBCL patients. Pretreatment and posttreatment anti-mCAR19 antibodies did not affect tisagenlecleucel cellular kinetics, including maximum concentration and persistence (r2 < 0.05), clinical response (day-28 response, duration of response, and event-free survival), and safety. T-cell responses were consistent over time, with net responses <1% at baseline and posttreatment time points in a majority of patients and no effect on transgene expansion or persistence or outcomes. Presence of baseline and/or posttreatment anti-mCAR19 antibodies or T-cell responses did not alter the activity of tisagenlecleucel in patients with r/r B-ALL or r/r DLBCL.
Introduction
Chimeric antigen receptor (CAR) T-cell therapies are an important clinical advancement for patients with advanced relapsed or refractory (r/r) B-cell malignancies. To date, little is known about the presence or potential effects of preexisting or posttreatment anti-murine CAR19 (mCAR19) antibodies, although early analyses suggest that neither preexisting nor posttreatment antibodies affect safety or efficacy of tisagenlecleucel.1,2
Similar to other biologics, CAR T-cell therapies can cause an immune response that may be more likely with murine-derived components3 and that may affect efficacy or result in adverse events.3-5 Posttreatment immune responses to therapeutic antibodies have been observed, and preexisting antibodies have been described for a variety of biopharmaceutical agents.6,7 Characterizing the incidence and prevalence of immunogenicity and their relationship to cellular kinetics, efficacy, and safety of CAR T-cell therapies is important because humoral and cellular immune responses have been implicated in reducing persistence and efficacy in early CAR T-cell therapy studies.8
Tisagenlecleucel, an anti-CD19 CAR T-cell therapy, reprograms patient T cells to identify and eliminate CD19-expressing B cells9 and is approved to treat patients up to age 25 years with r/r B-cell precursor acute lymphoblastic leukemia (B-ALL) and adults with r/r diffuse large B-cell lymphoma (DLBCL).10-12 Multicenter clinical studies have demonstrated high response rates, including 81% overall remission rate in pediatric and young adult patients with r/r B-ALL11 and 52% best overall response rate in adult patients with r/r DLBCL.12
The tisagenlecleucel CAR contains a murine single-chain variable fragment (mCAR19) antigen-binding extracellular domain that recognizes CD19, and intracellular 4-1BB costimulatory and CD3-ζ chain–signaling domains.9,13,14 Components of CARs (murine or human) have potential to elicit immune responses in treated patients.3-5 We evaluated the effects of humoral and cellular immune responses resulting from tisagenlecleucel treatment on cellular kinetics, efficacy, and safety in pediatric and young adult patients with r/r B-ALL and adult patients with r/r DLBCL.
Methods
Patients and clinical trial design
Data for pediatric and young adult patients with B-ALL were pooled from 2 clinical trials: ELIANA (registered at www.clinicaltrials.gov as #NCT02435849; data cutoff date, 1 July 2019) and ENSIGN (registered at www.clinicaltrials.gov as #NCT02228096; data cutoff date, 24 May 2019). Data for adult patients with DLBCL are based on 1 clinical trial: JULIET (registered at www.clinicaltrials.gov as #NCT02445248; data cutoff date, 11 December 2018). ELIANA and ENSIGN are single-arm, open-label, multicenter phase 2 studies examining tisagenlecleucel in patients with r/r B-ALL who were age 3 (at screening) to 21 years (at initial diagnosis).11,15 JULIET is a single-arm, open-label, multicenter phase 2 study of tisagenlecleucel in adult patients with r/r DLBCL.12 Details of the trials were previously described11,12,15 ; patient eligibility and prior therapies are summarized in supplemental Table 1. Trial protocols were reviewed and approved by local institutional review boards; all enrolled patients provided written informed consent.
Bioanalytic methods/analyses
Patient sample collection.
Patient samples for immunogenicity (serum for humoral and whole blood for cellular) and CAR transgene analyses were collected during the ELIANA and ENSIGN studies at enrollment; at days 14 and 28; at months 3, 6, 12, and 24 (collected at month 36 in ENSIGN only); and upon relapse in both trials. In the JULIET study, samples were collected at enrollment; at days 14 and 28; at months 3, 6, and 12; and upon relapse.
Humoral immunogenicity assay.
Humoral immunogenicity was determined by measuring anti-mCAR19 antibodies in serum using a validated flow cytometric assay. Briefly, anti-mCAR19–specific antibodies were detected via antibody binding to a Jurkat cell line expressing the tisagenlecleucel mCAR19 molecule, using antibody binding to the same cell line without mCAR19 (nontransduced) as a specificity control. A sample was considered anti-mCAR19+ if the signal difference between mCAR19-expressing cells and control cells was greater than or equal to an immunogenicity cut point (data supplement).16 Posttreatment anti-mCAR19 antibodies were considered positive when the level was greater than the individual patient’s baseline level (defined as 2.1578× anti-mCAR19 antibody median fluorescence intensity [MFI] at preinfusion, where 2.1578 corresponds to the cut point factor on mCAR19 Jurkat cells).16
Cellular immunogenicity assay.
T-cell responses to 2 different pools of mCAR19 peptides were measured by intracellular staining and subsequent flow cytometric analysis of interferon-γ (IFN-γ) production (no other cytokines were assessed; data supplement).
Clinical data analysis
Relationship between clinical parameters and immunogenicity.
The effect of anti-mCAR19 antibodies (humoral immunogenicity) at baseline or posttreatment on cellular kinetics, efficacy, and safety was assessed using summary statistics as well as graphical and model-based analyses. Summary statistics and graphical methods were used to assess the impact of cellular immune responses on cellular kinetics, efficacy, and safety using net mCAR19-specific, IFN-γ–producing T-cell response.
IVIG and humoral immunogenicity.
IV immunoglobulin (IVIG) is administered to some patients as treatment of hypogammaglobulinemia resulting from B-cell aplasia, an on-target effect of tisagenlecleucel.11,17 IVIG is prepared using sera from multiple healthy blood donors and likely contains alloreactive antibodies that could be detected as positive immunogenicity in the assay. Validation of the humoral assay included an additional step to evaluate levels of IVIG that would result in potential false-positive detection of anti-mCAR19 antibodies by binding to mCAR19 cells. IVIG concentrations were titrated across a dynamic range (0.0391-10 mg/mL) to evaluate binding on mCAR19 cells, using nontransduced and mCAR19 cells.16
Clinical response assessments
Efficacy end points included clinical response on day 28 (complete remission [CR] plus CR with incomplete blood count recovery) in r/r B-ALL or month-3 overall response (complete response plus partial response) in r/r DLBCL based on the timing of protocol-defined response assessments in the clinical trials. Clinical responses were categorized by anti-mCAR19 status (positive or negative) and presented as box plots. In patients with r/r B-ALL, best overall response was determined within 3 months of tisagenlecleucel infusion.11 Kaplan-Meier analyses were conducted for duration of response (DOR), progression-free survival, and overall survival (OS). Safety end points included cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome neurologic event grade.18
Statistical analysis
Summary statistics are provided for incidence and prevalence of humoral and cellular immunogenicity and for tisagenlecleucel cellular kinetic parameters as determined by quantitative polymerase chain reaction (categorized by whether the patient was positive or negative for anti-mCAR19 antibody posttreatment). Cellular kinetic parameters reported for tisagenlecleucel were calculated using conventional methods as previously described.1 Kaplan-Meier and Cox regressions were performed to evaluate the effect of humoral immunogenicity on DOR and event-free survival/progression-free survival end points. Waterfall plots summarize the effect of positive anti-mCAR19 antibodies posttreatment on DOR and OS.
Data sharing statement
Novartis is committed to sharing with qualified external researchers access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The data availability of these trials is according to the criteria and process described at www.clinicalstudydatarequest.com.
Results
Patient characteristics
Data from a total of 143 pediatric patients with r/r B-ALL (ELIANA, n = 79; ENSIGN, n = 64) and 115 adult patients with r/r DLBCL (JULIET) were analyzed. Patient demographics and baseline disease characteristics were described previously.11,12,15
Incidence and prevalence
Humoral (anti-mCAR19 antibodies) immunogenicity.
Baseline samples were collected after lymphodepletion but before tisagenlecleucel. Anti-mCAR19 antibodies were detected in 81% of patients with r/r B-ALL and in 94% of patients with r/r DLBCL at baseline. Previous therapy with murine or humanized monoclonal antibodies was infrequent (10%) in patients with B-ALL (ELIANA, n = 11 of 79; ENSIGN, n = 3 of 64; rituximab, n = 7; inotuzumab, n = 4; moxetumomab, n = 2; and alemtuzumab, n = 1). In DLBCL studies, patients commonly received rituximab, a chimeric human/mouse monoclonal antibody, before tisagenlecleucel infusion.12 Of note, a similar percentage (∼90%) of preexisting anti-mCAR19 antibodies was detected in samples from healthy volunteers during method validation.16
Cellular immunogenicity.
For a majority of patients with r/r B-ALL and patients with r/r DLBCL, both CD4+ and CD8+ T-cell responses to mCAR19 peptides (pools 1 and 2) were consistent and remained low over time, with net responses of <1% at baseline and posttreatment (supplemental Table 2). A total of 142 of 143 patients with r/r B-ALL and 107 of 115 patients with r/r DLBCL had T-cell responses <1% to the mCAR19 peptides.
To determine if T-cell response affected cellular kinetics or clinical outcomes, the T-cell responses in a subgroup of B-ALL patients in the 90th percentile for T-cell response were compared with those responses in the overall population. For B-ALL, 14 of 137 patients were in the 90th percentile for T-cell response at baseline. No notable differences in response category, CRS grade, maximum posttreatment transgene level, persistence, or DOR were observed for patients with the highest T-cell responses (supplemental Table 3; supplemental Figures 1-3). Similarly, no notable differences were observed among patients in the 90th percentile for highest maximum fold change in T-cell response from baseline in posttreatment samples (data not shown).
Cellular kinetics
The relationship between the presence of preexisting anti-mCAR19 antibodies and the maximum concentration (Cmax) after tisagenlecleucel administration was not significant in patients with r/r B-ALL (r2 < 0.001) or patients with r/r DLBCL (r2 = 0.005; Figure 1). Transgene exposure within the first 28 days (area under the curve from day 0 to 28 [AUC0-28d]) was also unaffected by preexisting humoral responses in r/r B-ALL and r/r DLBCL. We also evaluated whether tisagenlecleucel infusion resulted in posttreatment increase in anti-mCAR19 antibodies. Posttreatment increase in anti-mCAR19 antibodies was defined as increase in anti-mCAR19 antibodies above a patient-specific baseline sample. Overall, 42% of patients with r/r B-ALL and 9% of patients with r/r DLBCL experienced posttreatment increases in anti-mCAR19 antibodies. Importantly, the observed posttreatment increases in anti-mCAR19 antibodies did not affect tisagenlecleucel persistence or expansion (AUC0-28d) in patients with r/r B-ALL. There were insufficient r/r DLBCL patients with posttreatment increases in anti-mCAR19 antibodies above baseline levels to formally evaluate any potential correlation between anti-mCAR19 antibody levels and clinical end points.
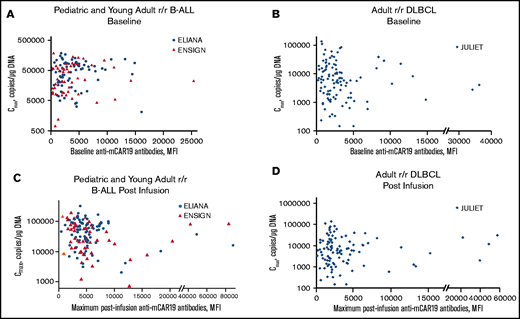
Preexisting and posttreatment anti-mCAR19 antibodies vs tisagenlecleucel expansion. Tisagenlecleucel expansion (Cmax) in pediatric and young adult patients with r/r B-ALL by preexisting (baseline) (A) and posttreatment (postinfusion) (C) anti-mCAR19 antibodies. Patients in the ELIANA trial are indicated by blue circles, and patients in the ENSIGN trial are indicated by red triangles. Tisagenlecleucel expansion (Cmax) in adult patients with r/r DLBCL by preexisting (baseline) (B) and posttreatment (postinfusion) (D) anti-mCAR19 antibodies. Patients in the JULIET trial are indicated by blue diamonds.
Preexisting and posttreatment anti-mCAR19 antibodies vs tisagenlecleucel expansion. Tisagenlecleucel expansion (Cmax) in pediatric and young adult patients with r/r B-ALL by preexisting (baseline) (A) and posttreatment (postinfusion) (C) anti-mCAR19 antibodies. Patients in the ELIANA trial are indicated by blue circles, and patients in the ENSIGN trial are indicated by red triangles. Tisagenlecleucel expansion (Cmax) in adult patients with r/r DLBCL by preexisting (baseline) (B) and posttreatment (postinfusion) (D) anti-mCAR19 antibodies. Patients in the JULIET trial are indicated by blue diamonds.
DLBCL-specific considerations
In JULIET (n = 111), most patients (98%) received prior rituximab therapy, ranging from >1 year before (15%) to within 1 month of tisagenlecleucel infusion (14%). Although rituximab exposure did not seem to increase the incidence of preexisting anti-mCAR19 antibodies compared with no prior exposure, only 2 patients had not received prior rituximab therapy (supplemental Table 4).
Effects of anti-mCAR19 antibodies on efficacy outcomes
Preexisting and posttreatment anti-mCAR19 antibodies did not affect the day-28 response (CR plus CR with incomplete blood count recovery) in patients with r/r B-ALL or month-3 response (complete response plus partial response) in patients with r/r DLBCL (Figure 2). In B-ALL, the overall response rate based on day-28 response was 11 (85%) of 13 for patients within the 90th percentile of MFI for anti-mCAR19 antibodies detected at any time point after tisagenlecleucel infusion and 94 (77%) of 122 for the remainder of the population. Best overall response rates in patients with r/r B-ALL were similar in the 90th percentile MFI compared with the remainder of the population (10 [77%] of 13 and 90 [74%] of 122, respectively). In addition, DOR and OS in patients with r/r B-ALL who received tisagenlecleucel were unaffected by preexisting and posttreatment anti-mCAR19 antibodies (Figure 3; supplemental Figure 3). The presence of preexisting and posttreatment anti-mCAR19 antibodies did not affect tisagenlecleucel maximal expansion (Cmax) or persistence (Figure 4).
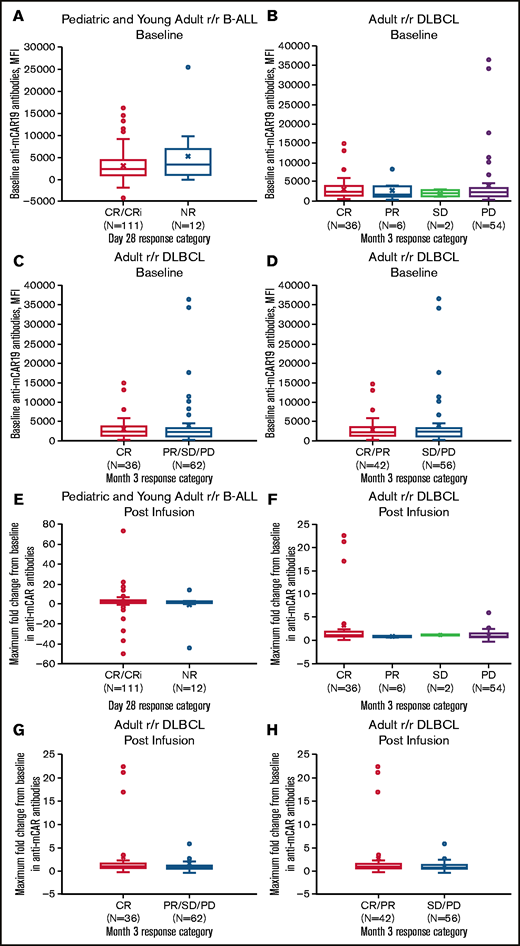
Pretreatment humoral immunogenicity and maximum fold change in anti-mCAR19 antibodies posttreatment by response category. Baseline anti-mCAR19 antibody levels (A-D) and maximum fold change in posttreatment anti-mCAR19 antibody levels (E-H) (by MFI) by response category. CRi, CR with incomplete blood count recovery; NR, no response; PR, partial response; PD, progressive disease; SD, stable disease.
Pretreatment humoral immunogenicity and maximum fold change in anti-mCAR19 antibodies posttreatment by response category. Baseline anti-mCAR19 antibody levels (A-D) and maximum fold change in posttreatment anti-mCAR19 antibody levels (E-H) (by MFI) by response category. CRi, CR with incomplete blood count recovery; NR, no response; PR, partial response; PD, progressive disease; SD, stable disease.
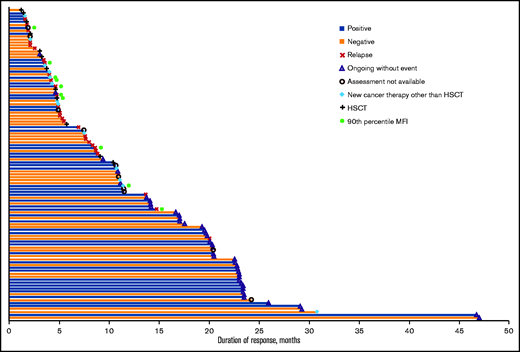
Effects of anti-mCAR19 antibodies on duration of response in pediatric and young adult patients with B-ALL. Duration of response among patients who were positive (blue bars) and negative (orange bars) for posttreatment anti-mCAR19 antibodies that increased above baseline levels. Censoring reasons are indicated by the symbols in the legend. Patients in the 90th percentile for anti-mCAR antibody MFI are shown (green circles). HSCT, hematopoietic stem cell transplantation.
Effects of anti-mCAR19 antibodies on duration of response in pediatric and young adult patients with B-ALL. Duration of response among patients who were positive (blue bars) and negative (orange bars) for posttreatment anti-mCAR19 antibodies that increased above baseline levels. Censoring reasons are indicated by the symbols in the legend. Patients in the 90th percentile for anti-mCAR antibody MFI are shown (green circles). HSCT, hematopoietic stem cell transplantation.
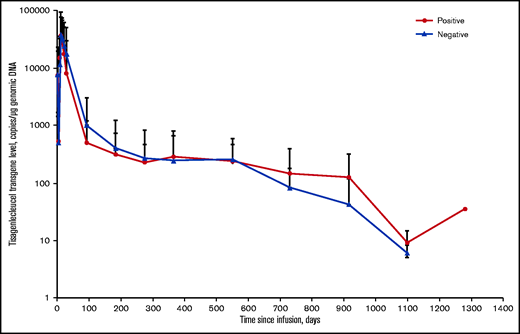
Effects of posttreatment anti-mCAR19 antibodies on tisagenlecleucel persistence in pediatric and young adult patients with r/r B-ALL. Solid red circles represent patients who were positive for posttreatment anti-mCAR19 antibodies. Solid blue triangles represent patients who were negative for posttreatment anti-mCAR19 antibodies. Data are presented as the arithmetic mean ± standard deviation.
Effects of posttreatment anti-mCAR19 antibodies on tisagenlecleucel persistence in pediatric and young adult patients with r/r B-ALL. Solid red circles represent patients who were positive for posttreatment anti-mCAR19 antibodies. Solid blue triangles represent patients who were negative for posttreatment anti-mCAR19 antibodies. Data are presented as the arithmetic mean ± standard deviation.
Effects of anti-mCAR19 antibodies on safety outcomes
Safety events (CRS or neurologic events) were not affected by preexisting or posttreatment anti-mCAR19 antibodies detected in patients with r/r B-ALL or patients with r/r DLBCL (supplemental Figure 4). Levels of anti-mCAR19 antibodies were similar across all grades of CRS and neurologic events. Patients with B-cell aplasia cannot mount a humoral immune response and are more susceptible to infections; we therefore examined whether there was any association between the presence or lack of anti-mCAR19 antibodies and infection. The incidence of anti-mCAR19 antibodies detected posttreatment was evaluated in patients who developed infections within 8 weeks postinfusion. Overall, 43% (34 of 79) of patients in ELIANA,11 41% (26 of 64) of patients in ENSIGN,15 and 37% (43 of 115) of patients in JULIET12 experienced posttreatment infections within 8 weeks postinfusion. However, there was no association between mCAR19 antibody levels and infection during this same 8-week period after infusion.
Hypogammaglobulinemia, B-cell aplasia, and immunogenicity
Model-based (Kaplan-Meier) analyses were performed to evaluate the impact of anti-mCAR19 antibody level on time to B-cell recovery. Baseline and maximum fold change of anti-mCAR19 antibodies did not show any apparent relationship with duration of B-cell aplasia (supplemental Figure 5). In addition, patients who received IVIG to overcome hypogammaglobulinemia showed no statistically significant differences in immunogenicity compared with patients who did not receive IVIG. It is important to note that most patients with DLBCL had received prior rituximab therapy, which can cause long-term B-cell aplasia, and most patients (79%) had B-cell aplasia at baseline.12 In fact, only 6 patients with DLBCL experienced B-cell recovery to within the normal range (80-616 cells per mL).19 Among patients with B-ALL, B-cell aplasia was present in 25 (18%) of 141 at baseline. After tisagenlecleucel infusion, patients with sustained B-cell aplasia had higher AUC0-28d and Cmax and longer median tisagenlecleucel maximal persistence than patients who experienced B-cell recovery in <6 months after infusion (supplemental Table 5).
Cellular immunogenicity.
Anti-mCAR19 CD4+ and CD8+ T-cell responses were measured by IFN-γ production in response to exposure to mCAR19 peptides. Overall, net T-cell responses to 2 pools of mCAR19 peptides were <1% in patients with r/r B-ALL (Figure 5). CD4+ T-cell responses did not affect tisagenlecleucel transgene expansion or persistence (Figure 5A-B) or patient outcomes (Figure 5C-D). Similarly, CD8+ T-cell responses to mCAR19 peptide pools did not affect tisagenlecleucel transgene expansion, persistence, or patient outcomes (data not shown). For most patients with r/r B-ALL and patients with r/r DLBCL, CD4+ and CD8+ T-cell responses to pool-1 mCAR19 peptides were consistent over time, with net responses of <1% at baseline and posttreatment (Figure 6). Similar responses to pool-2 mCAR19 peptides were observed (data not shown). In addition, cellular kinetic profiles were similar between patients in the 90th percentile for T-cell response and the overall patient population (supplemental Figures 1 and 2).
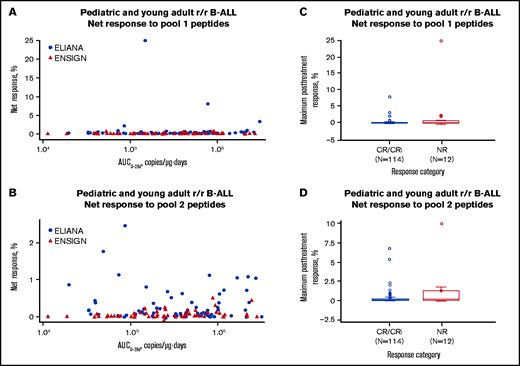
Effects of CD4+ T-cell responses to 2 pools of mCAR19 peptides on tisagenlecleucel transgene expansion and patient outcomes in pediatric and young adult patients with r/r B-ALL. Tisagenlecleucel transgene expansion (A-B) and patient outcomes (C-D). Patients from the ELIANA trial are indicated by blue circles, and patients from the ENSIGN trial are indicated by red triangles. Maximum T-cell responses are grouped by response category. Negative response data represent patients whose posttreatment T-cell responses were lower than their baseline responses. CRi, CR with incomplete blood count recovery; NR, no response.
Effects of CD4+ T-cell responses to 2 pools of mCAR19 peptides on tisagenlecleucel transgene expansion and patient outcomes in pediatric and young adult patients with r/r B-ALL. Tisagenlecleucel transgene expansion (A-B) and patient outcomes (C-D). Patients from the ELIANA trial are indicated by blue circles, and patients from the ENSIGN trial are indicated by red triangles. Maximum T-cell responses are grouped by response category. Negative response data represent patients whose posttreatment T-cell responses were lower than their baseline responses. CRi, CR with incomplete blood count recovery; NR, no response.
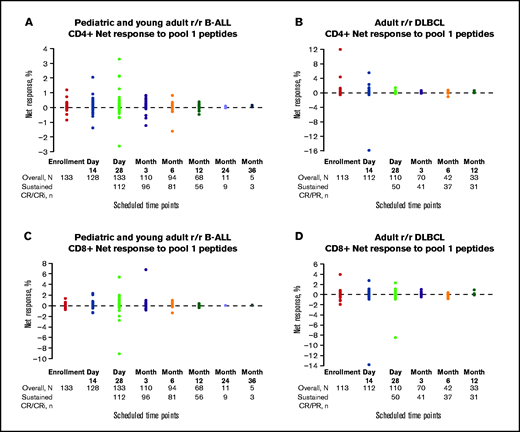
Net T-cell responses by scheduled time point. CD4+ and CD8+ net responses to pool-1 peptides in pediatric and young adult patients with r/r B-ALL (A,C) and adult patients with r/r DLBCL (B,D). Patients with sustained CR/CR with incomplete blood count recovery (CRi) or CR/partial response (PR) are indicated by solid color circles and are color-coded by scheduled time point for assessment. Negative net response data represent patients whose posttreatment T-cell responses were lower than the negative control (dimethylsulfoxide DMSO).
Net T-cell responses by scheduled time point. CD4+ and CD8+ net responses to pool-1 peptides in pediatric and young adult patients with r/r B-ALL (A,C) and adult patients with r/r DLBCL (B,D). Patients with sustained CR/CR with incomplete blood count recovery (CRi) or CR/partial response (PR) are indicated by solid color circles and are color-coded by scheduled time point for assessment. Negative net response data represent patients whose posttreatment T-cell responses were lower than the negative control (dimethylsulfoxide DMSO).
Discussion
These analyses represent the first comprehensive assessment of the effect of humoral and cellular immunogenicity and clinical end points for patients treated with tisagenlecleucel. We observed no clinically meaningful effect of preexisting or posttreatment immunogenicity (humoral or cellular) on the expansion or persistence of tisagenlecleucel, patient response, or safety. Low levels of preexisting anti-mCAR19 antibodies were highly prevalent, consistent with the prevalence of preexisting anti-mCAR19 antibodies detected in healthy volunteer sera used during method validation. As the field of CAR T-cell therapy evolves, optimization of immunogenicity assays should focus on identifying and quantifying functionally relevant anti-CAR antibodies. Preexisting antibodies that bind to biotherapeutic drugs have been detected in drug-naive individuals for a variety of biotherapeutic modalities, including single-chain variable fragments (scFvs), Fabs, (Fab′)2, nanobodies, and immunoglobulin G.20 In this case, our analyses show that these preexisting antibodies do not affect safety or efficacy; however, these data and interpretation apply only to tisagenlecleucel as examined in the clinical trial patient populations reported herein and should not be generalized to other CAR T-cell therapies or other patient populations.
Patients with prior exposure to murine antibodies could have preexisting or newly developing human anti-mouse immunoglobulin, which has been observed with antibody biotherapeutics.21,22 This could account for the slightly higher incidence of baseline anti-mCAR19 antibodies in patients with r/r DLBCL, of whom all but 2 had prior exposure to murine antibodies. However, prior exposure to rituximab did not affect the immunogenicity rate and did not increase the risk for cross-reactivity beyond that seen in patients with r/r B-ALL, suggesting that any anti-mouse antibodies generated by prior exposure to rituximab did not cross-react with mCAR19 or were short lived because of rituximab-induced B-cell aplasia.
Preexisting antibodies that bind to single-chain antibody therapeutics have been reported (eg, for anti-DR5 nanobody TAS26623 and anti-TNFR1 VH domain antibody24 ). Anti-immunoglobulin autoantibodies are known to be involved in the pathogenesis of some diseases and infections.25-30 Our analyses strongly discount the possibility that components of the natural antibody repertoire could also bind to the tisagenlecleucel CAR and exert clinical consequences. Indeed, we found no evidence that the presence of preexisting anti-mCAR19 antibodies or increases in posttreatment anti-mCAR19 antibodies affected the cellular kinetics, efficacy, or safety of tisagenlecleucel. This suggests that the anti-mCAR19 antibodies did not neutralize the activity of tisagenlecleucel therapy; however, a formal neutralizing antibody assessment was not conducted in this study. A neutralizing antibody assessment may allow further characterization of the anti-mCAR19 antibodies; however, this is not part of standard antidrug antibody assessment for CAR T-cell therapies and may be something to consider in the future on a case-by-case basis.
In this study, a higher proportion of patients with r/r B-ALL had posttreatment anti-mCAR19 antibodies compared with patients with r/r DLBCL. This may be due to greater use of IVIG to manage hypogammaglobulinemia in the r/r B-ALL population and the subsequent detection of anti-mCAR19 antibodies from IVIG preparations. The increase in posttreatment antibodies could be misinterpreted as boosted or induced immune response. Another reason for the difference in posttreatment antibodies between pediatric and adult patients could be due to rituximab exposure in most patients with r/r DLBCL, making them less able to mount a B cell–mediated response. Additional investigations or analyses in this area are needed to understand the role of immunogenicity in immunosuppressed patients.
During development and validation of the antibody detection assay, we evaluated adjusting the baseline threshold using sera from healthy volunteers; however, that would have resulted in a very high baseline threshold and could have resulted in false-negative results. This issue is similar to that seen with other biologics,20 and further characterization of preexisting humoral immunogenicity is warranted. Other CAR immunogenicity assays have been based on the CAR source antibody.31 In contrast, our cellular immunoglobulin assay is designed to also detect anti-scFv domain antibodies as well as antibodies to other extracellular domains of the tisagenlecleucel CAR molecule. One recent study demonstrated that preexisting anti-scFv antibodies exist in the majority of naive sera32 ; therefore, we believe that using the intrapatient control was the most appropriate option for determining whether exposure to tisagenlecleucel induced anti-mCAR19 antibodies above baseline levels. However, future analyses will be needed to determine whether certain regions of mCAR19 are favored for antibody binding and if the anti-mCAR19 antibody binding sites are outside the complementarity-determining regions of mCAR19 that bind CD19 on B cells. These and other assay enhancements are being evaluated for future refinement of the immunogenicity assay.
Our analysis demonstrated that T cell–mediated immunogenicity did not influence patient outcomes or cellular expansion in pediatric or young adult patients with r/r B-ALL or adult patients with r/r DLBCL. Consistent levels of preexisting and posttreatment anti-mCAR19 antibodies across response categories suggest that anti-mCAR19 antibodies did not affect outcomes. T-cell responses (as determined by IFN-γ secretion) were generally low (<1%) at all postinfusion time points, suggesting that for most patients, there was not a pronounced cellular response to mCAR19; however, our data on T-cell responses are based on IFN-γ secretion, and no other cytokines were assessed. A significant T-cell immune response against mCAR19 was estimated to be ≥1% activated CD4+ or CD8+ T cells, based on our experience with positive controls (data supplement). The observation that most patients were below this threshold suggests a limited cellular response to mCAR19. However, a complete understanding of the role of anti-mCAR19 immunogenicity in efficacy and safety will require future follow-up in clinical trials. Analysis of immunogenicity in ongoing and future clinical trials that allow reinfusion of CAR T-cell therapy may also provide insight into the potential effects of anti-CAR immune responses.
Tisagenlecleucel therapy targets both normal and malignant B cells and may result in immune dysfunction. B-cell aplasia and hypogammaglobulinemia may influence the incidence of anti-mCAR19 antibodies over time, as antibodies are degraded while B cells are depleted, and new antibodies may not be produced. In patients with DLBCL, prior rituximab therapy often results in long-term B-cell aplasia, and most patients in the JULIET study (79%) had B-cell aplasia at baseline.12 Therefore, it is likely that humoral immune responses in these patients may be attenuated, even at baseline. In addition, CAR T-cell therapy specifically targets CD19-expressing B cells, and the patients who respond to therapy develop B-cell aplasia after CAR T-cell infusion. It is not fully understood if tisagenlecleucel eliminates all plasma cells or whether preexisting plasma cells may continue to release some antibodies while antibody development against new antigens is blocked. In fact, the persistence of long-lived plasma cells after CD19-targeted CAR T-cell therapy was recently reported,33 suggesting that preexisting humoral immunity may indeed remain relatively stable for months to years after CAR T-cell therapy despite the development of B-cell aplasia. Conversely, if the presence or development of anti-mCAR19 antibodies results in a neutralizing immune response against the CD19-targeting CAR T cells, B-cell recovery may be observed.
In the B2101J clinical trial, which was part of the original regulatory submission for tisagenlecleucel, we observed a patient with r/r B-ALL who rapidly lost expression of the tisagenlecleucel (CTL019) transgene and may have developed immunity to the transgene (data supplement). The 6-year-old boy with B-ALL first relapsed after allogeneic stem cell transplantation, and his disease was refractory to subsequent salvage chemotherapy. He received lymphodepleting chemotherapy (cytarabine and etoposide) and then received a single infusion of CTL019 cells. At infusion, there was no evidence of blasts in the bone marrow or minimal residual disease by flow cytometry. Nine days after infusion, rapid transgene expansion (53 026 copies per μg at day 11) was observed, followed by rapid transgene loss on day 18, with no measurable transgene by day 28. Interestingly, this patient also had an approximate sixfold increase in posttreatment anti-mCAR19 antibodies by day 28 (supplemental Table 6), which would rank among the highest detected in the B2101J study. The patient achieved CR on day 28 but had an isolated skin relapse on day 185; he received localized radiation therapy and achieved remission of the skin lesion. The patient had a CD19+ bone marrow relapse on day 374 and went on to discontinue from the study on day 386. This is the only patient we have observed to date who had rapid expansion followed by rapid loss of the CAR transgene by day 28 and also had correspondingly high levels of anti-mCAR19 antibodies. Although this patient experienced CAR transgene loss, a durable CR was maintained until day 240, suggesting there was sufficient expansion to clear the measurable disease. In this case, it is not certain whether immunogenicity was the direct cause of rapid CAR transgene loss or the reason for relapse. It is hypothesized that immune-related loss of the CAR transgene would result in early relapse or relapse shortly after CAR transgene loss; however, this was not the case in this particular patient.
Currently, most CAR scFv domains, including tisagenlecleucel, are of murine origin,10,31 and individual patient reports have suggested that in some cases, the potential immunogenicity of CARs derived from murine antibodies can be a safety concern.34 Evidence from early studies suggested that humoral and cellular immune responses to murine-based CARs could potentially limit the persistence of CAR T cells in some patients.8 These reports involved CAR constructs that did not target normal B cells, unlike tisagenlecleucel. Future studies of immune responses to other CAR T-cell therapies that do not directly suppress the immune response (based upon the antigen being targeted) should be useful in determining potential immune responses to CAR T-cell therapies in general. In the future, it will be important to understand whether the lack of an observed effect of immunogenicity on tisagenlecleucel safety and efficacy is due to B-cell depletion or specific characteristics of the tisagenlecleucel CAR or whether prior reports represent rare events. Given that tisagenlecleucel targets both malignant and normal CD19+ B cells and therefore suppresses humoral immunity, it is not surprising that infused patients do not mount a meaningful immune response to tisagenlecleucel therapy. In addition, recipients of tisagenlecleucel therapy have also received prior B cell– and T cell–directed therapies, further reducing the likelihood of an immune response.
Anti-mouse immunoglobulin immune reactivity is a potential cause of immunogenicity that may be overcome by fully human or humanized CAR design. However, based on the results of this analysis, anti-mCAR19 immunogenicity does not seem to affect the safety or efficacy of tisagenlecleucel therapy. Still, there may be a role for humanized CARs for retreatment of patients who have previously been exposed to murine CARs. An early-phase study suggests that a humanized CAR may induce remission in patients who have relapsed or had partial or no response to previous murine-based CAR T-cell therapy.35 In fact, another study demonstrated immune-mediated rejection in a few patients who did not respond to a reinfusion of the same CAR T-cell therapy.36 The authors were able to detect CAR-specific T-cell responses in the patients who did not respond to a second infusion and found immunogenic epitopes within the murine scFv in 1 patient.36 Interestingly, a study of a fully human CAR T-cell therapy in patients with B-cell lymphoma reported anti-human CAR immune responses in 3 patients, but did not observe an effect on CAR T-cell expansion or persistence.37 Additional studies are needed to understand the potential benefits of using humanized CARs, perhaps in patients who do not respond to murine-based CAR T-cell therapy.
In conclusion, data from clinical trials of tisagenlecleucel in patients with r/r B-ALL and in patients with r/r DLBCL demonstrated that the presence of preexisting anti-mCAR19 antibodies or the development of posttreatment anti-mCAR19 antibodies did not affect tisagenlecleucel cellular kinetics, efficacy, or safety. Studies examining CAR T-cell therapy immunogenicity have been limited to date; therefore, it will be important to harmonize guidance for evaluating immunogenicity across CAR T-cell therapies.
Acknowledgments
The authors thank the patients and families enrolled in this study as well as the investigators and study site personnel. The authors also thank Grzegorz Terszowski and Denise Sickert for bioanalytic support and/or thoughtful discussions, Sebastian Spindeldreher at Integrated Biologix GmbH (Basel, Switzerland) for technical and scientific contributions to the immunogenicity assays, Maloy Mangada at Cambridge Biomedical for cellular immunogenicity analyses, and Janet Stegehuis and Henko Tadema at PRA for humoral immunogenicity analyses. Medical writing support was provided by Michael Hobert (Healthcare Consultancy Group).
Medical writing support was for this manuscript was funded by Novartis Pharmaceuticals Corporation. The studies presented herein were sponsored by Novartis Pharmaceuticals Corporation.
Authorship
Contribution: S.A.G., S.L.M, J.E.L., M.A.P., M.W.B., K.J.A., G.D.M, C.S.T., U.J., S.R.F., P.B., S.J.S., E.K.W., and T.W.L. enrolled patients, performed research, and contributed to data collection and interpretation; K.T.M., R.A., B.P., A.W., E.R.W., and F.M. performed cellular kinetic and/or statistical analyses and contributed to data interpretation; A.C.-A. contributed to data interpretation; K.T.M. wrote the first draft; all authors were involved in revising the manuscript and approved the final version; and all authors were involved in the clinical trials and had access to the primary data.
Conflict-of-interest disclosure: K.T.M. is an employee of and owns stock in Novartis. S.A.G. reports grants and personal fees from Novartis; personal fees from Adaptimmune, Jazz Pharmaceuticals, TCR2 Therapeutics, Eureka Therapeutics, Cellectis, Roche, Juno Therapeutics, and Vertex; and grants from Servier and Kite and has been issued a patent for toxicity management for antitumor activity of CARs. S.L.M. reports clinical trial support from Novartis; consulting, advisory boards, or study steering committee for Novartis, Kite, and Wugen. J.E.L., K.J.A., and C.S.T. report personal fees from Novartis. M.A.P. reports grants from Adaptive; personal fees from Novartis, Adaptive, CSL Behring, and Amgen; and nonfinancial support from Medac and Amgen. G.D.M. reports consultancy for Novartis. U.J. reports grants and personal fees from Novartis, Gilead, Celgene/Bristol-Myers Squibb, and Miltenyi. S.J.S. reports personal fees and honoraria from Acerta, AlloGene, AstraZeneca, BeiGene, Genentech/Roche, LoxoOncology, Novartis, and Tessa Therapeutics; consulting fees from Celgene/Juno; honoraria from Celgene, Nordic Nanovector, and Pfizer; steering committee participation for AbbVie, Celgene, Novartis, Juno, Nordic Nanovector, and Pfizer; and research support from AbbVie, Acerta, Celgene/Juno, DTRM Bio, Genentech, Incyte, Merck, Novartis, Portola, and TG Therapeutics and holds a patent for combination therapies of CAR and PD-1 inhibitors (royalties to Novartis). E.K.W. reports research grants from Novartis, Celldex, Partners Therapeutic, Juno, and Amgen and consulting fees from Cambium Medical Technologies, Novartis, Partners Therapeutics, Pharmacyclics, and CellDx and owns stock in Cerus, Chimerix, Cambium Medical Technologies, and Cambium Oncology. R.A. is an employee of Novartis Institutes for BioMedical Research and owns stocks in Novartis Pharmaceuticals. B.P. is an employee of and owns stock in Novartis. E.R.W. reports employment by Novartis and owns stock in Novartis. F.M. is an employee of and owns stock in Novartis. A.C.-A. is an employee of Novartis. T.W.L. reports personal fees from Novartis, Cellectis, Bayer, Loxo Oncology, and Eli Lilly; nonfinancial support from Novartis; and grants from Novartis, Pfizer, and Bayer. The remaining authors declare no competing financial interests.
Correspondence: Karen T. Mueller, Novartis Pharmaceuticals Corporation, One Health Plaza, East Hanover, NJ 07936; e-mail: karentmueller@gmail.com.

