

Key Points
GPX4 is expressed during human erythropoiesis and is required for the enucleation of orthochromatic erythroblasts.
The role of GPX4 in enucleation is independent of its function in ferroptosis regulation.

Abstract
The selenoprotein glutathione peroxidase 4 (GPX4), the only member of the glutathione peroxidase family able to directly reduce cell membrane–oxidized fatty acids and cholesterol, was recently identified as the central regulator of ferroptosis. GPX4 knockdown in mouse hematopoietic cells leads to hemolytic anemia and to increased spleen erythroid progenitor death. The role of GPX4 during human erythropoiesis is unknown. Using in vitro erythroid differentiation, we show here that GPX4-irreversible inhibition by 1S,3R-RSL3 (RSL3) and its short hairpin RNA–mediated knockdown strongly impaired enucleation in a ferroptosis-independent manner not restored by tocopherol or iron chelators. During enucleation, GPX4 localized with lipid rafts at the cleavage furrows between reticulocytes and pyrenocytes. Its inhibition impacted enucleation after nuclear condensation and polarization and was associated with a defect in lipid raft clustering (cholera toxin staining) and myosin-regulatory light-chain phosphorylation. Because selenoprotein translation and cholesterol synthesis share a common precursor, we investigated whether the enucleation defect could represent a compensatory mechanism favoring GPX4 synthesis at the expense of cholesterol, known to be abundant in lipid rafts. Lipidomics and filipin staining failed to show any quantitative difference in cholesterol content after RSL3 exposure. However, addition of cholesterol increased cholera toxin staining and myosin-regulatory light-chain phosphorylation, and improved enucleation despite GPX4 knockdown. In summary, we identified GPX4 as a new actor of human erythroid enucleation, independent of its function in ferroptosis control. We described its involvement in lipid raft organization required for contractile ring assembly and cytokinesis, leading in fine to nucleus extrusion.
Introduction
The glutathione peroxidase (GPX) enzymes are part of the protective system against oxidative damages that includes prevention of oxidation by iron-sequestering proteins and antioxidant molecules, scavengers of reactive oxygen species (ROS), and reduction of oxidants such as peroxides through enzymatic reactions. Experiments in mice revealed that the selenoprotein GPX4 was the only GPX family member essential for embryonic development.1 It is also the sole GPX able to directly reduce cell membrane cholesterol and polyunsaturated lipid peroxides into alcohols.2 Recent reports identified GPX4 as the central inhibitor of ferroptosis, a process during which iron-induced peroxidation of membrane lipids causes a specific cell death that can be reverted by lipophilic antioxidants such as tocopherol or by iron chelators.3-5 Ferroptosis was first described in ras-mutated cancer cells and was induced directly by 1S,3R-RSL3 (RSL3), a pharmacological inhibitor targeting GPX4 by covalent bond or indirectly by erastin that depleted the GPX4 cofactor glutathione by blocking cystine import through transporter Xc− inhibition.3,6 GPX4 has stimulated interest and is considered as a promising anticancer target.7 Many reports have since highlighted the implication of ferroptosis in multiple tissues and diseases including neurodegenerative diseases,8-12 acute kidney injury,13,14 cardiomyopathy,15 or, recently, hemochromatosis.16 Transferrin receptor I, highly expressed during erythropoiesis, was recently found upregulated in cancer cell lines treated with RSL3 and shown to be a specific ferroptosis marker.17 By potentially preventing from ferroptosis cells that contain the high iron level required for heme synthesis, GPX4 appears to be a good candidate for antioxidant defense in the erythroid lineage. The role of GPX4 in murine erythropoiesis has been recently revealed using a GPX4-knockout model restricted to hematopoietic cells. These mice developed anemia related to necroptotic death of erythroid precursors reverted in vivo by vitamin E and by Rip3 deletion.18 Late erythropoiesis was also impaired with defective reticulocyte maturation related to accumulation of large autophagosomes and impaired mitophagy.19 Although highly expressed at the RNA level at the orthochromatic stage,20 the role of GPX4 during human erythroid maturation has not so far been reported. Here, we aimed to study GPX4 expression and function during in vitro erythroid differentiation from human CD34+ cells obtained from apheresis. Using both an RSL3-mediated chemical inhibition and short hairpin RNA (shRNA)-mediated GPX4 knockdown, we describe for the first time a role for GPX4 in enucleation of human erythroblasts that is dependent on lipid raft clustering and myosin phosphorylation.
Methods
In vitro culture of human CD34+ progenitors
CD34+ progenitor cells were isolated by positive selection kit (Miltenyi Biotec) using an immunomagnetic bead cell-sorting system (AutoMacs; Miltenyi Biotec) from healthy leukapheresis samples, in agreement with our institutional ethics committee (Amiens Centre Hospitalo-Universitaire). From day 1 to day 6, sorted cells (purity >95%) were cultured in Iscove modified Dulbecco medium (Biochrom) supplemented with stabilized glutamine (Eurobio), containing 2% human AB serum (H2B), 100 U/mL penicillin (Eurobio), 100 µg/mL streptomycin (Eurobio), 500 µg/mL human holotransferrin (Sigma-Aldrich), 10 µg/mL recombinant human insulin (Sigma-Aldrich), 2 IU/mL heparin (Braun), 100 ng/mL human stem cell factor (Miltenyi Biotec), and 3 IU/mL erythropoietin (Roche). From day 7 to day 20, stem cell factor was removed to the erythroid-differentiation medium.
Clonogenic potential
Cultured cells were plated in triplicate at a density of 2000 cells in 1 mL of semisolid MethoCult H4435–enriched medium (Stem Cell Technologies) and incubated at 37°C in a humidified atmosphere with 5% CO2 after addition of dimethyl sulfoxide (DMSO) or 1 µM RSL3. The erythroid burst-forming unit (BFU-E) and erythroid colony-forming unit (CFU-E) colonies were cultured for 14 days according to the criteria described by Dover et al.21
Lentiviral particle production and cell transduction
Three shRNAs against GPX4 and 1 scramble shRNA cloned in pLKO.1-CMV-tGFP vector were purchased from Sigma-Aldrich and designed using the Mission shRNA tool (detailed sequences in supplemental Table 1). Viral production and transduction were ensured as recently described.22
Flow cytometry
Cells were harvested sequentially from day 1 to day 20 of differentiation, washed, and stained with appropriate panels of conjugated antibodies in phosphate-buffered saline plus 2 mM EDTA and 0.5% bovine serum albumin on ice (supplemental Table 2). In all conditions, exclusion of nonviable cells was assessed using 7-amino-actinomycin D or 4′,6-diamidino-2-phenylindole (DAPI). AlexaFluor 647 cholera toxin subunit b (CTB; ThermoFisher) was used to stain lipid microdomains rich in ganglioside GM1. Lipid-peroxidative damage was assessed by staining cells for 30 minutes with 10 µM lipophilic fluorophore probe 11C-BODIPY581/591(ThermoFisher) as previously described.23 Filipin (Sigma-Aldrich) was used at 0.1 mg/mL to assess membrane cholesterol content after cell fixation using 1% paraformaldehyde.24
Fluorescent microscopy
Cells (5 × 104/mL) treated with vehicle or RSL3 were fixed with 1% paraformaldehyde (ThermoFisher), permeabilized (Permwash; BD), washed by centrifugation (1500 rpm, 5 minutes), resuspended in 100 μL of 1× phosphate-buffered saline–5% bovine serum albumin, then stained with AlexaFluor 647 GPX4 (Santa Cruz Biotechnology) and AlexaFluor 488–conjugated Flotillin-2 (Santa Cruz Biotechnology) or AlexaFluor 488–conjugated CTB (ThermoFisher). After gentle washing followed by cytospin (300g, 5 minutes), coverslip-attached erythroblasts were mounted onto glass plates using ProLong Gold Antifade Mountant with DAPI (ThermoFisher). Images were taken using an Axio Imager M2 ApoTome microscope running ZEN software (Zeiss).
Reagents
RSL3, simvastatin, ferrostatin, and necrostatin were purchased from Selleckchem and α-tocopherol, Qvd-Oph, 3-MA, deferoxamine, mercaptosuccinic acid (MSA), cholesterol, and isopentenyl pyrophosphate were obtained from Sigma-Aldrich.
Statistical analysis
Statistical analyses were performed using 2-tailed P value and parametric tests. Statistical significance used was α = 0.05. For quantitative variables, we used the Student t test or 1-way analysis of variance test and Tukey post hoc analysis for multiparametric analysis. All numeric values were as mean values plus or minus standard error of the mean (SEM).
Methods for imaging flow cytometry (IFC), immunoblots, real-time quantitative polymerase chain reaction, and neutral lipid quantification are detailed in supplemental Methods.
Results
Following GPX4-inhibition effects throughout human erythroid differentiation
GPX4 expression at the RNA and protein levels was first investigated during ex vivo erythropoiesis from human CD34+ cells. The GPX4 messenger RNA level was highly expressed from day 6 to day 11 compared with early progenitors (supplemental Figure 1A), then decreased during terminal differentiation. At the protein level, GPX4 expression was also highest from day 6 to day 11, then decreased from day 14 to day 17 during terminal stages when orthochromatic erythroblasts endured nucleus extrusion, in agreement with published proteomic data (supplemental Figure 1B).25 In an attempt to verify whether GPX4 expression correlated with a specific function during human erythropoiesis, we performed pharmacological inhibition throughout our differentiation model using RSL3 that binds GPX4 specifically and covalently, resulting in its posttranslational degradation.5,26,27 For ex vivo CD34+ cell differentiation, we tested 3 RSL3 concentrations: 0.5 µM, 1 µM, and 2 µM and measured, from day 1 to day 18, cell count and viability (data not shown). We chose 1 µM RSL3 as the optimal dose with no lethality and an efficient GPX4 knockdown, in agreement with other reports.5,28 The GPX4 protein level assessed by immunoblot decreased drastically after 1 µM RSL3 exposure for 24 hours and 96 hours (supplemental Figure 1C-D). In contrast, RSL3 did not affect expression of GPX1, another GPX family member expressed in the erythroid lineage (supplemental Figure 1C). At 1 µM, RSL3 added at day 0 affected neither cell proliferation nor viability, highlighting a potential resistance of primary erythroblasts to ferroptosis driven by the GPX4 inhibitor (Figure 1A). We then wondered whether the successive steps of erythroid differentiation could be affected by RSL3-mediated GPX4 inhibition. The progenitor compartment, assessed using erythroid colony-forming assay, was not impacted by RSL3 exposure (Figure 1B). We performed flow cytometry (FCM) to follow erythroid maturation from hematopoietic progenitors to nucleated orthoerythroblasts (OrthoE) at day 15. No significant changes in CD34, CD36, CD71, glycophorin A (GPA), Band 3, and CD49d expression were observed between RSL3-exposed cells and controls (Figure 1C and supplemental Figure 2A for FCM strategy). We followed erythroid maturation more precisely, assessing characteristics of late erythropoiesis, that is, loss of CD49d assessed by FCM between day 15 and day 18. We also assessed cell downsizing, and decrease in nucleocytoplasmic ratio using IFC. We found no differences between DMSO and RSL3 conditions, only a trend toward increased cell size but below the threshold of significance (Figure 1D-F), arguing for a nonessential role of GPX4 from progenitor to orthochromatic stage.
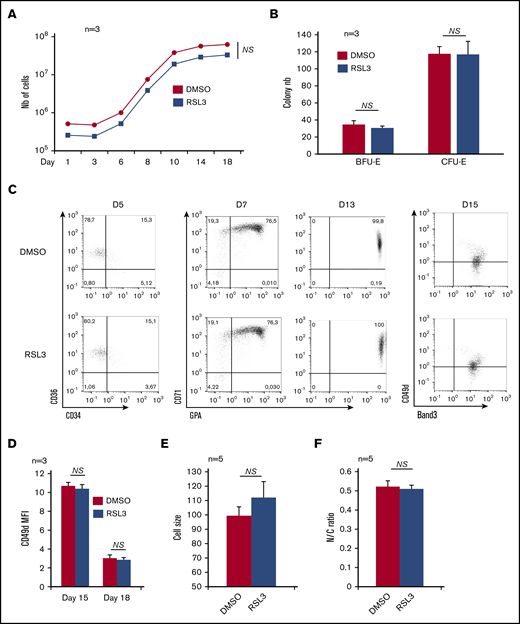
Effects of GPX4 inhibition on human in vitro erythropoiesis from CD34+cells to orthochromatic erythroblasts. (A) Effect of GPX4 inhibitor, RSL3 at 1 µM, vs DMSO on cell counts from day 1 to day 18 during in vitro erythroid differentiation from peripheral blood CD34+ cells. Dead cells were excluded using trypan blue staining (n = 3). (B) Effect of GPX4 inhibitor, RSL3 at 1 µM, vs DMSO on clonogenic activity of erythroid progenitors. No differences in BFU-E and CFU-E counts were noticed (n = 3). (C) FCM dot plots assessing erythroid differentiation at early day 5 (CD34/CD36), intermediate day 7 and day 13 (CD71/GPA), and late day 15 (CD49d/Band 3) stages, in DMSO and 1 µM RSL3 conditions. Dot plots were obtained from 1 representative experiment (n = 3). (D-F) Comparative assessment of 3 features of orthochromatic erythroblast maturation between 1 µM RSL3 and DMSO-treated cells: mean fluorescence intensity (MFI) of CD49d assessed by FCM showing its kinetics of decrease between day 15 and day 18 (n = 3) (D), IFC analysis of cell size (E) and nucleocytoplasmic ratio (F), at day 18 to day 20 (depending on the kinetics of differentiation) (n = 5). None of these parameters were statistically different between DMSO and RSL3 conditions. A trend toward a higher cell size after RSL3 exposure was observed, but below significance threshold (P = .09). Statistical significance determined by Student t test. Error bars are SEM. NS, nonsignificant.
Effects of GPX4 inhibition on human in vitro erythropoiesis from CD34+cells to orthochromatic erythroblasts. (A) Effect of GPX4 inhibitor, RSL3 at 1 µM, vs DMSO on cell counts from day 1 to day 18 during in vitro erythroid differentiation from peripheral blood CD34+ cells. Dead cells were excluded using trypan blue staining (n = 3). (B) Effect of GPX4 inhibitor, RSL3 at 1 µM, vs DMSO on clonogenic activity of erythroid progenitors. No differences in BFU-E and CFU-E counts were noticed (n = 3). (C) FCM dot plots assessing erythroid differentiation at early day 5 (CD34/CD36), intermediate day 7 and day 13 (CD71/GPA), and late day 15 (CD49d/Band 3) stages, in DMSO and 1 µM RSL3 conditions. Dot plots were obtained from 1 representative experiment (n = 3). (D-F) Comparative assessment of 3 features of orthochromatic erythroblast maturation between 1 µM RSL3 and DMSO-treated cells: mean fluorescence intensity (MFI) of CD49d assessed by FCM showing its kinetics of decrease between day 15 and day 18 (n = 3) (D), IFC analysis of cell size (E) and nucleocytoplasmic ratio (F), at day 18 to day 20 (depending on the kinetics of differentiation) (n = 5). None of these parameters were statistically different between DMSO and RSL3 conditions. A trend toward a higher cell size after RSL3 exposure was observed, but below significance threshold (P = .09). Statistical significance determined by Student t test. Error bars are SEM. NS, nonsignificant.
Defective enucleation due to RSL3- or shRNA-mediated GPX4 knockdown
We then studied the last step of erythroblast maturation, that is, enucleation, which can be assimilated to an asymmetric cytokinesis leading to genesis from 1 erythroblast (GPAhigh, Hoechst+) of 1 enucleated reticulocyte (GPAhigh, Hoechst−) and 1 extruded nucleus, that is, pyrenocyte (GPAlow, Hoechst+) (supplemental Figure 2A).29-31 We cultured cells as described in “Methods” from day 0, then added DMSO and RSL3 at day 14 and carefully followed the enucleation process by FCM, cytology, and IFC (day 18 to day 20). In control cells, enucleation reached 75% at day 20. In contrast, RSL3-exposed cells enucleated less efficiently, showing 30% and 50% reduction at day 18 and day 20, respectively, in comparison with DMSO (Figure 2A). This enucleation defect was confirmed using cytology after May-Grünwald-Giemsa (MGG) staining and IFC, showing with both techniques a higher proportion of nucleated erythroblasts after RSL3 exposure (Figure 2B; supplemental Figure 2B). To verify that enucleation defect was not related to an off-target effect of RSL3, we performed GPX4 knockdown using 3 GPX4-specific shRNA sequences cloned into a lentiviral vector. After transduction, GFP+ erythroblasts were sorted and GPX4 expression quantified at RNA and protein levels. Using real-time quantitative polymerase chain reaction, we observed 65%, 25%, and 63% decreases in GPX4 messenger RNA level in SH1-, SH2-, and SH3-transduced cells, respectively (supplemental Figure 3A). Using western blot, SH1 and SH3 induced 78% and 80% downregulation of GPX4 protein expression, respectively, whereas SH2 was less efficient (59% decrease) (supplemental Figure 3B). shRNA-mediated GPX4 knockdown did not impact erythroid differentiation until enucleation (supplemental Figure 3C). As for RSL3, the percentage of Hoechst+ cells at day 20 was significantly higher in GPX4-knockdown conditions than in controls and depended on the level of GPX4 knockdown (Figure 2C). Taken together, our data showed that enucleation was significantly impaired after GPX4 knockdown.
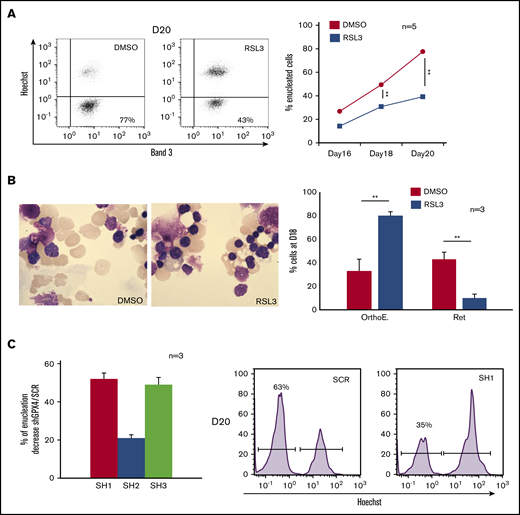
Enucleation impairment induced by GPX4 inhibition and gene silencing. (A, left) Representative Band 3/Hoechst dot plots obtained at day 20 in GPA+ control and 1 µM RSL3-treated cells using FCM (n = 5). (A, right) Curves showing quantification of enucleated GPA+/Hoechst− cells (%) from day 16 to day 20 in 1 µM RSL3 and control treatments (n = 5). Percentage of enucleated cells. DMSO vs RSL3: day 18, 49% ± 4% vs 31% ± 3%, P < .01; day 20, 78% ± 5% vs 39% ± 5%, P < .01. (B, left) May-Grünwald-Giemsa (MGG) staining (magnification ×100) of DMSO or 1 µM RSL3-treated cells. Images shown were taken at day 18 and were representative of 3 independent experiments. (B, right) Cytological count after MGG staining of orthochromatic erythroblasts and reticulocytes (%) in 1 µM RSL3 and control conditions (n = 3). Percentage of RET. DMSO vs RSL3: 43% ± 6% vs 10% ± 3%, P < .01. (C, left) histogram representing the percentage of decrease in day 20 enucleation observed after transduction of 3 different shRNA against GPX4 (SH1, SH2, SH3) in comparison with control (ShSCR) (n = 3). Percentage of enucleation decrease in comparison with SCR. SH1: 52% ± 3%; SH2: 21% ± 2%; SH3: 49% ± 4%, SCR vs each SH: P < .01. (C, right) A representative histogram of Hoechst staining at day 20 in SCR and SH1-transduced cells. **P < .01. Error bars are SEM.
Enucleation impairment induced by GPX4 inhibition and gene silencing. (A, left) Representative Band 3/Hoechst dot plots obtained at day 20 in GPA+ control and 1 µM RSL3-treated cells using FCM (n = 5). (A, right) Curves showing quantification of enucleated GPA+/Hoechst− cells (%) from day 16 to day 20 in 1 µM RSL3 and control treatments (n = 5). Percentage of enucleated cells. DMSO vs RSL3: day 18, 49% ± 4% vs 31% ± 3%, P < .01; day 20, 78% ± 5% vs 39% ± 5%, P < .01. (B, left) May-Grünwald-Giemsa (MGG) staining (magnification ×100) of DMSO or 1 µM RSL3-treated cells. Images shown were taken at day 18 and were representative of 3 independent experiments. (B, right) Cytological count after MGG staining of orthochromatic erythroblasts and reticulocytes (%) in 1 µM RSL3 and control conditions (n = 3). Percentage of RET. DMSO vs RSL3: 43% ± 6% vs 10% ± 3%, P < .01. (C, left) histogram representing the percentage of decrease in day 20 enucleation observed after transduction of 3 different shRNA against GPX4 (SH1, SH2, SH3) in comparison with control (ShSCR) (n = 3). Percentage of enucleation decrease in comparison with SCR. SH1: 52% ± 3%; SH2: 21% ± 2%; SH3: 49% ± 4%, SCR vs each SH: P < .01. (C, right) A representative histogram of Hoechst staining at day 20 in SCR and SH1-transduced cells. **P < .01. Error bars are SEM.
GPX4 inhibition impaired enucleation in a ferroptosis-, mitophagy-, and necroptosis- independent manner
GPX4 is in many cell types a central regulator of ferroptosis.5 However, the level of lipid membrane peroxidation after GPX4 knockdown in primary erythroblasts, measured by 11C-BODIPY581/591 staining, was low, affecting <5% of erythroblasts after 12-hour 1 µM RSL3 exposure. Increasing RSL3 to 3 µM impacted cell viability but weakly increased 11C-BODIPY581/591 zfluorescence (<10%) (Figure 3A). Erythroleukemic UT7 cells were also 10-fold more resistant to RSL3-induced lipid peroxidation than the myeloid cell line U937 and lymphoid cell line RAMOS (supplemental Figure 4A). This weak 11C-BODIPY581/591staining argues for an enucleation defect being ferroptosis-independent. Indeed, (i) the indirect ferroptosis inducer erastin, which acts upstream of GPX4, did not impact enucleation (supplemental Figure 4B) and (ii) the enucleation rate at day 20 was not recovered by pretreatment with ferroptosis inhibitors such as tocopherol, ferrostatin, and deferoxamine (Figure 3B). We also investigated involvement of other cell-death pathways: necroptosis (Necrox), apoptosis (Qvd-Oph), or mitophagy (3-MA) inhibitors failed to revert the GPX4 inhibition–related defect of enucleation (Figure 3B). We then assessed whether the relative resistance of primary erythroblasts to ferroptosis might be due to compensatory mechanisms by other enzymes harboring GPX activity such as peroxiredoxin-6 (PRDX6), the only enzyme of the PRDX family having both phospholipase and GPX activity.32 PRDX6 expression is maximal at the end of erythroid maturation according to the transcriptomic database.33 We used MSA, a selective inhibitor of PRDX6 peroxidase activity.34 We observed a synergistic effect of RSL3 and MSA on the induction of membrane lipid peroxidation at day 15, reflecting the fact that GPX4 and PRDX6 may compensate each other’s deficiency to protect human erythroblasts from lipid peroxidation (Figure 3C).
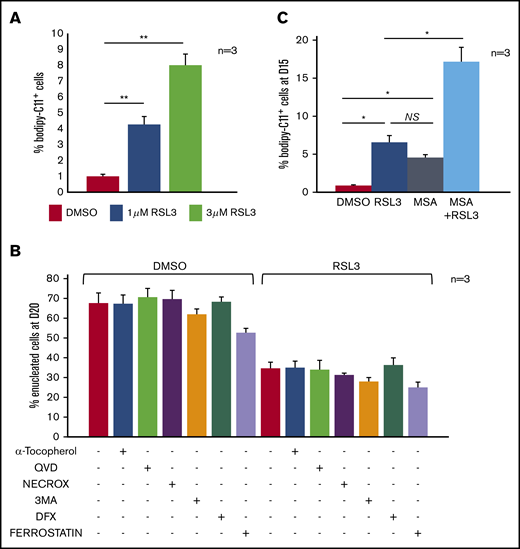
Enucleation defect induced by GPX4 inhibition was not related to ferroptosis in human primary erythroblasts. (A) FCM quantification of lipid ROS using 11C-BODIPY581/591 staining in primary erythroblasts treated with RSL3 at 2 concentrations (1 µM and 3 µM) during 12 hours. H2O2 was used as positive control inducing a shift >70% (not shown). RSL3 induced a dose-dependent, statistically significant but weak increase in the percentage of positive cells (n = 3). Percentage of 11C-BODIPY581/591+ cells. DMSO vs 1 µM RSL3: 1% ± 0.1% vs 4% ± 0.5%, P < .05; 1 µM RSL3 vs 3 µM RSL3: 4% ± 0.5% vs 8% ± 0.7%, P < .01. (B) Histogram showing quantification of enucleated GPA+/Hoechst− cells (%) at day 20 in DMSO and 1 µM RSL3-treated cells, with and without ferroptosis inhibitors (100 µM α-tocopherol, 2 µM ferrostatin, 5 µM deferoxamine), 20 µM apoptosis inhibitor (QVD), 10 µM necroptosis inhibitor (Necrox), and 5 mM autophagy inhibitor (3MA). There was no statistically significant difference in the enucleation rate between DMSO and all of the different inhibitors tested (n = 3). (C) Induction of membrane lipid peroxidation assessed by FCM and 11C-BODIPY581/591 staining after inhibition of GPX4 using 1 µM RSL3 and inhibition of the GPX activity of PRDX6 using 40 µM MSA (n = 3). DMSO vs RSL3 and MSA, P < .05; RSL3 vs MSA, P: NS; RSL3 or MSA vs RSL3 + MSA, P < .05. *P < .05; **P < .01. Error bars are SEM.
Enucleation defect induced by GPX4 inhibition was not related to ferroptosis in human primary erythroblasts. (A) FCM quantification of lipid ROS using 11C-BODIPY581/591 staining in primary erythroblasts treated with RSL3 at 2 concentrations (1 µM and 3 µM) during 12 hours. H2O2 was used as positive control inducing a shift >70% (not shown). RSL3 induced a dose-dependent, statistically significant but weak increase in the percentage of positive cells (n = 3). Percentage of 11C-BODIPY581/591+ cells. DMSO vs 1 µM RSL3: 1% ± 0.1% vs 4% ± 0.5%, P < .05; 1 µM RSL3 vs 3 µM RSL3: 4% ± 0.5% vs 8% ± 0.7%, P < .01. (B) Histogram showing quantification of enucleated GPA+/Hoechst− cells (%) at day 20 in DMSO and 1 µM RSL3-treated cells, with and without ferroptosis inhibitors (100 µM α-tocopherol, 2 µM ferrostatin, 5 µM deferoxamine), 20 µM apoptosis inhibitor (QVD), 10 µM necroptosis inhibitor (Necrox), and 5 mM autophagy inhibitor (3MA). There was no statistically significant difference in the enucleation rate between DMSO and all of the different inhibitors tested (n = 3). (C) Induction of membrane lipid peroxidation assessed by FCM and 11C-BODIPY581/591 staining after inhibition of GPX4 using 1 µM RSL3 and inhibition of the GPX activity of PRDX6 using 40 µM MSA (n = 3). DMSO vs RSL3 and MSA, P < .05; RSL3 vs MSA, P: NS; RSL3 or MSA vs RSL3 + MSA, P < .05. *P < .05; **P < .01. Error bars are SEM.
RSL3-induced enucleation defect was restored by cholesterol or its precursor IPP but was not directly related to decreased cholesterol content
Isopentenyl pyrophosphate (IPP), product of the mevalonate pathway, is an intermediate metabolite of cholesterol synthesis, and also a source of isopentenyl transfer RNA (tRNA), which is required for selenoprotein translation,35,36 highlighting the cross talk between GPX4/ferroptosis and cholesterol pathways37 (supplemental Figure 5A). We wondered whether RSL3 could favor the use of IPP toward isopentenyl-tRNA synthesis as a compensatory mechanism to GPX4 knockdown, at the expense of cholesterol production. Indeed, cholesterol is essential for cytokinesis38 and nucleus expulsion in mammalian erythropoiesis.39,40 Simvastatin, a specific inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase, significantly decreased the enucleation rate, mimicking the effect of GPX4 knockdown (supplemental Figure 5B). When 10 µM cholesterol was added in the medium at day 14, enucleation at day 20 was partially restored in RSL3-treated erythroblasts as shown by FCM (Figure 4A) and cytology (Figure 4B). We quantified total cholesterol using a filipin fluorescent probe41 and found no difference between control and RSL3 conditions at day 20 (Figure 4C). We also performed neutral lipid extraction and quantification by analytic gas chromatography with flame-ionization detection and found no difference in the cholesterol/total neutral lipid ratio between the 2 conditions (Figure 4D). Notably, adding 5 μM IPP at day 14 in the medium partially restored enucleation at day 20 (Figure 4E) but not GPX4 expression as assessed by western blot (Figure 4F). Taken together, our results evidenced that RSL3-mediated enucleation impairment was not related to a cholesterol quantitative defect by mevalonate pathway derivation. However, the recovery of the phenotype, by increased cholesterol uptake, evocated its involvement in the enucleation defect, but more likely in a qualitative way, through an anomaly of its repartition within the cell membrane.
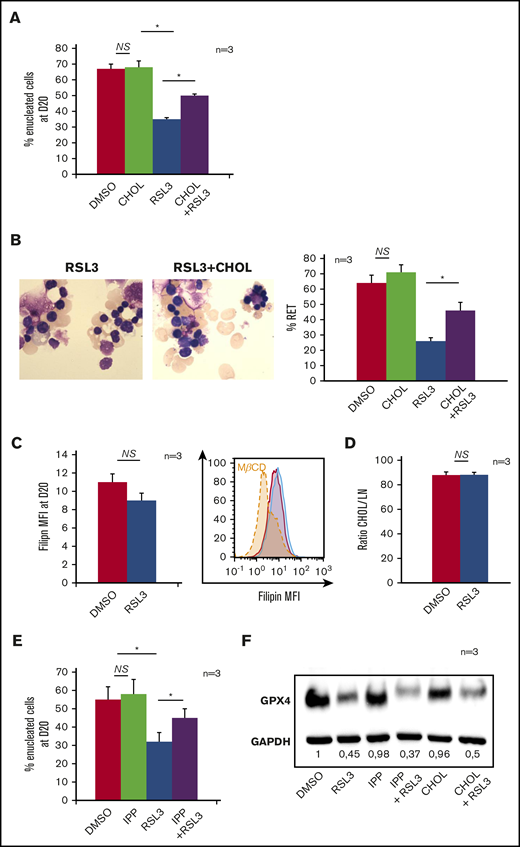
Mevalonate pathway molecules partially reverted the enucleation defect induced by GPX4 inhibition. (A) FCM histogram showing quantification of enucleated day 20 GPA+/Hoechst− cells treated with DMSO or 1 µM RSL3, with and without adding 10 μM cholesterol (CHOL) at day 14 in the culture medium (n = 3). Percentage of enucleated cells. RSL3 vs RSL3 + CHOL, 35% ± 1% vs 50% ± 1.6%, P < .01. (B, left) Day 20 MGG staining (magnification ×100) of DMSO, 10 µM CHOL, 1 µM RSL3 and RSL3 + CHOL conditions. (B, right) MGG staining count of orthochromatic erythroblasts and reticulocytes (%) (n = 3). Percentage of RET. RSL3 vs RSL3 + cholesterol: 26% ± 4% vs 46% ± 6%, P < .05. (C) Day 20 cholesterol quantification in RSL3- and DMSO-treated cells using filipin staining on FCM after cell fixation. Left, Filipin MFI DMSO vs 1 µM RSL3: 11 ± 0.9 vs 9 ± 0.2, n = 3, P = NS. Right, A representative histogram of 3 independent experiments; methyl-β-cyclodextrin (MBCD) was used as a control of cholesterol depletion. (D) Left, Histogram showing quantification of cholesterol/total neutral lipids ratio measured by the gas chromatography with flame-ionization detection method as described in “Methods” and normalized by protein quantity in day 20 erythroblasts (n = 3). Percentage of CHOL/NL DMSO vs 1 µM RSL3: 87.9% ± 2.6% vs 88.2% ± 2%, n = 3, P = NS. (E) Histogram showing FCM quantification of enucleated day 20 GPA+/Hoechst− cells treated with DMSO or 1 µM RSL3, with and without 5 µM IPP, at day 20 (n = 3). IPP partially restored the enucleation defect induced by RSL3. Percentage of enucleated cells RSL3 vs RSL3 + IPP: 31% ± 5% vs 45% ± 5%, P < .05. (F) Representative immunoblot from 3 independent experiments showing that the 1 µM RSL3-related GPX4 knockdown at the protein level was not reverted by 10 µM cholesterol or 5 µM IPP. Numbers represent mean GPX4/GAPDH ratio of each condition relative to DMSO (n = 3). *P < .05. Error bars are SEM.
Mevalonate pathway molecules partially reverted the enucleation defect induced by GPX4 inhibition. (A) FCM histogram showing quantification of enucleated day 20 GPA+/Hoechst− cells treated with DMSO or 1 µM RSL3, with and without adding 10 μM cholesterol (CHOL) at day 14 in the culture medium (n = 3). Percentage of enucleated cells. RSL3 vs RSL3 + CHOL, 35% ± 1% vs 50% ± 1.6%, P < .01. (B, left) Day 20 MGG staining (magnification ×100) of DMSO, 10 µM CHOL, 1 µM RSL3 and RSL3 + CHOL conditions. (B, right) MGG staining count of orthochromatic erythroblasts and reticulocytes (%) (n = 3). Percentage of RET. RSL3 vs RSL3 + cholesterol: 26% ± 4% vs 46% ± 6%, P < .05. (C) Day 20 cholesterol quantification in RSL3- and DMSO-treated cells using filipin staining on FCM after cell fixation. Left, Filipin MFI DMSO vs 1 µM RSL3: 11 ± 0.9 vs 9 ± 0.2, n = 3, P = NS. Right, A representative histogram of 3 independent experiments; methyl-β-cyclodextrin (MBCD) was used as a control of cholesterol depletion. (D) Left, Histogram showing quantification of cholesterol/total neutral lipids ratio measured by the gas chromatography with flame-ionization detection method as described in “Methods” and normalized by protein quantity in day 20 erythroblasts (n = 3). Percentage of CHOL/NL DMSO vs 1 µM RSL3: 87.9% ± 2.6% vs 88.2% ± 2%, n = 3, P = NS. (E) Histogram showing FCM quantification of enucleated day 20 GPA+/Hoechst− cells treated with DMSO or 1 µM RSL3, with and without 5 µM IPP, at day 20 (n = 3). IPP partially restored the enucleation defect induced by RSL3. Percentage of enucleated cells RSL3 vs RSL3 + IPP: 31% ± 5% vs 45% ± 5%, P < .05. (F) Representative immunoblot from 3 independent experiments showing that the 1 µM RSL3-related GPX4 knockdown at the protein level was not reverted by 10 µM cholesterol or 5 µM IPP. Numbers represent mean GPX4/GAPDH ratio of each condition relative to DMSO (n = 3). *P < .05. Error bars are SEM.
Enucleation impairment related to GPX4 knockdown occurs at the pyrenocyte-reticulocyte cytokinesis step
Enucleation is a well-coordinated multistep process that requires (i) chromatin condensation, (ii) nucleus migration at the cell surface on the pole of OrthoE, and (iii) contractile actin ring (CAR) formation leading to cytokinesis. OrthoE nucleus compactness assessed by IFC was not impacted by RSL3 exposure (Figure 5A). A close examination of cytospin after MGG staining revealed that RSL3-treated cells presented a dense polar nucleus, revealing that nucleus migration was not impacted (Figure 5B). We confirmed these data using IFC42,43 by quantifying the “delta centroid” corresponding to the distance between the center of the nucleus mask (DAPI) and the center of the cytoplasmic mask (GPA).44 RSL3-treated cells exhibited a δ centroid slightly higher than control erythroblasts (Figure 5C). By defining 4 gates corresponding to successive steps in nucleus extrusion (for mask strategy, cf supplemental Figure 6A-B), we observed that RSL3 impacted enucleation during the separation between reticulocyte and pyrenocyte (Figure 5D). Because membrane lipid reorganization is known to play a central role in enucleation through clustering of lipid rafts at the cleavage furrow between pyrenocyte and reticulocyte,45 we assessed GPX4 localization at day 15 before enucleation using immunofluorescence and observed a perinuclear staining with polar reinforcement, evocating its potential role at this cleavage furrow (Figure 6A). Moreover, we observed that GPX4 colocalized with 2 lipid raft markers: Flotillin-2, a major protein component46 (Figure 6B), and CTB, a specific marker of GM1 ganglioside47 (Figure 6C), arguing for a GPX4 role in lipid raft clustering at the cleavage furrow.
![Enucleation impairment induced by GPX4 inhibition occurred after nucleus condensation and polarization. (A) IFC analysis of nuclear compactness in GPA+/Hoechst+ day 18 to day 20 erythroblasts (performed when enucleation reached 50% in controls) treated with DMSO or 1 µM RSL3 (n = 5). To measure nuclear compactness, we used the dedicated compactness feature from IDEAS software applied on the nuclear image channel 01 as identified by Hoechst staining. Briefly, it reveals the degree of how well the object is packed together: the higher the value, the more condensed is the object. (B) MGG staining (magnification ×40) showing nucleus polarization in DMSO and 1 µM RSL3 conditions in day 18 to day 20 erythroblasts (arrow shows polar nucleus in orthochromatic erythroblasts; pyrenocytes [P], reticulocytes [R]). (C) Left, IFC measuring δ centroid in GPA+/Hoechst+ erythroblasts treated with DMSO or 1 µM RSL3 (n = 5). δ centroid indicates the eccentricity level of the nucleus inside cell. Right, A representative IFC image showing OrthoE-polarized nucleus in DMSO and 1 µM RSL3 conditions. δ centroid DMSO vs RSL3: 0.93 ± 0.03 vs 0.99 ± 0.05, P < .05. (D) IFC analysis and quantification (%) of GPA+/Hoechst+ erythroblasts at different enucleation steps in DMSO or 1 µM RSL3 conditions (n = 5). Gates of the successive steps in nucleus polarization and extrusion are defined and detailed in supplemental Figure 6. Briefly, Init corresponded to initiation of polarization with loss of the central position. Enuc 1 to 3 corresponds to progressive increase of the nucleus δ centroid associated with a decrease in the Brightfield aspect ratio, Enuc 3 corresponding to extruding nuclei. Percentage of Enuc2. DMSO vs RSL3: 7.9% ± 3% vs 4.8% ± 3.6%, P < .05; % Enuc 3: 1.2% ± 0.6% vs 0.4 ± 0.2 ±: P < .05. *P < .05. Error bars are SEM.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/22/10.1182_bloodadvances.2020003100/3/m_advancesadv2020003100r1f5.png?Expires=1769174520&Signature=K-jbcMUeiaU7lw~hFNT9is-LfejOx7l-rJrIQAvAP5fQkSn2sixw9rhVpL~RkWHSpgCx4krZGJ82AvouR32~PrzlIpkO4G2ReNVOsSlQxJjPoeggl0OTPVSnnrMrnu8QhzmReXmM6s21McL0C9MmZn~ObJcW5CzCRos7wTGxKqJRGBXP8o42DVkpxUKqURcd-qTtYcY~sMmXkurQ8QGEMVkH4sqLoE1GSSCIAlwVNtVAqIl3n4fSNq7MH2z6PARdvR7YO8moanPuWlUkhk0qhEi7-3yNRjBSeVSsA60kvcv4Zs0KhruTlv6MGY8Rs7~j9e-u6DIRhrk~JL0kuhGR4A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Enucleation impairment induced by GPX4 inhibition occurred after nucleus condensation and polarization. (A) IFC analysis of nuclear compactness in GPA+/Hoechst+ day 18 to day 20 erythroblasts (performed when enucleation reached 50% in controls) treated with DMSO or 1 µM RSL3 (n = 5). To measure nuclear compactness, we used the dedicated compactness feature from IDEAS software applied on the nuclear image channel 01 as identified by Hoechst staining. Briefly, it reveals the degree of how well the object is packed together: the higher the value, the more condensed is the object. (B) MGG staining (magnification ×40) showing nucleus polarization in DMSO and 1 µM RSL3 conditions in day 18 to day 20 erythroblasts (arrow shows polar nucleus in orthochromatic erythroblasts; pyrenocytes [P], reticulocytes [R]). (C) Left, IFC measuring δ centroid in GPA+/Hoechst+ erythroblasts treated with DMSO or 1 µM RSL3 (n = 5). δ centroid indicates the eccentricity level of the nucleus inside cell. Right, A representative IFC image showing OrthoE-polarized nucleus in DMSO and 1 µM RSL3 conditions. δ centroid DMSO vs RSL3: 0.93 ± 0.03 vs 0.99 ± 0.05, P < .05. (D) IFC analysis and quantification (%) of GPA+/Hoechst+ erythroblasts at different enucleation steps in DMSO or 1 µM RSL3 conditions (n = 5). Gates of the successive steps in nucleus polarization and extrusion are defined and detailed in supplemental Figure 6. Briefly, Init corresponded to initiation of polarization with loss of the central position. Enuc 1 to 3 corresponds to progressive increase of the nucleus δ centroid associated with a decrease in the Brightfield aspect ratio, Enuc 3 corresponding to extruding nuclei. Percentage of Enuc2. DMSO vs RSL3: 7.9% ± 3% vs 4.8% ± 3.6%, P < .05; % Enuc 3: 1.2% ± 0.6% vs 0.4 ± 0.2 ±: P < .05. *P < .05. Error bars are SEM.
Enucleation impairment induced by GPX4 inhibition occurred after nucleus condensation and polarization. (A) IFC analysis of nuclear compactness in GPA+/Hoechst+ day 18 to day 20 erythroblasts (performed when enucleation reached 50% in controls) treated with DMSO or 1 µM RSL3 (n = 5). To measure nuclear compactness, we used the dedicated compactness feature from IDEAS software applied on the nuclear image channel 01 as identified by Hoechst staining. Briefly, it reveals the degree of how well the object is packed together: the higher the value, the more condensed is the object. (B) MGG staining (magnification ×40) showing nucleus polarization in DMSO and 1 µM RSL3 conditions in day 18 to day 20 erythroblasts (arrow shows polar nucleus in orthochromatic erythroblasts; pyrenocytes [P], reticulocytes [R]). (C) Left, IFC measuring δ centroid in GPA+/Hoechst+ erythroblasts treated with DMSO or 1 µM RSL3 (n = 5). δ centroid indicates the eccentricity level of the nucleus inside cell. Right, A representative IFC image showing OrthoE-polarized nucleus in DMSO and 1 µM RSL3 conditions. δ centroid DMSO vs RSL3: 0.93 ± 0.03 vs 0.99 ± 0.05, P < .05. (D) IFC analysis and quantification (%) of GPA+/Hoechst+ erythroblasts at different enucleation steps in DMSO or 1 µM RSL3 conditions (n = 5). Gates of the successive steps in nucleus polarization and extrusion are defined and detailed in supplemental Figure 6. Briefly, Init corresponded to initiation of polarization with loss of the central position. Enuc 1 to 3 corresponds to progressive increase of the nucleus δ centroid associated with a decrease in the Brightfield aspect ratio, Enuc 3 corresponding to extruding nuclei. Percentage of Enuc2. DMSO vs RSL3: 7.9% ± 3% vs 4.8% ± 3.6%, P < .05; % Enuc 3: 1.2% ± 0.6% vs 0.4 ± 0.2 ±: P < .05. *P < .05. Error bars are SEM.
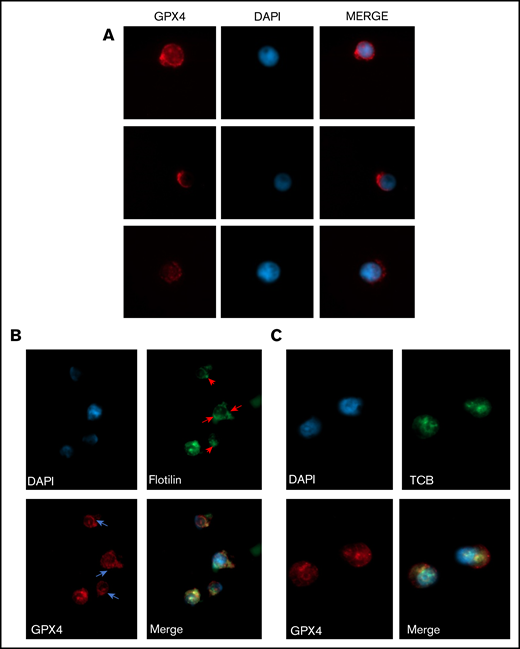
GPX4 had a perinuclear distribution during enucleation and localized with lipid raft markers flotillin and cholera toxin b subunit. (A) Representative image of AlexaFluor 647–conjugated GPX4 immunolabeling and DAPI staining during erythroblast enucleation (n = 3). These images were taken at day 15 of differentiation at 60× original magnification used with oil immersion. (B-C) Representative image of DAPI staining and AlexaFluor 647–conjugated GPX4 and AlexaFluor 488–conjugated Flotillin-2 immunolabeling (B) or AlexaFluor 647–conjugated GPX4 and AlexaFluor 488–conjugated CTB immunolabeling (C) on day 15 at the beginning of enucleation at 60× magnification used with oil immersion (n = 3).
GPX4 had a perinuclear distribution during enucleation and localized with lipid raft markers flotillin and cholera toxin b subunit. (A) Representative image of AlexaFluor 647–conjugated GPX4 immunolabeling and DAPI staining during erythroblast enucleation (n = 3). These images were taken at day 15 of differentiation at 60× original magnification used with oil immersion. (B-C) Representative image of DAPI staining and AlexaFluor 647–conjugated GPX4 and AlexaFluor 488–conjugated Flotillin-2 immunolabeling (B) or AlexaFluor 647–conjugated GPX4 and AlexaFluor 488–conjugated CTB immunolabeling (C) on day 15 at the beginning of enucleation at 60× magnification used with oil immersion (n = 3).
GPX4 knockdown–mediated enucleation defect is related to an impaired lipid raft clustering and CAR formation that can be restored by adding exogenous cholesterol
Clustering of lipid rafts containing GM1 gangliosides at the cleavage furrow is crucial for CAR formation between the nascent reticulocyte and pyrenocyte border.45 First, we studied CTB distribution using immunofluorescence microscopy and observed that, in control erythroblasts, CTB staining was strong and more intense around the nucleus, at the pole of nascent reticulocyte as previous reported.45 In contrast, a diffuse and weak CTB signal was observed in RSL3-treated cells (Figure 7A; supplemental Figure 7). This decreased CTB staining was confirmed using FCM (Figure 7B). These data supported that GPX4 inhibition was strongly deemed to perturb lipid raft integrity, resulting in impaired enucleation that may reflect a defect in CAR formation/function. Indeed, enucleation in mice requires CAR assembly and anchorage at the furrow, which depends on myosin-regulatory light-chain (MRLC) phosphorylation.45 We evaluated then whether GPX4 inhibition could affect MRLC phosphorylation. IFC revealed, as expected, phosphorylated MRLC (pMRLC) reinforcement at the cleavage furrow between the reticulocyte and the pyrenocyte (supplemental Figure 8A). The staining was, however, strongly decreased in the RSL3 condition, using western blot and FCM (Figure 7C-D), even if still predominant at the pyrenocyte/reticulocyte interface (supplemental Figure 8B). At last, because cholesterol is required for lipid raft accumulation and stabilization during cytokinesis,48 we hypothesized that the partial reversion of the RSL3-induced enucleation defect observed after adding cholesterol in the medium (Figure 4A) was due to its ability to stabilize lipid rafts despite GPX4 knockdown. Indeed, we observed that adding cholesterol from day 14 restored lipid raft formation as assessed by CTB staining at day 20 (Figure 7E) and CAR assembly as assessed by pMRLC quantification using immunoblot at day 18 (Figure 7F), in parallel with the restoration of enucleation in a context of GPX4 knockdown.
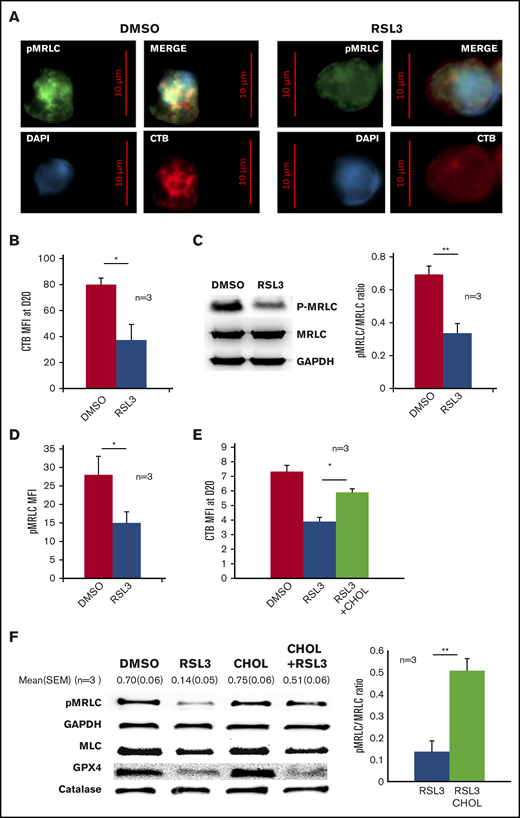
GPX4 inhibition led to disruption of lipid rafts at cleavage furrow and to decrease in myosin phosphorylation that was partially restored by cholesterol. (A) Representative image of DAPI, AlexaFluor 488–conjugated pMRLC, AlexaFluor 647–conjugated CTB staining using fluorescence microscopy in DMSO and 1 µM RSL3-treated cells at day 18 (n = 3). (B) Histogram showing the significant decrease in CTB MFI measured by FCM at day 20 in 1 µM RSL3-treated erythroblasts in comparison with DMSO (n = 3). Mean MFI: 80 ± 5 (DMSO) vs 37 ± 12 (RSL3), P < .05. (C, left) Representative pMRLC immunoblot of erythroblasts treated with DMSO or 1 µM RSL3 during the enucleation stage at day 18. (C, right) Quantification of the decrease in pMRLC induced by 1 µM RSL3 exposure, using the pMRLC/MRLC vs GAPDH ratio (n = 3): 0.69 ± 0.05 vs 0.34 ± 0.06; P < .001. (D) Histogram showing MFI of pMRLC, measured by FCM in fixed and permeabilized erythroblasts in DMSO and 1 µM RSL3 conditions, during the enucleation stage (n = 3). P < .05. (E) Addition of 10 µM cholesterol at day 14 corrected the CTB decrease induced by RSL3 exposure, as assessed by FCM (n = 3). Day 20 erythroblasts, CTB MFI RSL3 vs RSL3 + CHOL: 3.9 ± 0.3 vs 5.9 ± 0.2, P < .05. (F, left) Representative pMRLC and GPX4 immunoblot of erythroblasts treated with DMSO or 1 µM RSL3, with and without 10 µM cholesterol, during the enucleation stage (n = 3). Catalase, MRLC, and GAPDH were used as load controls. (B, right) Densitometric pMRLC/MRLC vs GAPDH ratio between RSL3 and RSL3 + CHOL from 3 independent experiments: 0.137 ± 0.05 vs 0.507 ± 0.06, P < .001. *P < .05; **P < .01. Error bars are SEM.
GPX4 inhibition led to disruption of lipid rafts at cleavage furrow and to decrease in myosin phosphorylation that was partially restored by cholesterol. (A) Representative image of DAPI, AlexaFluor 488–conjugated pMRLC, AlexaFluor 647–conjugated CTB staining using fluorescence microscopy in DMSO and 1 µM RSL3-treated cells at day 18 (n = 3). (B) Histogram showing the significant decrease in CTB MFI measured by FCM at day 20 in 1 µM RSL3-treated erythroblasts in comparison with DMSO (n = 3). Mean MFI: 80 ± 5 (DMSO) vs 37 ± 12 (RSL3), P < .05. (C, left) Representative pMRLC immunoblot of erythroblasts treated with DMSO or 1 µM RSL3 during the enucleation stage at day 18. (C, right) Quantification of the decrease in pMRLC induced by 1 µM RSL3 exposure, using the pMRLC/MRLC vs GAPDH ratio (n = 3): 0.69 ± 0.05 vs 0.34 ± 0.06; P < .001. (D) Histogram showing MFI of pMRLC, measured by FCM in fixed and permeabilized erythroblasts in DMSO and 1 µM RSL3 conditions, during the enucleation stage (n = 3). P < .05. (E) Addition of 10 µM cholesterol at day 14 corrected the CTB decrease induced by RSL3 exposure, as assessed by FCM (n = 3). Day 20 erythroblasts, CTB MFI RSL3 vs RSL3 + CHOL: 3.9 ± 0.3 vs 5.9 ± 0.2, P < .05. (F, left) Representative pMRLC and GPX4 immunoblot of erythroblasts treated with DMSO or 1 µM RSL3, with and without 10 µM cholesterol, during the enucleation stage (n = 3). Catalase, MRLC, and GAPDH were used as load controls. (B, right) Densitometric pMRLC/MRLC vs GAPDH ratio between RSL3 and RSL3 + CHOL from 3 independent experiments: 0.137 ± 0.05 vs 0.507 ± 0.06, P < .001. *P < .05; **P < .01. Error bars are SEM.
Discussion
GPX4 was highly expressed in our culture system of human erythroid differentiation, reaching a peak at day 6 that corresponds to erythroblast exponential amplification associated with higher transferrin receptor expression and the beginning of hemoglobin synthesis.49-51 Because erythroblasts require a high bioavailable iron concentration to synthesize hemoglobin, a seducing hypothesis would be that GPX4 would protect these cells with an elevated cellular iron pool from ferroptosis. However, no significant impact of GPX4 inhibition in terms of cell death was observed during erythroblast differentiation. Moreover, the lipid peroxide level was weakly increased in RSL3-treated primary erythroblasts. This is in contrast with mouse models, because Gpx4−/− mouse erythroblasts presented increased lipid peroxidation, ROS level, and perturbed reticulocyte maturation.18,19 Such a discrepancy between human and mouse erythropoiesis has been described, notably when using knockout mice to model human diseases such as Diamond-Blackfan anemia52 or congenital dyserythropoietic anemia type II.53 The relative resistance of human erythroblasts to ferroptosis and their ability to overcome the negative effects of GPX4 inhibition, if surprising, has been described in other cell models.28,54 Our findings showed that erythroblasts, for protecting themselves against ferroptosis, may rely on other enzymes with GPX activity that compensate each other deficiency. Our data identified PRDX6, expressed in erythroblasts33 and recently identified as a regulator of ferroptosis,55 as a potential candidate. Alternatively, other mechanisms of resistance could be also investigated, including ferroptosis suppressor protein1 (FSP1) as a lipid peroxyl radical trap56 or Prominin2-dependent multivesicular body formation and iron-containing exosomes release.57
Enucleation is probably an evolutionary process that leaves more space for hemoglobin inside red cells and increases their deformability, which is crucial for red cell survival in the red pulp of the spleen.58 It is a complex process that requires reorganization of the red cell lipid membrane and cytoplasmic cytoskeleton, leading to nucleus expulsion as well as asymmetric sorting of membrane proteins and lipids.30,59,60 Enucleation can be assimilated to an asymmetric cytokinesis61-63 with well-defined hallmarks: (i) a decrease in nucleus size and chromatin condensation related to posttranscriptional modifiers of gene expression,63-66 (ii) nuclear polarization,44,67 (iii) membrane lipid raft clustering,46 (iv) formation of a contractile ring through a Rac-GTPase pathway,68,69 and calcium-dependent activation of calmodulin inducing myosin II phosphorylation,70 and (v) accumulation at the furrow of clathrin-dependent vesicles that provide membrane material.71,72 Our main finding was that GPX4 knockdown altered human erythroblast enucleation in vitro during steps (iii) and (iv), as shown by the arrest of nucleus extrusion after its polarization at the cell surface. Interestingly, this phenotype was ferroptosis- and necroptosis-independent although these cell deaths were associated with GPX4 deficiency in mice erythroid cells.18 Instead, an enucleation defect observed after GPX4 knockdown was associated with an alteration of lipid raft clustering. Lipid rafts are dynamic microdomains involved in membrane trafficking and signaling.73,74 Many reports highlighted their crucial role during cytokinesis,48 a process to which enucleation can be compared.61 During cytokinesis, lipid rafts cluster at the cleavage furrow between the 2 daughter cells, allowing recruitment of small GTPases and progression of cytokinesis through a CAR formation. Konstantidinis et al confirmed using IFC that mouse erythroblast enucleation had the same requirement in terms of lipid raft confluence and CAR assembly.45 Our results show that in human OrthoE, GPX4, the only GPX family member located at the cell membrane localizes at the cleavage furrow with lipid raft markers and seems required for the reorganization of membrane lipid domains. Interestingly, an enucleation defect could be partially restored by adding an excess of cholesterol. Exogenous cholesterol is known to integrate the outer leaflet of the plasma membrane and concentrates during cytokinesis at the cleavage furrow, allowing sphingomyelin accumulation75 and probably restoring lipid raft signaling.76 Indeed, increasing the membrane cholesterol content could partially overcome GPX4 deficiency in RSL3-treated cells by increasing lipid raft markers and myosin phosphorylation. However, the underlying mechanisms linking GPX4 to lipid rafts remain unclear. We first thought that GPX4 knockdown would induce a decrease in membrane cholesterol content by driving IPP toward selenocysteyl-tRNA synthesis and GPX4 translation at the expense of the cholesterol pathway. However, total cholesterol content was equivalent between DMSO- and RSL3-treated cells. GPX4 knockdown may induce a qualitative defect with abnormal cholesterol distribution and destabilization of lipid rafts. High-resolution techniques that allow precise analysis of lipid distribution during the enucleation process would be of great interest in that matter.
Finding a new protein involved in erythropoiesis raises the question of its potential implication in pathophysiology of hematological diseases and whether it could be used as a target for therapeutics. Ineffective erythropoiesis leading to anemia is observed in benign and malignant hemopathies such as thalassemia and myelodysplastic syndromes. In these diseases, erythroid defects leading to inaccurate reticulocyte production are heterogeneous and occur at multiple steps, including apoptosis of erythroid precursors, defective HSP70/GATA1 interaction, translation defect, aberrant splicing, oxidative stress, and anomalies in terminal differentiation involving mitophagy and nuclear condensation.77-80 Enucleation is directly impaired during congenital dyserythropoietic anemia type (CDA) IV (OMIM #613673), a rare type of ineffective erythropoiesis leading to numerous circulating nucleated red cells. This autologous dominant disease is related to E325K mutation in KLF1, leading to expression of a variant with a strong dominant-negative effect on KLF1 regulation network.81,82 Ex vivo culture of CD34+ cells from a CDA IV patient revealed no defect in proliferation nor erythroid maturation until the orthoE stage but an alteration in enucleation reproducing the same phenotype observed after RSL3 exposure or GPX4 knockdown.83 Defect in enucleation could then be related to decreased GPX4 expression because (i) the GPX4 enhancer/promoter region carries an KLF1-binding consensus site,84 (ii) GPX4 downregulation is observed in fetal liver cells of Klf1−/− mice,85 and in polyE and orthoE of the E339D KLF1-mutant Nan mice.86 Moreover, when studied in vitro, Eklf−/− erythroid cells undergo normal nuclear condensation and polarization but are blocked at the last step of enucleation.87,88 Considering these data, studying GPX4 expression, lipid raft clustering, and CAR formation in erythroid cells from CDA IV patients to identify their potential involvement in the phenotype in addition to the already described cell cycle defect would be of great interest.87
Deciphering human enucleation mechanisms is also a cornerstone for improving ex vivo red cell generation for transfusion purposes: protocols for large-scale production of enucleated erythrocytes from CD34+ or peripheral mononuclear cells have been well described, with an up to 90% enucleation rate, but are still donor-dependent.89,90 Attempts to use an inexhaustible source of stem cells such as human embryonic stem cells/human pluripotent stem cells or immortalized cell lines are challenged by the insufficient enucleation rate obtained after driving these cells into erythroid differentiation.91 Pathophysiological mechanisms leading to this defective enucleation remain not totally understood, despite recent data showing persistent vimentin expression having a negative effect on enucleation.92 It has been shown that adding exogenous cholesterol improved the osmotic resistance of ex vivo–generated red blood cells from CD34+ cells. In this culture system with high enucleation efficiency, cholesterol supplementation could even increase the reticulocyte count and fasten erythroid maturation.93 Therefore, previous data describing the importance of lipid rafts for CAR formation during enucleation, in addition to our results on involvement of GPX4 and cholesterol in this process, may represent a new way to study, understand, and improve enucleation of erythroblasts obtained from these donor-independent stem cell sources.
In summary, our data identify GPX4 as a new actor in the vast family of proteins involved in human erythroblast enucleation. We could in this report specify the precise step during which it is involved, that is, after nuclear condensation and polarization in the OrthoE stage. We also demonstrated its involvement in lipid raft clustering and myosin phosphorylation, which are known to be essential for CAR assembly and cytokinesis between pyrenocytes and reticulocytes at the last step of erythroblast maturation.
Data-sharing requests may be e-mailed to the corresponding author, Loïc Garçon, at garcon.loic@chu-amiens.fr.
Acknowledgments
The authors are grateful to University of Picardie-Jules Verne and to Centre Hospitalier Universitaire Amiens Picardie for technical support. The authors thank Justine Bertrand-Michel and Aurélie Batut (MetaToul-Lipidomique Core Facility [I2MC, INSERM 1048, Toulouse, France], MetaboHUB-ANR-11-INBS-0010) for lipidomic analysis, advice, and technical support. The authors are grateful to Marie-Noelle Lacassagne, Christele Ossart, and Aline Regnier (Cellular Therapy Laboratory of Centre Hospitalier Universitaire Amiens) who provided human blood samples. The authors thank Lydie Da Costa for scientific advice.
This work was supported by the Réseau Hématologie Picardie (RHEPI) Association and by the Club du Globule Rouge et du Fer (CGRF).
Authorship
Contribution: H.O.-H., K.M., Y.D., R.L.-D.-S., C.C., A.C., P.V., A.W., N.J., D.L., and J.D. performed the experiments; H.O.-H., K.M., J.R., and L.G. designed the study and wrote the paper; and T.B. and J.P.M. provided intellectual input.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Loïc Garçon, EA4666 HEMATIM, CURS, Université Picardie Jules Verne, Centre Hospitalier Universitaire Amiens, 80054 Amiens Cedex 1 France; e-mail: garcon.loic@chu-amiens.fr.

