

Key Points
Rps14-deficient zebrafish demonstrate features of MDS and impaired stress responses.
Rps14-induced anemia is rescued on ligation of Tlr7, via paradoxical suppression of inflammatory signalling and enhanced differentiation.

Abstract
Myelodysplastic syndrome (MDS) is a hematological malignancy characterized by blood cytopenias and predisposition to acute myeloid leukemia (AML). Therapies for MDS are lacking, particularly those that have an impact in the early stages of disease. We developed a model of MDS in zebrafish with knockout of Rps14, the primary mediator of the anemia associated with del(5q) MDS. These mutant animals display dose- and age-dependent abnormalities in hematopoiesis, culminating in bone marrow failure with dysplastic features. We used Rps14 knockdown to undertake an in vivo small-molecule screening, to identify compounds that ameliorate the MDS phenotype, and we identified imiquimod, an agonist of Toll-like receptor-7 (TLR7) and TLR8. Imiquimod alleviates anemia by promoting hematopoietic stem and progenitor cell expansion and erythroid differentiation, the mechanism of which is dependent on TLR7 ligation and Myd88. TLR7 activation in this setting paradoxically promoted an anti-inflammatory gene signature, indicating cross talk via TLR7 between proinflammatory pathways endogenous to Rps14 loss and the NF-κB pathway. Finally, in highly purified human bone marrow samples from anemic patients, imiquimod led to an increase in erythroid output from myeloerythroid progenitors and common myeloid progenitors. Our findings have both specific implications for the development of targeted therapeutics for del(5q) MDS and wider significance identifying a potential role for TLR7 ligation in modifying anemia.
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of myeloid malignancies associated with cytopenias and evolution to acute myeloid leukemia (AML). MDS with loss of all or part of the long arm of chromosome 5 (del(5q) MDS) is the most common subtype,1 and, importantly, 5q loss has been shown to be an initiating event in MDS development in most such cases.2,3
The only curative therapy for patients with MDS remains stem cell transplantation, for which many older patients with MDS are unfit. Thus, there remains an unmet need for novel therapies for del(5q) MDS.
Ribosomal protein of the small subunit, RPS14 (also now known as uS114 ), located in the critically deleted region of chromosome 5q, is one of several genes identified as a haploinsufficient tumor suppressor gene in del(5q) MDS.5 ‐10 We and others have shown haploinsufficient levels of Rps14 recapitulate features of del(5q) MDS in zebrafish, mice, and primary human cells.5,11,12
Proposed mechanisms for Rps14-mediated hematopoietic defects include p53-dependent apoptosis and increased proinflammatory innate-immune signaling via Toll-like receptor-4 (TLR4), resulting from an increase in translation of the cognate ligands S100A8 and S100A9.11 In addition, activation of TLR-MyD88-NF-κB signaling in del(5q) MDS results from haploinsufficiency of other genes and microRNAs contained within the 5q critically deleted region.7,9 However, signaling through TLR4 is also necessary for the generation of hematopoietic stem and progenitor cells (HSPCs) in both steady-state and stressed hematopoiesis.13 ‐15 This finding indicates that homeostatic regulation of inflammatory signaling through TLR pathways is critical to maintaining normal hematopoiesis.
We created a novel zebrafish model of del(5q) MDS, using transcription activator–like effector nucleases (TALENs) to mutate rps14. We used Rps14-deficient zebrafish to identify small molecules that alleviate anemia in this model and identified the TLR7/8 agonist imiquimod. This effect was dependent on TLR7 and resulted in improved hemoglobinization and an increase in HSPCs. Importantly, imiquimod led to an increase in erythroid output in both fish and primary human cells from anemic patients, an effect not restricted to, but enhanced by, the haploinsufficiency of Rps14. Analysis of Rps14-deficient HSPCs showed upregulation of negative regulators of canonical WNT signaling, which was reversed after imiquimod exposure. Furthermore, there was paradoxical downregulation of inflammatory signaling and NF-κB target genes in Rps14+/− embryos exposed to imiquimod, that was dependent on Myd88. These data suggest cross talk between the endogenous proinflammatory effects of Rps14 haploinsufficiency, and TLR7 activation led to attenuation of anemia in our model.
Methods
Zebrafish husbandry
Zebrafish (Danio rerio) AB, AB/TL, and transgenic strains Tg(gata1:DsRed), Tg(itga2b:GFP), and Tg(NFκB:GFP)sh235 were maintained according to standard procedures and UK Home Office guidelines.16 ‐19 The embryos were staged according to Kimmel et al20 and expressed in postfertilization hours (hpf) or days (dpf).
Generation of a zebrafish rps14 mutant line
TALENs targeting exon 2 of the zebrafish rps14 gene were made by using FLASH.21
Morpholinos
Morpholinos (MOs) targeting the 5'UTR/ATG codon (gata1) or splice donor sites (rps14, myd88) or Gene Tools standard control were injected into 1- to 2-cell–stage embryos at doses previously described.12,22,23
Immunohistochemistry
o-Dianisidine staining of hemoglobin and Sudan black (SB) staining to visualize granulocytes were performed as previously described.24,25 SB+ cells were quantified from the distal end of the yolk extension to the tail tip in the caudal hematopoietic tissue (CHT; fetal liver equivalent in zebrafish).
Induction of stress
Hemolytic stress was induced by exposure of the embryos to phenylhydrazine (PHZ).26 They were incubated in 1 µg/mL PHZ from 24 to 48 hpf and then washed. Cold stress was induced after gastrulation by placing 6 somite stage embryos at 22°C.
Microscopy
Microscopy was performed with a Leica M205 FA stereomicroscope equipped with a Leica DFC310 FX camera and LAS 4.0 software. All images were processed with Fiji version 2.0.0-rc- 43/1.51f or Photoshop CS6.
Statistical analysis
Data are presented as means ± standard deviation. Analysis was performed with Prism (GraphPad Software, La Jolla CA). Specific statistical tests are shown in the figure legends.
Zebrafish analysis by flow cytometry
Individual embryos and adult zebrafish kidneys were analyzed by flow cytometry, as described previously.12
Cell sample preparation and imaging
The cells (1 × 105) were centrifuged onto slides, fixed with methanol, and stained with May-Grunwald-Giemsa stain. Images were taken with the Nanozoomer 2.0 RS (Hamamatsu).
Western blot analysis
Protein lysates were obtained from pooling 6-dpf genotyped embryos. Blots were probed with phospho-eIF2α (Ser51) (119A11) rabbit mAb (3597; Cell Signaling) and total EIF2α (ab26197; Abcam).
Results
Rps14 loss results in dose-dependent hematopoietic abnormalities
We have shown that knockdown of Rps14 using MOs results in anemia that models the erythroid defect of del(5q) MDS.12 We used TALENs to generate a stable Rps14 mutant zebrafish with an early frameshift mutation (rps14E8fs; supplemental Figure 1). Using whole-mount immunofluorescence, we demonstrated loss of Rps14 protein in an allelic dose-dependent manner, indicating that Rps14E8fs/+ animals have haploinsufficient protein levels that model those in del(5q) MDS (supplemental Figure 2). Rps14E8fs/E8fsembryos, herein referred to as Rps14−/−, showed profound developmental anomalies that were lethal by 5 dpf. o-Dianisidine staining at 4 dpf showed that Rps14−/− embryos have markedly reduced hemoglobinization (Figure 1C) compared with that of Rps14+/+embryos (Figure 1A). However, developmental morphology and hemoglobinization in the Rps14+/− embryos was indistinguishable from that of their Rps14+/+ siblings (Figure 1A-B). To quantify erythroid cells, we used Rps14E8fs carrying Tg(gata1:dsRed). Importantly, flow cytometry of the embryos demonstrated a decrease in the number of dsRed-expressing erythroid cells at 3 dpf (Figure 1D; supplemental Figure 3A). This finding indicates that the Rps14+/− embryos had anemia associated with a reduced number of red cells, yet maintained adequate hemoglobinization.
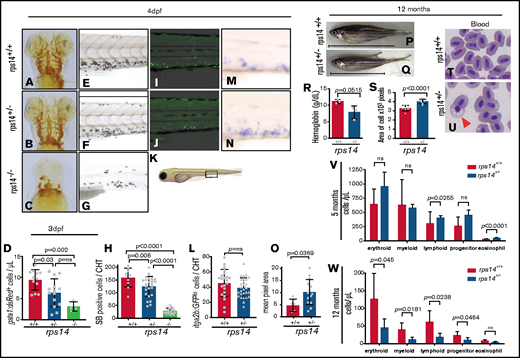
Rps14 stable mutants show dose-dependent effects on hematopoiesis in embryos. (A-C) Assessment of hemoglobinized cells with o-dianisidine staining. Ventral views of the head in 4-dpf embryos. Rps14−/− zebrafish showed profound loss of hemoglobinized cells (C) and developmental anomalies. Rps14+/− were indistinguishable from their WT siblings by microscopy (A-B). (D) Number of erythroid cells by flow cytometry of individual Tg(gata1:dsRed);Rps14+/− embryos at 3 dpf. There was an allelic dose-dependent effect on the dsRed-expressing number of cells. (E-G) SB-stained 4-dpf embryos for granulocytes. (H) Quantification of SB+ cells. (K) Region of CHT depicted in cartoon (adapted from Lizzy Griffiths with permission). SB staining shows an allelic dose-dependent effect of Rps14. (I-J) Lateral views of the CHT of 4dpf Rps14 mutant fish carrying the itga2b:GFP transgene, labeling HSPCs that reside in the CHT. (L) Quantitation of stationary GFPlo cells in the CHT. (M-N) Expression of c-myb by in situ hybridization in 4-dpf embryos. (O) In contrast to itga2b-GFP, c-myb expression is increased in the CHT, quantified by median expression intensity in CHT.58 (P-Q) Lateral views of 12-month-old adult fish show the decreased size of heterozygotes. The anterior faces left and the dorsal side faces upward. The horizontal line shows the body length excluding the tail. (R) Quantification of hemoglobin concentration. (S) Analysis of cell size. Red blood cells were significantly larger in Rps14+/− mutants compared with siblings. (T-U) Micrographs of blood smears from Rps14+/− and WT siblings; the arrowhead indicates poorly hemoglobinized erythroid cell in Rps14+/−. (V-W) Absolute number of cells per microliter of different cell types in the kidney marrow of 5- and 12-month-old fish, showing progressive differences between heterozygous rps14 mutants and their WT siblings. Statistical comparison by 1-way ANOVA with Tukey’s multiple-comparisons test (D,H) or unpaired Student t test. Original magnification x100 for panels T and U.
Rps14 stable mutants show dose-dependent effects on hematopoiesis in embryos. (A-C) Assessment of hemoglobinized cells with o-dianisidine staining. Ventral views of the head in 4-dpf embryos. Rps14−/− zebrafish showed profound loss of hemoglobinized cells (C) and developmental anomalies. Rps14+/− were indistinguishable from their WT siblings by microscopy (A-B). (D) Number of erythroid cells by flow cytometry of individual Tg(gata1:dsRed);Rps14+/− embryos at 3 dpf. There was an allelic dose-dependent effect on the dsRed-expressing number of cells. (E-G) SB-stained 4-dpf embryos for granulocytes. (H) Quantification of SB+ cells. (K) Region of CHT depicted in cartoon (adapted from Lizzy Griffiths with permission). SB staining shows an allelic dose-dependent effect of Rps14. (I-J) Lateral views of the CHT of 4dpf Rps14 mutant fish carrying the itga2b:GFP transgene, labeling HSPCs that reside in the CHT. (L) Quantitation of stationary GFPlo cells in the CHT. (M-N) Expression of c-myb by in situ hybridization in 4-dpf embryos. (O) In contrast to itga2b-GFP, c-myb expression is increased in the CHT, quantified by median expression intensity in CHT.58 (P-Q) Lateral views of 12-month-old adult fish show the decreased size of heterozygotes. The anterior faces left and the dorsal side faces upward. The horizontal line shows the body length excluding the tail. (R) Quantification of hemoglobin concentration. (S) Analysis of cell size. Red blood cells were significantly larger in Rps14+/− mutants compared with siblings. (T-U) Micrographs of blood smears from Rps14+/− and WT siblings; the arrowhead indicates poorly hemoglobinized erythroid cell in Rps14+/−. (V-W) Absolute number of cells per microliter of different cell types in the kidney marrow of 5- and 12-month-old fish, showing progressive differences between heterozygous rps14 mutants and their WT siblings. Statistical comparison by 1-way ANOVA with Tukey’s multiple-comparisons test (D,H) or unpaired Student t test. Original magnification x100 for panels T and U.
We next determined the impact of Rps14 loss on definitive myelopoiesis. Using SB to stain granulocytes at 4 dpf, we identified a reduction in SB+ granulocytes located within the CHT (Figure 1K) in a dose-dependent manner, with allelic loss of Rps14 (Figure 1 E-H).
We then assessed the effects of Rps14 loss on HSPC in the Tg(itga2b:GFP) strain, in which GFPlo cells in the CHT label HSPCs.18 HSPC quantification in the CHT at 4 dpf of Rps14+/− mutants compared with that in Rps14+/+embryos did not demonstrate a significant difference (Figure 1I-L; supplemental 3B). HSPCs and extensive autofluorescence were virtually absent in the Rps14−/− embryos and were not assessable. We further recorded the number of HSPCs by using whole-mount in situ hybridization (WISH) of c-myb in the CHT at 4 dpf. By contrast to the Tg(itga2b:GFPlo) cells, c-myb expression in the CHT increased in Rps14+/− embryos compared with that in Rps14+/+embryos (Figure 1M-O). Itga2b-GFPlo-expressing cells are some of the first to arise during definitive hematopoiesis and are restricted to HSPC- and megakaryocytic lineage–committed cells, whereas c-myb is expressed in a broader subset of HSPCs, as well as more committed myeloid lineage cells (http://servers.binf.ku.dk/bloodspot/).27 Therefore the increase in expression of c-myb with normal itga2b and reduced mature myeloid cells suggests an increase in myeloid-lineage–restricted progenitor cells in Rps14+/− embryos, with a concomitant block in differentiation.
Rps14+/− adult mutants have anemia and features of MDS
Rps14+/− adult zebrafish are significantly smaller than Rps14+/+ ones (Figure 1P-Q; supplemental Figure 4A-B). Hemoglobin levels showed a lower concentration of hemoglobin in Rps14+/− than in Rps14+/+ zebrafish (Figure 1R). Furthermore, examination of hematopoietic cell morphology from Rps14+/− adults showed cells that have defective hemoglobinization (Figure 1T-U; arrowhead), large red cells (Figure 1S), and dyserythropoiesis in the kidney marrow (supplemental Figure 4C-J).
We next used flow cytometric examination of kidney marrow (the site of adult hematopoiesis) to assess the effects of Rps14 loss on hematopoiesis. At 5 months of age, an increase in eosinophils and lymphocytes was observed in Rps14+/− compared with Rps14+/+ zebrafish by forward and side scatter examination of the cell populations (Figure 1V; supplemental Figure 4K). We observed an increase in Tg(itga2b:GFPlo) cells in the progenitor cell fraction (supplemental Figure 4K-L), a feature observed in murine Rps14 conditional knockouts.19,28 By 12 months, the kidney marrow of Rps14+/− mutants showed defects in all lineages, indicating features of bone marrow (BM) failure (Figure 1W; supplemental Figure 4M).
Stress markedly exacerbates the hematopoietic defects in Rps14 heterozygous animals
To refine the effects of Rps14 haploinsufficiency on hematopoiesis, we exposed Rps14+/− embryos to hematopoietic stress.11 First, we used cold stress, incubating embryos at 22°C from 10 hpf for 4 days to the prim-22 stage (supplemental Figure 5A).29 Rps14+/− embryos were significantly more anemic after cold stress than their Rps14+/+ siblings, with no clear difference in the developmental stage (supplemental Figure 5B-D). To determine the effects of stress specifically on definitive hematopoietic cells, we used hemolytic stress induced by incubating embryos in PHZ for 24 hours (24-48 hpf) and assessed the recovery of hematopoiesis (Figure 2A).30 Embryos exposed to PHZ showed loss of hemoglobinized erythroid cells by apoptosis (supplemental Figure 6). At 6 dpf, wild-type (WT) animals had completely recovered, with o-dianisidine staining showing normal hemoglobinization, whereas Rps14+/− siblings remained markedly anemic (Figure 2B-C, quantified in Figure 2D). We further assessed the effect of hemolytic stress on HSPCs using the Tg(itga2b:GFP;rps14+/−) strain. As mentioned previously, Rps14+/− embryos showed no difference in the number of Tg(itga2b:GFP) HSPCs in the absence of PHZ. PHZ stress resulted in an increase in HSPCs compared with nonstressed animals in WT; however, Rps14+/− larvae did not elicit this response, leading to a significant decrease in HSPCs compared with stressed WT siblings (Figure 2E).
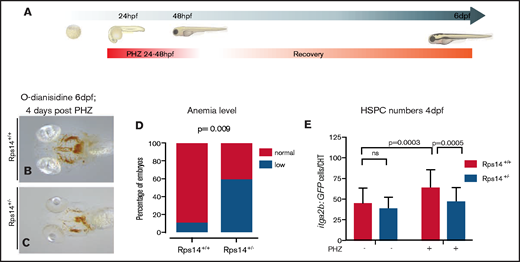
Hemolytic stress augments the hematopoietic phenotype in Rps14+/− mutants and is rescued by imiquimod. (A) Hemolytic stress experiment. (B-C) Representative views (ventral) of rps14+/+ and rps14+/− siblings treated as in panel A and stained with o-dianisidine at 6 dpf. Rps14+/− mutants demonstrate a clear anemic phenotype, quantified in panel D. (E) Effect of hemolytic stress on HSPCs. Statistical comparisons were performed with Fisher’s exact test (D) or ANOVA (E).
Hemolytic stress augments the hematopoietic phenotype in Rps14+/− mutants and is rescued by imiquimod. (A) Hemolytic stress experiment. (B-C) Representative views (ventral) of rps14+/+ and rps14+/− siblings treated as in panel A and stained with o-dianisidine at 6 dpf. Rps14+/− mutants demonstrate a clear anemic phenotype, quantified in panel D. (E) Effect of hemolytic stress on HSPCs. Statistical comparisons were performed with Fisher’s exact test (D) or ANOVA (E).
Small-molecule screenings identify imiquimod as a modifier of anemia in Rps14-deficient embryos
To use our system to assess for potential novel therapeutic agents in del(5q) MDS, we developed a small-molecule screen for modifiers of anemia (Figure 3A). We used MO knockdown of Rps14, as the morphant phenotype is comparable to mutants, to obtain an increased number of molecules to be tested.12 We screened the Spectrum collection for compounds that could alleviate the characteristic morphological defects and/or anemia resulting from Rps14 MO injection. Treatment of Rps14 morphants with the TLR7/8 pathway agonist imiquimod strikingly rescued the anemia phenotype of the Rps14 morphants (Figure 3D) compared with dimethyl sulfoxide (DMSO)-treated negative controls (Figure 3C) to a level similar to that observed in experiments using l-leucine.12

Small-molecule screening for modifiers of anemia in Rps14 deficiency identifies imiquimod. (A) The screening design. (B-E) Rps14 4-dpf morphants and controls treated with DMSO, l-leucine, or imiquimod were stained for hemoglobin with o-dianisidine. Ventral views of the head (top) and lateral (bottom) views, with the anterior facing left and the dorsal facing upward. (F) Semiquantitative analysis of the effects of imiquimod on the severity of hemoglobinization. Imiquimod improved the level of hemoglobinization compared with the DMSO control. Statistical comparisons performed by Fisher’s exact test. Original maginfication x80 for panels B-E.
Small-molecule screening for modifiers of anemia in Rps14 deficiency identifies imiquimod. (A) The screening design. (B-E) Rps14 4-dpf morphants and controls treated with DMSO, l-leucine, or imiquimod were stained for hemoglobin with o-dianisidine. Ventral views of the head (top) and lateral (bottom) views, with the anterior facing left and the dorsal facing upward. (F) Semiquantitative analysis of the effects of imiquimod on the severity of hemoglobinization. Imiquimod improved the level of hemoglobinization compared with the DMSO control. Statistical comparisons performed by Fisher’s exact test. Original maginfication x80 for panels B-E.
We next assessed the improvement in hemoglobinization with DMSO or 5 µM imiquimod treatment by classifying each larva by the severity of their anemia. Imiquimod markedly decreased the proportion of larvae with severe anemia (Figure 3F).
Imiquimod rescues stress-induced anemia in Rps14 heterozygotes
We next assessed the effects of imiquimod on Rps14+/− mutants exposed to PHZ. As observed in Rps14 morphants, imiquimod rescued the anemia observed in stressed Rps14+/− mutants (Figure 4A-E). We also observed a dose-dependent effect of imiquimod on the proportion of anemic animals at 6 days (Figure 4F).

Imiquimod exerts its effect on Rps14-deficient anemic embryos via on-target activation of TLR7 (A-D) Ventral views of 6-dpf embryos stained with o-dianisidine, treated with PHS to induce hemolytic stress or with DMSO (A,C) or with PHZ and imiquimod (B,D). Imiquimod rescued stress-induced anemia. (F) Quantified as the normal/total ratio of embryos across 3 replicates at a concentration of 20 µM. (E) Effect of imiquimod analyzed across the dose range, analyzed by nonlinear regression. (G) Flow cytometric analysis of rps14E8fs;Tg(itga2b:GFP) single embryos exposed to hemolytic stress and then treated with DMSO or imiquimod. Imiquimod enhanced the itga2b:GFPlo cells, and the effect was most marked in the Rps14+/− embryos, where there was a significant interaction between the drug and genotype. (H-M) Ventral views of 6-dpf embryos stained with o-dianisidine, treated to induce hemolytic stress (PHZ) and with DMSO (H,K), gardiquimod (I,L), or motolimod (J,M). Gardiquimod, but not motolimod, rescued the stress-induced anemia in Rps14+/− embryos. (N) The gardiquimod rescue effect analyzed across dose range by nonlinear regression. Tlr7 knockout was validated using Miseq (O) and restriction enzyme digest with BplI (P), which digested only the WT. (Q) Knockout of Tlr7 abrogated the rescue of anemia by imiquimod. Original magnification x80.
Imiquimod exerts its effect on Rps14-deficient anemic embryos via on-target activation of TLR7 (A-D) Ventral views of 6-dpf embryos stained with o-dianisidine, treated with PHS to induce hemolytic stress or with DMSO (A,C) or with PHZ and imiquimod (B,D). Imiquimod rescued stress-induced anemia. (F) Quantified as the normal/total ratio of embryos across 3 replicates at a concentration of 20 µM. (E) Effect of imiquimod analyzed across the dose range, analyzed by nonlinear regression. (G) Flow cytometric analysis of rps14E8fs;Tg(itga2b:GFP) single embryos exposed to hemolytic stress and then treated with DMSO or imiquimod. Imiquimod enhanced the itga2b:GFPlo cells, and the effect was most marked in the Rps14+/− embryos, where there was a significant interaction between the drug and genotype. (H-M) Ventral views of 6-dpf embryos stained with o-dianisidine, treated to induce hemolytic stress (PHZ) and with DMSO (H,K), gardiquimod (I,L), or motolimod (J,M). Gardiquimod, but not motolimod, rescued the stress-induced anemia in Rps14+/− embryos. (N) The gardiquimod rescue effect analyzed across dose range by nonlinear regression. Tlr7 knockout was validated using Miseq (O) and restriction enzyme digest with BplI (P), which digested only the WT. (Q) Knockout of Tlr7 abrogated the rescue of anemia by imiquimod. Original magnification x80.
Inflammatory signaling through other TLRs has been shown to affect the emergence of HPSCs. To determine at which point during hematopoiesis imiquimod exerted its effect and to assess the effect of TLR7 signaling on HSPC, we used Tg(itga2b:GFP);rps14+/− exposed to hemolytic stress and then treated with imiquimod or control. In PHZ-stressed control mutants, GFPlo HSPCs were reduced in Rps14+/− compared with Rps14+/+embryos. Exposure to imiquimod resulted in an increase in GFPlo cells in both Rps14+/− and Rps14+/+embryos; however, the effect was more marked in Rps14+/− compared with Rps14+/+embryos. A 2-way analysis of variance (ANOVA) demonstrated an interaction between the effects of the Rps14 heterozygosity and imiquimod treatment (Figure 4G).
These findings indicate that imiquimod leads to both improved hemoglobinization and increased HSPCs.
Imiquimod affects erythropoiesis through TLR7 ligation
Imiquimod is an imidazoquinoline, which binds to TLR7 and TLR8 on their dimerization interfaces. This binding is enhanced for TLR7 when single-stranded RNA (its cognate ligand) is bound at a different site.31 ‐34 To determine whether the hematopoietic effects of imiquimod that we observed were obtained through ligation of TLR7, we used additional small molecules that selectively activate these receptors. Gardiquimod, a specific TLR7 agonist rescued the anemia associated with Rps14 heterozygosity (Figure 4H-L,N). In contrast, the TLR8-specific agonist motolimod did not rescue the effects (Figure 4J,M). This result indicates that the effects we observed were specific to ligation of TLR7 but not TLR8. To further confirm this notion, we used a Tlr7 crispant knockout (Figure 4O-P). Rps14+/− x Rps14+/+ clutches were injected with Tlr7 CRIPSR guide or control and then exposed to PHZ. TLR7 crispants abrogated the imiquimod-mediated rescue of anemia in Rps14 heterozygotes (Figure 4Q). These results show that the effects of imiquimod occur specifically through TLR7.
Imiquimod-treated Rps14+/− HSPCs show reversal of WNT signaling and paradoxical downregulation of inflammation and an increase in erythroid differentiation
We sought to define the mechanism by which stress from PHZ led to more marked anemia in Rps14+/− embryos and whether imiquimod alleviated the anemia and reversed the effect. We have observed an increase in phosphorylation of Eif2α at serine 51 (Eif2αP) in Rps19 morphants,35 which occurs in response to stressors, including free heme, resulting in global translation reduction. We hypothesized that Eif2αP would also occur in Rps14+/− embryos under PHZ-induced hemolytic stress. At 6 dpf, the Eif2αP/total ratio was increased in Rps14+/− compared with Rps14+/+ embryos (P = .02); however, this was not clearly influenced by the presence of PHZ or imiquimod (Figure 5A-B). Therefore, we concluded that Rps14+/− results in Eif2αP independent of stress, which contributes to defective translation in our model.
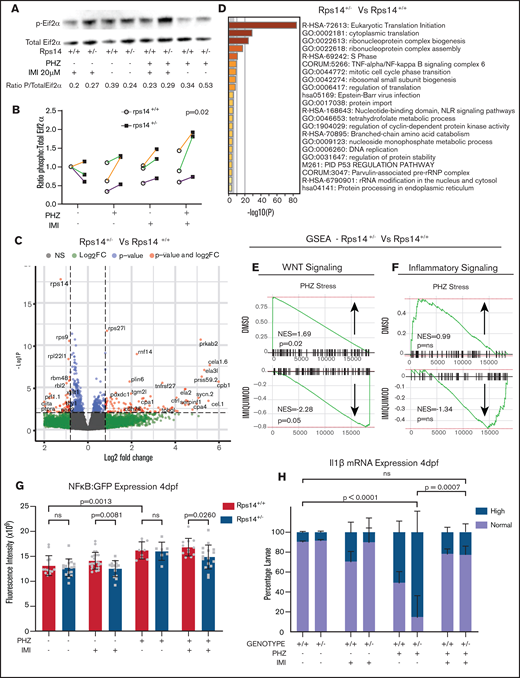
RNA sequencing analysis of HSPCs shows the effects on WNT and inflammatory signaling pathways. (A) Representative western blot of p-Eif2α (phosphoserine 51) and Eif2α total in Rp14 mutants at 6-dpf exposed to PHZ and/or imiquimod. (B) Normalized ratio (to untreated WT) of p-Eif2α/total Eif2α shown in 3 experiments, represented by different colored lines. The P-value refers to the effect of genotype on the ratio. (C) Differential gene expression of Rps14+/− vs Rps14+/+ embryos shown as a volcano plot for all conditions combined. Rps14 is shown in bold. (D) Metascape Pathway Analysis of Rps14+/− vs Rps14+/+ differentially expressed genes showing the top 20 enriched pathways. (E-F) GSEA analysis comparing Rps14+/− with Rps14+/+ in DMSO-treated vs imiquimod-treated HSPCs. (E) Negative regulation of WNT signaling was enriched in Rps14+/− vs Rps14+/+ DMSO-treated HSPCs, and the effect was reversed in imiquimod-treated HSPCs. (F) Similarly, inflammatory signaling was enriched in Rps14+/− vs Rps14+/+ DMSO-treated HSPCs but suppressed in imiquimod-treated HSPCs. (G) Quantification of total fluorescence of NFKβ/GFP at 4 dpf shows an increase with PHZ stress but a decrease with imiquimod in Rps14+/− compared with Rps14+/+. (H) PHZ causes an increase in il1b expression by in situ hybridization in Rps14+/− which is rescued by imiquimod. Statistical comparisons are by 2-way ANOVA (B,H; genotype and condition) or ANOVA (G).
RNA sequencing analysis of HSPCs shows the effects on WNT and inflammatory signaling pathways. (A) Representative western blot of p-Eif2α (phosphoserine 51) and Eif2α total in Rp14 mutants at 6-dpf exposed to PHZ and/or imiquimod. (B) Normalized ratio (to untreated WT) of p-Eif2α/total Eif2α shown in 3 experiments, represented by different colored lines. The P-value refers to the effect of genotype on the ratio. (C) Differential gene expression of Rps14+/− vs Rps14+/+ embryos shown as a volcano plot for all conditions combined. Rps14 is shown in bold. (D) Metascape Pathway Analysis of Rps14+/− vs Rps14+/+ differentially expressed genes showing the top 20 enriched pathways. (E-F) GSEA analysis comparing Rps14+/− with Rps14+/+ in DMSO-treated vs imiquimod-treated HSPCs. (E) Negative regulation of WNT signaling was enriched in Rps14+/− vs Rps14+/+ DMSO-treated HSPCs, and the effect was reversed in imiquimod-treated HSPCs. (F) Similarly, inflammatory signaling was enriched in Rps14+/− vs Rps14+/+ DMSO-treated HSPCs but suppressed in imiquimod-treated HSPCs. (G) Quantification of total fluorescence of NFKβ/GFP at 4 dpf shows an increase with PHZ stress but a decrease with imiquimod in Rps14+/− compared with Rps14+/+. (H) PHZ causes an increase in il1b expression by in situ hybridization in Rps14+/− which is rescued by imiquimod. Statistical comparisons are by 2-way ANOVA (B,H; genotype and condition) or ANOVA (G).
To refine further the mechanism by which imiquimod increases HSPCs and mature erythroid cells more potently in Rps14+/− than in WT embryos, we performed RNA sequencing analysis of HSPCs, with or without PHZ stress and treated with imiquimod or vehicle control (supplemental Figure 3C). Systems level analysis of genes confirmed highly significant knockdown of Rps14 at the RNA level in HSPCs in heterozygotes across all conditions (Figure 5C).36 Pathway analysis of genes differentially regulated in Rps14+/− compared with Rps14+/+ embryos demonstrated enrichment of pathways involving ribosome biogenesis, translation, and p53 and TNFα/NF-κB signaling (Figure 5D), in keeping with known Rps14-associated pathways in hematopoiesis.36,37 The number of differentially regulated genes was relatively few (n = 35-225 between conditions), suggesting that the observed phenotypes most likely arise posttranscriptionally or through non–cell-autonomous effects. To further assess the changes in differentially expressed genes, we undertook Gene Set Enrichment Analysis (GSEA).38,39 We observed that negative regulators of canonical WNT signaling were upregulated in PHZ-stressed Rps14+/− HSPCs compared with those of siblings, but were markedly downregulated after exposure to imiquimod (Figure 5E). We also observed reciprocal changes in inflammatory signaling signatures in stressed Rps14+/− compared with siblings, with exposure to imiquimod resulting in a change from pro- to anti-inflammatory signaling (Figure 5F). We validated this observation by using quantitative polymerase chain reaction to identify downstream mediators of TLR-signaling (supplemental Figure 7A). Consistent with our GSEA findings, we showed that the presence of imiquimod reversed the expression level of key NF-κB proinflammatory target genes in Rps14 morphants. To further investigate these findings in Rps14+/− mutants, we used a Tg(NFκB:GFP) reporter line. We found that PHZ stress led to an increased expression of GFP in Tg(NFκB:GFP) cells at 4 dpf, but this effect was reversed in Rps14+/− compared with Rps14+/+ embryos in the presence of imiquimod (Figure 5G). We also assessed levels of interleukin-1β (IL1β) by WISH. PHZ exposure resulted in an increased number of embryos with high levels of IL1β expression, and this effect was again reversed by the presence of imiquimod (Figure 5H). Finally, we used an Myd88 MO to show that loss of Myd88 abrogated the imiquimod-mediated rescue of anemia in Rps14+/− embryos (supplemental Figure 7B), in a way similar to the loss of Tlr7 (Figure 4Q). These data combined show that Rps14+/− mutants have a proinflammatory state through activation of the TLR-MyD88-NF-κB signaling complex, and treatment of Rps14+/− embryos with imiquimod paradoxically reverses this effect.
Imiquimod enhances erythroid differentiation
Our data suggest that treating Rps14+/− mutants with imiquimod resulted in an increase in HSPCs but also induced an increase in more mature erythroid cells. To assess whether this increase is due to general expansion of the cell pool or a specific effect on erythroid differentiation, we assessed erythroid differentiation in Rps14+/− mutants with and without imiquimod. A GSEA showed that stressed HSPCs have a downregulated erythroid differentiation signature in Rps14+/− compared with that of siblings. Imiquimod reversed this effect, showing a proerythroid differentiation signature (supplemental Figure 8A-B). We used WISH to assess the expression of gata1 at 3 dpf and showed that, during recovery from PHZ stress, there was an increase in gata1 expression that was more pronounced in Rps14+/+ than in Rps14+/− embryos and that this effect was reduced in the presence of imiquimod (supplemental Figure 8C). To further assess these effects, we ablated all mature erythroid cells using MO knockdown of the master erythropoiesis regulator Gata1 in Tg(gata1:dsRed) transgenic animals.19,40 GATA1 has been shown to be central to the mechanism of anemia associated with RPS-14 and other ribosomal proteins.41 ‐46 Treatment of Gata1 morphants with imiquimod increased the numbers of circulating and static erythroid cells (supplemental Figure 8E-I; supplemental Movies 1 ‐4 ), and centrifuging of sorted dsRed cells showed increased numbers of mature erythroid and myeloid cells (supplemental Figure 8J-L). Therefore, our data support that imiquimod not only increases HSPCs but promotes both erythroid and myeloid differentiation on the background of anemia associated with Rps14 or Gata1 deficiency.
In vitro hematopoietic colony output is enhanced in anemic human primary cells treated with imiquimod
Our zebrafish studies showed that imiquimod increases HSPCs and can alleviate anemia by enhancing erythroid differentiation in Rps14- and Gata1-deficient anemia. To determine whether these effects were also observed in human cells, we sought to analyze BM of patients with MDS. Unfortunately, no samples from patients with MDS del(5q) were available for analysis. However, we think that the effect of imiquimod is most likely not limited to patients with ribosomal protein abnormalities, but more to those who have a basal inflammatory state that is common to loss of Rps14 and other MDS subtypes.47,48 Therefore, we used patient samples of anemic dysplasia in the BM (supplemental Table 1). HSPC output from patients with anemia was extremely low, with less than 0.02% of plated cells giving rise to any colonies (Figure 6A). This contrasts with the nonanemic control where 8% of plated cells gave rise to colonies (supplemental Figure 9A) and where the overall colony output was enhanced by imiquimod.
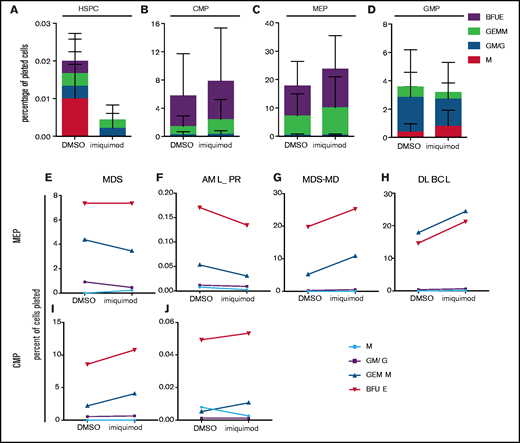
Imiquimod enhances erythroid output of human primary cells in vitro. (A-D) Primary cells obtained from BMs of anemic patients detailed in the supplemental Table 1 (n = 4) underwent fluorescence-activated cell sorting into different populations. Sort purity was verified as greater than 95% for all conditions. CFU-assays were performed in triplicate in Methocult without serum. Colonies were scored at 12 days, and output is shown as percentages of cells input. HSPCs (A), CMPs (B), MEPs (C), and granulocyte macrophage progenitors (GMPs) (D). (E-G) Individual patient plots show the change in colony output associated with imiquimod for MEPs (E-H) and CMPs (I-J). BFUE, burst-forming unit erythroid; GEMM, granulocyte, erythrocyte, macrophage, and megakaryocyte; GM/G, granulocyte-macrophage/granulocyte; M, macrophage; AML_PR, acute myeloid leukemia in partial remission; MDS-MD, myelodysplastic syndrome with multilineage dysplasia; DLBCL, diffuse large B-cell lymphoma.
Imiquimod enhances erythroid output of human primary cells in vitro. (A-D) Primary cells obtained from BMs of anemic patients detailed in the supplemental Table 1 (n = 4) underwent fluorescence-activated cell sorting into different populations. Sort purity was verified as greater than 95% for all conditions. CFU-assays were performed in triplicate in Methocult without serum. Colonies were scored at 12 days, and output is shown as percentages of cells input. HSPCs (A), CMPs (B), MEPs (C), and granulocyte macrophage progenitors (GMPs) (D). (E-G) Individual patient plots show the change in colony output associated with imiquimod for MEPs (E-H) and CMPs (I-J). BFUE, burst-forming unit erythroid; GEMM, granulocyte, erythrocyte, macrophage, and megakaryocyte; GM/G, granulocyte-macrophage/granulocyte; M, macrophage; AML_PR, acute myeloid leukemia in partial remission; MDS-MD, myelodysplastic syndrome with multilineage dysplasia; DLBCL, diffuse large B-cell lymphoma.
In both common myeloid progenitors (CMPs) and megakaryocyte/erythroid progenitors (MEPs) from anemic patients we observed an increase in colony output in the imiquimod-treated cells compared with the controls (Figures 6B-C). This finding reflects an increase in both colony-forming unit-granulocytes, erythrocytes, macrophages, and megakaryocytes (GEMMs) and burst-forming unit-erythroids. In contrast, there was no effect on overall colony output from granulocyte-monocyte progenitors, rather a lineage bias toward more mature myeloid (Granulocyte-Macrophage (GM) and granulocyte (G)) colonies rather than GEMMs (Figure 6D). To further highlight the effects at the MEP and CMP level, Figure 6E-J shows the output from each patient with or without the addition of imiquimod. Although these results did not reach significance, taken together the data indicate that the effects of imiquimod treatment on erythropoiesis may result from effects at the level of both CMPs and MEPs in human cells. Our data also indicate that imiquimod has variable effects at different stages of lineage commitment in both the myeloid and erythroid compartments and that these are not limited to Rps14 deficiency, suggesting that cross talk between mediators of inflammation may occur more widely.
Discussion
In this study, we generated a model of del(5q) MDS, using TALENS to introduce a frameshift mutation in Rps14. Embryos deficient in Rps14 showed dose-dependent defects in the number of mature myeloid cells. Erythropoiesis was similarly reduced; however, the effects on the erythroid lineage as well as HSPCs were much more striking after exposure to stress. These findings suggest that Rps14 haploinsufficiency has significant effects on steady-state hematopoiesis, but that Rps14 also has a specific role in the maintenance of stress-induced hematopoiesis. As PHZ results in stress to the hematopoietic system by hemolysis, we assessed whether the Rps14-specific effects of PHZ is mediated by stress-response pathways sensitive to free heme via Eif2αP. Interestingly, although we did not show an effect of PHZ on Eif2αP, Rps14+/− mutants, regardless of treatment, showed an increase in Eif2αP compared with their siblings, suggesting that a global reduction in translation arising from Eif2αP contributed to the phenotypes observed.
To identify potential novel therapeutic agents for del(5q) MDS, we conducted an in vivo small-molecule screening for compounds that could alleviate anemia in Rps14-deficient embryos. The screening identified a striking rescue of the anemic phenotype with the TLR7/8 agonist imiquimod. Our data showed that imiquimod exerted its effect on erythroid cells via TLR7 signaling. We also showed a marked effect of imiquimod on itga2b:GFP-expressing HPSCs. Effects were observed in Rps14+/− as well as Rps14+/+embryos, but notably the effect in Rps14+/− was more pronounced.
Proinflammatory signaling through TLR4 has been identified as a mechanism of anemia in Rps14-deficient mice, and blocking this signaling pathway can rescue this effect.11 Rps14+/− HSPCs showed upregulation of the TNF/NF-κB signaling complex in our model, indicating conservation of this pathway. However, TLR7 ligation also activated this pathway through Myd88. Therefore, our findings that activation of this pathway alleviated anemia appeared counterintuitive. RNA sequencing data support that TLR7 activation in Rps14+/− HSPCs paradoxically results in downregulation of inflammation, which we validated by measuring NF-κB and IL1β expression levels. A paradigm for such cross talk between activation of different TLRs and the effects of this on downstream signaling and homeostasis of inflammatory signaling has been elucidated for TLR3 and TLR7.49 Coactivation of TLR3 and TLR7 leads to an enhanced production of some inflammatory cytokines; however, production of several key pathway intermediates, such as TRAF6, is reduced. Induction of inflammatory tolerance has also been described between TLR4 and TLR7/8 in monocytes, which requires microRNA 146a.50 This suggests that, although some components of the innate immune response act in concert to enhance inflammatory signaling, there are underlying mechanisms in place to halt excessive immune activation, or even specifically reduce inflammation.
The effects of imiquimod in our model were not limited to increased HSPCs. Using the differentiation arrest observed in Gata1 morphants, we demonstrated an effect of imiquimod on erythroid (as well as myeloid) differentiation. Interestingly, Gata1 not only has a central role in the pathogenesis of ribosomal protein–mediated anemias, but is also thought to be key in cytopenias associated with activation of the inflammasome in general.
When comparing Rps14+/−-stressed HSPCs with those of siblings, stressed HSPCs showed a significant increase in negative regulators of the canonical WNT pathway. Regulation of HSPC specification, emergence, expansion, and differentiation have all been shown to be tightly regulated, in part, by components of the WNT/β-catenin pathway.51,52 However, temporally, both imiquimod treatment and the effects we observed occurred after specification and emergence of HSPCs, suggesting in this context that WNT pathway inhibition in stressed Rps14+/− may impede expansion or differentiation of HSPCs and that this effect is alleviated by imiquimod. Furthermore, cross talk between NF-κB inflammatory signaling and WNT pathway activation and/or inhibition has been reported in several cell types (reviewed in Ma and Hottiger53 ), including evidence that prolonged TLR4-mediated inflammation suppresses WNT/β-catenin in bone, resulting in apoptosis and necrosis.54 Our study showed that possible cross talk may occur between these pathways.
Although, TLRs, their ligands, and the inflammatory consequences of signaling have been shown to differ among species,55 TLR7 and TLR8 are the most highly conserved of all the TLRs, and inflammatory responses to R848 are preserved in fish.56 Nonetheless, to establish the relevance of our findings in humans, we used BM cells from anemic patients to determine the effects of imiquimod. Our findings suggest that imiquimod can enhance erythroid output in anemic individuals at the level of both CMPs and MEPs, supporting the data from the fish. In these experiments, very few colonies were derived from HSCs of anemic patients; therefore, assessment of the effects of imiquimod on HSCs was not possible. One possibility is that the effects observed in our system are non–cell autonomous via other inflammatory cells. Most of functional effects of TLR7 signaling have been described via innate immune cells. Furthermore, we observed a larger inflammatory signaling response when analyzing downstream effector genes of TLR signaling in whole-embryo RNA extracts (Figure 5; supplemental Figure 7). Thus, the cell of origin of the effects observed in our study is not yet defined and is the focus of ongoing work.
Finally, a key unanswered question is how Rps14 deficiency and the associated effects on ribosome assembly and translation lead to activation of inflammatory signaling. Murine studies defined increased production of S100 proteins to be the mediators of increased inflammatory signaling via TLR4.11 However, it is attractive to speculate that, given the structural knowledge that TLR7 activation by imiquimod is enhanced by single-stranded RNA species, the production of aberrant ribosomal RNA species observed in Rps14-knockout cells may contribute to our observation that the effects of imiquimod are more marked in Rps14-deficient cells.5,57 To date, no such phenomenon has been observed for endogenous RNA species outside of autoimmune disease.
In summary, we describe a model of del(5q) MDS in zebrafish and define a novel role for TLR7 in enhancing hematopoiesis in this model through modulation of the inflammatory response and differentiation.
Acknowledgments
The authors thank the CRUK-UCL flow cytometry core and UCL zebrafish facility for their assistance; and Catherine Loynes, Dave Drew, and Stephen Renshaw for facilitating the acquisition of new animal lines and reagents during the COVID lockdown period.
E.M.P. was supported by a CRUK Advanced Clinician Scientist Fellowship and is a former recipient of a Wellcome-Beit Intermediate Clinical Fellowship and the Leuka John Goldman Fellowship for future science. C.H. was supported by a Medical Research Council Clinical Research Training Fellowship. O.P. was supported by BECAS Chile (CONICYT) and Overseas Research Scholarship (UCL). L.E.V. was supported by a FONDECYT grant (11160951) and Medical Research Council grants G0900994 and MR/L003775/1 (Steve W. Wilson and Gaia Gestri, Department of Cell and Developmental Biology, UCL). C.B. was supported by the Swedish Research Council (grant 2015-00135) and Marie Sklodowskan Curie Actions, Cofund, and Project INCA (grant 600398).
Authorship
Contribution: E.M.P., O.A.P., and A.L. designed and performed the experiments; J.R., C.H., A.L., P.D., and Y.H. performed the experiments and contributed to scientific analysis and discussions; Y.J. conducted the screening with assistance from M.C.V. and O.A.P.; L.E.V. and K.T. made TALENS targeting rps14; and C.B. and S.R. provided essential technical assistance for human hematopoietic cell studies and analysis.
Conflict-of-interest disclosure: E.M.P. has received honoraria for consultancy for Novartis, Celgene, and Takeda that are not related to the work in this study. The remaining authors declare no competing financial interests.
Correspondence: Elspeth M. Payne, UCL Cancer Institute–314, 72 Huntley St, London WC1E6BT, United Kingdom; e-mail: e.payne@ucl.ac.uk.

