

Key Points
There is a dose–response relationship between Lman1 expression level and secretion levels of LMAN1 cargoes in mice (FV, FVIII, and A1AT).
Therapeutic targeting of LMAN1 to reduce FV and FVIII as an anticoagulant strategy may only require partial inhibition of LMAN1 function.

Abstract
Combined deficiency of coagulation factors V and VIII (F5F8D) is an autosomal recessive bleeding disorder caused by loss-of-function mutations in either LMAN1 or MCFD2. The latter genes encode 2 components of a mammalian cargo receptor that facilitates secretion of coagulation factor V (FV) and factor VIII (FVIII) from the endoplasmic reticulum (ER) to the Golgi via coat protein complex II vesicles. F5F8D patients exhibit FV and FVIII levels that are ∼10% to 15% of normal. We report herein a comparative analysis for a series of murine Lman1 alleles. Consistent with previous reports, mice completely deficient in LMAN1 (Lman1−/−) exhibit ∼50% FV and FVIII levels. In contrast, mice carrying a hypomorphic Lman1 allele (Lman1cgt/cgt) that expresses ∼6% to 8% of wild-type Lman1 mRNA levels exhibit intermediate plasma FV and FVIII reductions (∼70% of wild-type levels). Lman1−/− mice exhibit ER accumulation of another LMAN1 cargo, alpha-1 antitrypsin (A1AT), with an intermediate level of A1AT ER retention observed in Lman1cgt/cgt mice. Finally, the previously reported strain-specific, partially penetrant, perinatal lethality of LMAN1-deficient mice (Lman1gt1/gt1) was confirmed in Lman1−/− mice, although it was not observed in Lman1cgt/cgt mice. Taken together, these results show a dose-dependent effect of residual LMAN1 on the secretion of its cargo proteins. The results also suggest that human subjects with hypomorphic LMAN1 mutations might present with mild bleeding phenotypes resulting from more modest reductions in FV and FVIII, which could be missed by routine clinical evaluation. Finally, these findings suggest that therapeutic targeting of LMAN1 to reduce FV and FVIII as an anticoagulant strategy may only require partial inhibition of LMAN1 function.
Introduction
The LMAN1/MCFD2 cargo receptor is required for efficient secretion of coagulation factors V (FV) and VIII (FVIII).1 FV is synthesized in hepatocytes2,3 (humans and mice) and megakaryocytes4 (mice), whereas FVIII is synthesized exclusively by endothelial cells.5,6 Human mutations in either LMAN1 or MCFD2 cause combined deficiency of coagulation FV and FVIII (F5F8D), an autosomal recessive bleeding disorder characterized by reduction of both FV and FVIII to ∼10% to 15% of normal levels.7,8
LMAN1 is a type 1 transmembrane protein that cycles between the endoplasmic reticulum (ER) and the ER Golgi intermediate compartment (ERGIC) in the mammalian secretory pathway; it belongs to a family of homologous l-type mannose-binding lectins that function in the trafficking of N-linked glycoproteins.9-11 LMAN1 contains an ER luminal carbohydrate recognition domain that binds cargo proteins,12,13 as well as a short cytosolic tail that interacts with the SEC24 component of the coat protein complex II vesicle coat (for anterograde transport to the ERGIC).14,15 LMAN1 forms a 1:1 complex with MCFD2, and together these proteins function as a cargo receptor complex. Approximately 70% of human F5F8D patients exhibit mutations in LMAN1, with the remainder in MCFD2.16 Nearly all reported human LMAN1 mutations are null mutations predicted to result in complete absence of the LMAN1 protein, with 2 interesting exceptions that are missense mutations (W67S and C475R) resulting in disruption of protein oligomerization and protein structural integrity.7,13,17-21 The marked preponderance of null LMAN1 mutations in F5F8D patients suggests that even small residual levels of LMAN1 expression may be sufficient to maintain physiological levels of FV and FVIII secretion.
LMAN1 and MCFD2 are ubiquitously expressed, and their orthologs are present in invertebrates (including Caenorhabditis elegans) before the appearance of FV and FVIII, suggesting a broader role for LMAN1 in protein secretion. Other potential LMAN1 secretory cargoes include alpha-1 antitrypsin (A1AT),22 cathepsin C,23 cathepsin Z,24 Mac-2 binding protein,25 matrix metalloproteinase-9,26 and γ-aminobutyric acid type A receptors.27 Among these potential cargo proteins, only A1AT has been confirmed in vivo (in the mouse),28 although γ-aminobutyric acid type A receptors are decreased in LMAN1-deficient mouse brain.27 In addition, LMAN1 has been implicated in the production of infectious particles from several highly pathogenic RNA virus families29 and may also contribute to photoreceptor homeostasis.30 Furthermore, mutations resulting in LMAN1 inactivation have been identified at high frequencies in colorectal tumors with microsatellite instability.31
Mice homozygous for a gene trap insertion in Lman1 intron 10 (Lman1gt1/gt1) exhibit FV and FVIII activity levels that are reduced to ∼50% of wild-type littermate levels.28 These mice also exhibit retention of A1AT in the hepatocyte ER, with normal steady-state plasma A1AT levels in female mice and only a slight reduction in plasma A1AT levels in male mice.28,32 The less severe reductions of FV and FVIII in Lman1gt1/gt1 mice relative to those of F5F8D humans could be explained by low levels of residual LMAN1 expression from the gene trap allele, or differences in FV and FVIII secretion between mice and humans. Lman1gt1/gt1 mice also exhibit a partially penetrant, perinatal lethality that is restricted to certain genetic strain backgrounds.28 In contrast, Mcfd2−/− mice exhibit slightly lower FV and FVIII levels than Lman1gt1/gt1 mice but no perinatal lethality.32
We report here an analysis of mice homozygous for a second, independent Lman1 null allele with complete LMAN1 deficiency (Lman1−/−), as well as hypomorphic LMAN1-deficient mice (Lman1cgt/cgt) that retain ∼7% of normal Lman1 expression. We describe a dose–response relationship between the level of Lman1 gene expression and the secretion of LMAN1 cargoes, suggesting that LMAN1 may be a potential therapeutic target for anticoagulation. We also confirm a strain-specific, perinatal lethality in LMAN1-deficient mice, indicating that additional LMAN1-dependent secretory cargoes remain to be identified.
Methods
Lman1 and Mcfd2 mutant mice
Lman1gt1 mice carrying a gene trap insertion in Lman1 intron 10 (Figure 1A) were described previously28 and are maintained on a C57BL/6J genetic background. Polymerase chain reaction (PCR) genotyping for the Lman1gt1 allele was performed as previously described.
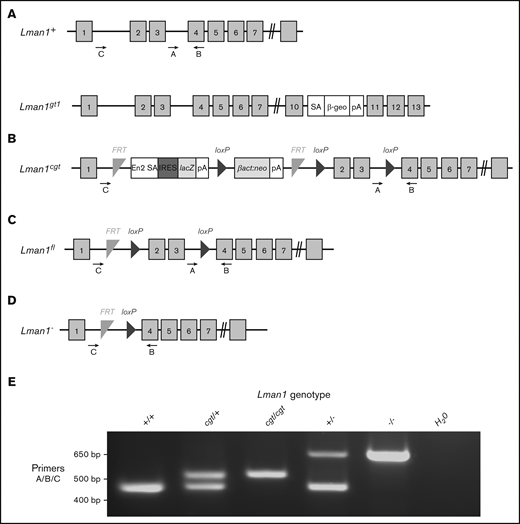
Schematic of Lman1 mutant alleles. (A) Lman1gt1 allele carries a gene trap insertion in intron 10.28 (B) The Lman1 conditional gene trap allele (Lman1cgt) contains a gene trap insertion in intron 1 flanked by 2 FRT sites. Mice carrying this allele were crossed to β-actin FLP transgenic mice.5 Mice heterozygous for the resulting Lman1 “floxed” allele (Lman1fl/+) (C) were crossed to EIIA-Cre+ transgenic mice to excise exons 2 and 3, generating the Lman1 null allele (Lman1–) (D). Gray blocks represent exons. A, B, and C represent genotyping primers. (E) A 3-primer PCR assay (primers A, B, and C) distinguishes the Lman1+ (444 bp), Lman1cgt (508 bp), and Lman1– (635 bp) alleles. The Lman1fl allele (not shown in this image) also generates the same PCR product band as the Lman1cgt allele. βact:neo, human β-actin promoter-driven neomycin cassette; β-Geo, β-galactosidase-neo fusion; cgt, conditional gene trap; En2 SA, splice acceptor of mouse En2 exon 2; fl, floxed; IRES, encephalomyocarditis virus (EMCV) internal ribosomal entry site; lacZ, Escherichia coli β-galactosidase gene; pA, poly-adenylation signal; SA, splice acceptor cassette.
Schematic of Lman1 mutant alleles. (A) Lman1gt1 allele carries a gene trap insertion in intron 10.28 (B) The Lman1 conditional gene trap allele (Lman1cgt) contains a gene trap insertion in intron 1 flanked by 2 FRT sites. Mice carrying this allele were crossed to β-actin FLP transgenic mice.5 Mice heterozygous for the resulting Lman1 “floxed” allele (Lman1fl/+) (C) were crossed to EIIA-Cre+ transgenic mice to excise exons 2 and 3, generating the Lman1 null allele (Lman1–) (D). Gray blocks represent exons. A, B, and C represent genotyping primers. (E) A 3-primer PCR assay (primers A, B, and C) distinguishes the Lman1+ (444 bp), Lman1cgt (508 bp), and Lman1– (635 bp) alleles. The Lman1fl allele (not shown in this image) also generates the same PCR product band as the Lman1cgt allele. βact:neo, human β-actin promoter-driven neomycin cassette; β-Geo, β-galactosidase-neo fusion; cgt, conditional gene trap; En2 SA, splice acceptor of mouse En2 exon 2; fl, floxed; IRES, encephalomyocarditis virus (EMCV) internal ribosomal entry site; lacZ, Escherichia coli β-galactosidase gene; pA, poly-adenylation signal; SA, splice acceptor cassette.
Lman1cgt (Figure 1B) and Lman1fl (Figure 1C) mice were previously described5 and are maintained on a C57BL/6J genetic background. The Lman1fl allele was converted to the null Lman1– allele [Lman1tm1d(KOMP)Wtsi] (Figure 1D) by Cre-mediated excision of exons 2 and 3. Transgenic C57BL/6J mice carrying Cre recombinase driven by a ubiquitous EIIA promoter [B6.FVB-Tg(EIIa-cre)C5379Lmgd/J; stock no. 003724] were obtained from The Jackson Laboratory, as were 129S1/SvImJ mice (stock no. 002448). Primer trio A/B/C was used to distinguish between the Lman1+, Lman1cgt, and Lman1– alleles (Figure 1E) as previously described5 with the primers listed in Table 1. Mcfd2– mice were previously generated by deleting exons 2 and 3 of the Mcfd2 gene,32 with genotyping performed as previously described. The Mcfd2– allele was maintained on a C57BL/6J genetic background. To assess Lman1−/− lethality on a mixed genetic background, Lman1+/− F1 mice were generated [by crossing an Lman1+/− mouse on C57BL/6J strain to a wild-type (Lman1+/+)129S1/SvImJ mouse] and subsequently intercrossed to produce F2 offspring on a mixed genetic background (designated B6;129).
Primer sequences
| Primer | 5′ → 3′ Sequence |
|---|---|
| Genotyping primers | |
| Lman1 primer A | GGCTTTCTTGACACCTTCAATTTAA |
| Lman1 primer B | CCAAGTGAAGGGAAGACCATCAAGC |
| Lman1 primer C | GACCCCTAGTGACGGGTTCTTGTC |
| EIIA-Cre transgene Fwd | CCGCTGGAGATGACGTAGTT |
| EIIA-Cre transgene Rev | CGCATAACCAGTGAAACAGCATTGC |
| qRT-PCR primers | |
| Gapdh Fwd | TGTGTCCGTCGTGGATCTGA |
| Gapdh Rev | ACCACCTTCTTGATGTCATCATACTT |
| β-actin Fwd | CTAAGGCCAACCGTGAAAAG |
| β-actin Rev | GGGGTGTTGAAGGTCTCAAA |
| F5 | ACGTGGTGAAGAGAGATATCGAC |
| R5 | ACAAAGTGGATCGTGGATAGACA |
| F6 | GCCCAGGCGGGGAATGCTATTCCAAG |
| R6 | TCTCGAAGGCTGCTTTTGCTTTGGT |
| Aft6 Fwd | CTTCCTCCAGTTGCTCCATC |
| Aft6 Rev | CAACTCCTCAGGAACGTGCT |
| BiP Fwd | CATGGTTCTCACTAAAATGAAAGG |
| BiP Rev | GCTGGTACAGTAACAACTG |
| Chop Fwd | CTGCCTTTCACCTTGGAGAC |
| Chop Rev | CGTTTCCTGGGGATGAGATA |
| XBP total Fwd | AAGAACACGCTTGGGAATGG |
| XBP total Rev | ACTCCCCTTGGCCTCCAC |
| XBP spliced Fwd | GAGTCCGCAGCAGGTG |
| XBP spliced Rev | GTGTCAGAGTCCATGGGA |
| Primer | 5′ → 3′ Sequence |
|---|---|
| Genotyping primers | |
| Lman1 primer A | GGCTTTCTTGACACCTTCAATTTAA |
| Lman1 primer B | CCAAGTGAAGGGAAGACCATCAAGC |
| Lman1 primer C | GACCCCTAGTGACGGGTTCTTGTC |
| EIIA-Cre transgene Fwd | CCGCTGGAGATGACGTAGTT |
| EIIA-Cre transgene Rev | CGCATAACCAGTGAAACAGCATTGC |
| qRT-PCR primers | |
| Gapdh Fwd | TGTGTCCGTCGTGGATCTGA |
| Gapdh Rev | ACCACCTTCTTGATGTCATCATACTT |
| β-actin Fwd | CTAAGGCCAACCGTGAAAAG |
| β-actin Rev | GGGGTGTTGAAGGTCTCAAA |
| F5 | ACGTGGTGAAGAGAGATATCGAC |
| R5 | ACAAAGTGGATCGTGGATAGACA |
| F6 | GCCCAGGCGGGGAATGCTATTCCAAG |
| R6 | TCTCGAAGGCTGCTTTTGCTTTGGT |
| Aft6 Fwd | CTTCCTCCAGTTGCTCCATC |
| Aft6 Rev | CAACTCCTCAGGAACGTGCT |
| BiP Fwd | CATGGTTCTCACTAAAATGAAAGG |
| BiP Rev | GCTGGTACAGTAACAACTG |
| Chop Fwd | CTGCCTTTCACCTTGGAGAC |
| Chop Rev | CGTTTCCTGGGGATGAGATA |
| XBP total Fwd | AAGAACACGCTTGGGAATGG |
| XBP total Rev | ACTCCCCTTGGCCTCCAC |
| XBP spliced Fwd | GAGTCCGCAGCAGGTG |
| XBP spliced Rev | GTGTCAGAGTCCATGGGA |
Timed matings were performed by intercrossing Lman1+/− mice. Embryos from each cross were harvested at 18.5 days postcoitus. A tail biopsy was obtained from each embryo for genotyping, and embryos were fixed in Z-Fix solution (Anatech LTD) for histology. All animals were housed according to the University of Michigan Unit of Laboratory Animal Medicine guidelines, and the University Committee on the Use and Care of Animals approved all studies.
Messenger RNA isolation and gene expression analysis
Total messenger RNA (mRNA) was isolated from tissues as previously described.33 Reverse transcription and quantitative real-time PCR amplification were performed as previously described,34 with primers listed in Table 1. Relative gene expression was calculated by using the 2−ΔΔCT method. β-actin and glyceraldehyde-3-phosphate dehydrogenase were used as internal controls. Two samples of each genotype were analyzed in triplicate for Lman1 expression levels. The Lman1 primers used for these assays were chosen based on the differences in the Lman1cgt and Lman1gt1 alleles. The Lman1cgt allele has a gene trap in Lman1 intron 1, whereas the Lman1gt1 allele has a gene trap in intron 10. The 5′ end of the primer F6 matches exon 1 and the 3′ end matches exon 2, and thus it should only anneal to mRNA, correctly splicing these 2 exons (around the Lman1cgt gene trap). Similarly, the downstream F5-R5 primer pair anneals to any mRNA transcript in which splicing around the Lman1gt1 gene trap occurs. Primer sequences for unfolded protein response genes were previously described and are listed in Table 1.35
Western blots
Western blot analysis of mouse tissues and plasma samples was performed as previously described.5 Tissue lysate concentrations were determined by DC Protein Assay (Bio-Rad). Equal volumes of mouse plasma from individual animals were used for western blot analysis of plasma proteins. Antibodies used in the western blot experiments were: a mouse monoclonal antibody against murine glyceraldehyde-3-phosphate dehydrogenase (EMD Millipore), a mouse monoclonal antibody against murine RALA (Sigma-Aldrich), a chicken polyclonal antibody against murine A1AT (GenWay Biotech), a rabbit polyclonal antibody against human A1AT (Proteintech), and a rabbit polyclonal antibody against murine LMAN1 (Sigma-Aldrich). Horseradish peroxidase was detected by using an enhanced chemiluminescence kit (PerkinElmer). The fraction of A1AT retained in the ER (upper band:lower band in the same western blot lane) in liver lysates was quantified by using an IRDye-conjugated secondary antibody (LI-COR Biosciences) and the Odyssey Infrared Imaging System, with analysis performed on the Odyssey CLx Image Studio program (LI-COR Biosciences). Densitometry quantification of the LMAN1 and A1AT western blots was performed with ImageJ software (https://imagej.nih.gov), with normalization to the respective control band in each lane.
Tissue histology
Tissues for histologic examination were placed in Z-Fix solution (Anatech LTD) and prepared as previously described.36,37 Histologic surveys (hematoxylin and eosin staining) were performed on adult Lman1−/− and Lman1+/+ mouse tissues, as well as on embryos harvested from timed matings and newborn pups from Lman1+/− intercrosses, by a pathologist blinded to mouse genotype at the University of Michigan Pathology Cores for Animal Research, as previously described.36,38 Images were taken by using an Olympus BX43 microscope and an Olympus DP72 camera. Kidney and lung images are magnified 40×, and heart and liver images are magnified 60×.
Measurement of FV and FVIII levels
Blood collection via inferior vena cava puncture and platelet-poor plasma isolation were performed as previously described.5 Plasma FV and FVIII activity levels were determined in prothrombin time–based and partial thromboplastin time–based assays, respectively, as previously described.
Statistical analysis
A χ2 test was used to evaluate statistical deviation from expected Mendelian ratios for the genotypes of offspring from the matings listed in Table 2 and for the genotype distributions listed in Table 3. Analysis of the FV and FVIII activity assays was performed by using the Student t test for comparison between 2 different genotypes and by one-way analysis of variance for comparisons of >2 groups. Statistical analysis of the ratios of post-ER A1AT:ER-retained A1AT for Lman1 mutant mice and wild-type control mice was performed by using the Student t test for comparison between different genotypes. P < .05 is considered statistically significant for these analyses; P values that are not statistically significant are abbreviated as “n.s.” in the figures and legends.
Intercrosses of Lman1 mutant mice
| Crosses | Strain | Genotype distribution at 3 wk, % (no. observed) | P, χ2 | |
|---|---|---|---|---|
| +/+ and +/− | −/− | |||
| Expected % | 75 | 25 | ||
| Lman1gt1/+ x Lman1gt1/+ | C57BL/6J | 83 (201) | 17 (40) | <.003 |
| Lman1cgt/+ x Lman1cgt/+ | C57BL/6J | 80 (107) | 20 (27) | >.19 |
| Lman1+/− x Lman1+/− | C57BL/6J | 83 (184) | 17 (38) | <.007 |
| Dead pups (<24 h) | C57BL/6J | 50 (11) | 50 (11) | <.007 |
| Lman1+/− x Lman1+/− | B6;129 mixed | 76 (95) | 24 (30) | >.79 |
| Crosses | Strain | Genotype distribution at 3 wk, % (no. observed) | P, χ2 | |
|---|---|---|---|---|
| +/+ and +/− | −/− | |||
| Expected % | 75 | 25 | ||
| Lman1gt1/+ x Lman1gt1/+ | C57BL/6J | 83 (201) | 17 (40) | <.003 |
| Lman1cgt/+ x Lman1cgt/+ | C57BL/6J | 80 (107) | 20 (27) | >.19 |
| Lman1+/− x Lman1+/− | C57BL/6J | 83 (184) | 17 (38) | <.007 |
| Dead pups (<24 h) | C57BL/6J | 50 (11) | 50 (11) | <.007 |
| Lman1+/− x Lman1+/− | B6;129 mixed | 76 (95) | 24 (30) | >.79 |
The χ2 test was based on an expected genotype ratio of 3:1, with Lman1−/− mice expected to represent 25% of the offspring from each mating, and all other genotypes cumulatively accounting for 75% of offspring.
Genotype distribution of pups from intercrosses of Lman1+/− and Mcfd2+/− mice
| Genotype | P, χ2 | ||
|---|---|---|---|
| Lman1−/− | Lman1+/− or Lman1+/+ | ||
| Observed | 48 | 217 | <.01 |
| Expected | 66 | 199 | |
| Mcfd2−/− | Mcfd2+/− or Mcfd2+/+ | ||
| Observed | 61 | 204 | >.45 |
| Expected | 66 | 199 | |
| Genotype | P, χ2 | ||
|---|---|---|---|
| Lman1−/− | Lman1+/− or Lman1+/+ | ||
| Observed | 48 | 217 | <.01 |
| Expected | 66 | 199 | |
| Mcfd2−/− | Mcfd2+/− or Mcfd2+/+ | ||
| Observed | 61 | 204 | >.45 |
| Expected | 66 | 199 | |
Intercrosses between mice doubly heterozygous for Lman1-– and Mcfd2– were performed to generate double-null Lman1−/− mice, Mcfd2−/− mice, and littermate controls; 265 mice of the 9 possible expected genotypes were generated (Table 3).
Results
Reduced Lman1 expression results in dose-dependent decreases in FV and FVIII
Lman1 mRNA expression in the liver of Lman1cgt/cgt mice was reduced to ∼4% to 6% of wild-type levels as measured by quantitative reverse transcription PCR (qRT-PCR) (Figure 2A-B), compared with <1% of wild type in liver mRNA from Lman1gt1/gt1 mice. This low level of Lman1 mRNA expression likely results from a low level of alternative splicing around the Lman1cgt gene trap construct. Reduced levels of the expected truncated mRNA were detected in Lman1gt1/gt1 mice when tested with the F6/R6 primer pair, consistent with nonsense-mediated decay of the Lman1gt1 β-geo fusion mRNA compared with wild-type Lman1 mRNA.39 Less than 1% of Lman1 mRNA expression distal to intron 10 was detected in the liver of Lman1gt1/gt1 mice (primer pair F5/R5), consistent with a complete block of mRNA transcription distal to the gene trap insertion. An identical qRT-PCR study for Lman1 mRNA expression in kidney lysates of Lman1cgt/cgt mice was consistent with the liver expression levels, with reduction to ∼6% to 8% of wild-type levels (Figure 2C-D). LMAN1 protein levels detected in liver lysates (Figure 3A) were consistent with the qRT-PCR results, with reduced levels in Lman1cgt/cgt (∼15% of wild-type levels by densitometry of the western blot bands) and undetectable levels in Lman1−/− and Lman1gt1/gt1 mice. Analysis of additional tissues from Lman1cgt/cgt mice exhibited consistently reduced LMAN1 relative to wild-type mice (Figure 3B).

Lman1 mRNA expression levels in Lman1cgt/cgtand Lman1gt1/gt1mice. (A-B) qRT-PCR of liver complementary DNA from Lman1cgt/cgt and Lman1gt1/gt1 mice. (C-D) qRT-PCR of kidney complementary DNA from Lman1cgt/cgt mice. Three mice per genotype were used for qRT-PCR analysis. Horizontal bars represent means, and error bars represent standard error of the mean.
Lman1 mRNA expression levels in Lman1cgt/cgtand Lman1gt1/gt1mice. (A-B) qRT-PCR of liver complementary DNA from Lman1cgt/cgt and Lman1gt1/gt1 mice. (C-D) qRT-PCR of kidney complementary DNA from Lman1cgt/cgt mice. Three mice per genotype were used for qRT-PCR analysis. Horizontal bars represent means, and error bars represent standard error of the mean.
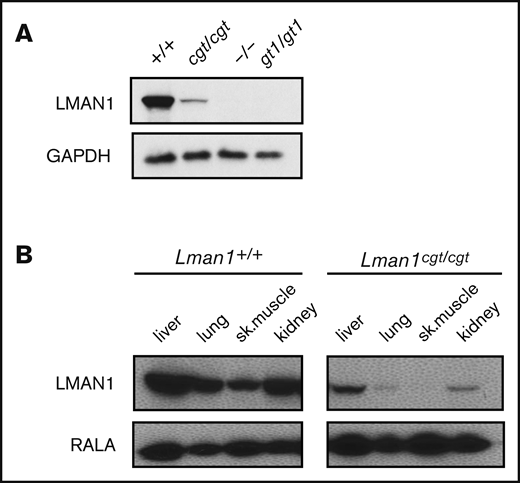
Western blot of LMAN1 expression in Lman1 mutant mice. (A) Western blot of liver lysates from homozygote carriers of the Lman1 mutant alleles: Lman1cgt, Lman1–, and Lman1gt1. (B) Western blot of multiple tissue lysates from wild-type and Lman1cgt/cgt mice. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Western blot of LMAN1 expression in Lman1 mutant mice. (A) Western blot of liver lysates from homozygote carriers of the Lman1 mutant alleles: Lman1cgt, Lman1–, and Lman1gt1. (B) Western blot of multiple tissue lysates from wild-type and Lman1cgt/cgt mice. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To test the hypothesis that Lman1 expression levels correlate with secretion levels of LMAN1-dependent cargoes in a dose-dependent manner, we measured plasma activity levels of FV and FVIII, as well as relative levels of plasma and intracellular (hepatocyte) A1AT in mice homozygous for each of the Lman1 alleles. Lman1−/− and Lman1gt1/gt1 mice on a C57BL/6J genetic background or Lman1−/− mice on a mixed B6;129 genetic background (supplemental Figure 1) exhibited plasma FV and FVIII activity levels that are ∼50% of wild-type levels, consistent with previous reports.28 Lman1−/− mice (C57BL/6J) exhibit FV activity levels that are 48.7% ± 4.0% of wild-type levels (P < .0001) but which are indistinguishable from the levels in Lman1gt1/gt1 mice (54.4% ± 3.9%) (Figure 4A). FV activity levels of Lman1+/− mice are indistinguishable from wild-type mice, consistent with the autosomal recessive inheritance pattern of F5F8D. In contrast, the hypomorphic Lman1cgt/cgt mice exhibit intermediate levels of FV activity (67.1% ± 5.3%), significantly lower than those of wild-type mice (P < .0004) but significantly higher than those of the complete null Lman1−/− mice (P < .011). Data for FVIII activity levels in these different mouse genotypes were similar, as shown in Figure 4B. Lman1−/− mice (C57BL/6J) exhibit FVIII activity levels that are 49.2% ± 4.6% of wild-type levels (P < .0001) but which are indistinguishable from the levels in Lman1gt1/gt1 mice (49.5% ± 2.2%). FVIII activity levels of Lman1+/− mice are indistinguishable from those of wild-type mice. The hypomorphic Lman1cgt/cgt mice exhibit intermediate levels of FVIII activity (62.8% ± 4.2%), significantly lower than those of wild-type mice (P < .0001) but significantly higher than those of the complete null Lman1−/− mice (P < .042). Heterozygous carriers of the Lman1cgt, Lman1gt1, or the Lman1– alleles exhibit FV and FVIII activity levels indistinguishable from those of control mice or from one another (supplemental Figure 2A-D), consistent with the autosomal recessive inheritance of F5F8D in humans, and with previously published reports in mice.28

Plasma FV and FVIII activity levels for Lman1 mutant mice (C57BL/6J background). (A) Plasma FV activity as percentage of wild-type. (B) Plasma FVIII activity as percentage of wild-type. Each symbol represents an individual animal. Horizontal lines indicate mean, and error bars indicate standard error of the mean for each genotype.
Plasma FV and FVIII activity levels for Lman1 mutant mice (C57BL/6J background). (A) Plasma FV activity as percentage of wild-type. (B) Plasma FVIII activity as percentage of wild-type. Each symbol represents an individual animal. Horizontal lines indicate mean, and error bars indicate standard error of the mean for each genotype.
A1AT accumulates in the ER of Lman1-deficient mice
It was previously shown that Lman1gt1/gt1 liver extracts contain higher levels of intracellular A1AT, compared with wild-type control mice, despite their comparable steady-state plasma A1AT levels.28 It was similarly shown, through endoglycosidase H digestion experiments, that the increased intracellular accumulation of A1AT in Lman1gt/gt mice is the result of ER retention of A1AT. Similar experiments were performed for the current study using liver extracts from wild-type, Lman1cgt/cgt, Lman1gt1/gt1, and Lman1−/− mice to determine if there is also a dose–response relationship between levels of Lman1 expression and ER-to-Golgi transport of A1AT. As shown in Figure 5A, western blots were performed to identify A1AT levels in 3 independent samples for each Lman1 genotype. The A1AT antibody produces a doublet, with the top band corresponding to the A1AT that has been processed in the Golgi (the same size as the secreted plasma A1AT band); the lower band in the doublet corresponds to the ER-retained A1AT. This lower band was previously shown to be sensitive to endoglycosidase H digestion, consistent with retention in the ER.28
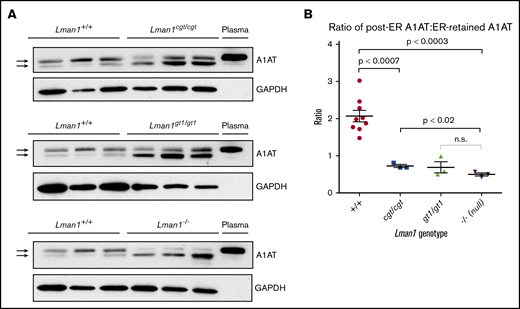
Western blot for A1AT in liver lysates. (A) Liver lysates prepared from Lman1cgt/cgt, Lman1gt1/gt1, Lman1−/−, and Lman1+/+ mice were analyzed by western blotting using an anti-A1AT antibody. The lower A1AT band corresponds to ER-retained A1AT (and was previously shown to be endoglycosidase H sensitive28 ). The upper A1AT band corresponds to post-ER A1AT and is comparable in molecular weight to secreted plasma A1AT. Each lane corresponds to an individual mouse. (B) The ratio of post-ER A1AT:ER-retained A1AT (upper band:lower band) in the hepatic lysates of each mouse as determined by densitometry; a ratio >1 indicates that the majority of the A1AT has been secreted beyond the ER, whereas a ratio <1 indicates that the majority of the A1AT is retained in the ER. Horizontal lines indicate the mean, and error bars indicate the standard error of the mean for each genotype.
Western blot for A1AT in liver lysates. (A) Liver lysates prepared from Lman1cgt/cgt, Lman1gt1/gt1, Lman1−/−, and Lman1+/+ mice were analyzed by western blotting using an anti-A1AT antibody. The lower A1AT band corresponds to ER-retained A1AT (and was previously shown to be endoglycosidase H sensitive28 ). The upper A1AT band corresponds to post-ER A1AT and is comparable in molecular weight to secreted plasma A1AT. Each lane corresponds to an individual mouse. (B) The ratio of post-ER A1AT:ER-retained A1AT (upper band:lower band) in the hepatic lysates of each mouse as determined by densitometry; a ratio >1 indicates that the majority of the A1AT has been secreted beyond the ER, whereas a ratio <1 indicates that the majority of the A1AT is retained in the ER. Horizontal lines indicate the mean, and error bars indicate the standard error of the mean for each genotype.
Consistent with the prior report for Lman1gt1/gt1 mice, intracellular A1AT accumulation in hepatic lysates was observed in Lman1cgt/cgt, Lman1gt1/gt1, and Lman1−/− hepatocytes (Figure 5A). Compared with the high proportion of post-ER A1AT (upper band) in wild-type mice, LMAN1-deficient mouse hepatic lysates have lower levels of post-ER A1AT and higher levels of ER-retained A1AT (lower band). The upper and lower bands in each doublet were quantified, and the ratio of post-ER A1AT to ER-retained A1AT was calculated for each sample (Figure 5B). A ratio >1 indicates that the majority of the intracellular A1AT has reached the Golgi, whereas a ratio <1 indicates that the majority of the intracellular A1AT is retained in the ER. These ratios follow a similar dose–response relationship to the level of Lman1 expression as observed for FV and FVIII plasma activity levels. The Lman1−/− mice exhibit a statistically significantly lower ratio compared with wild-type mice (mean 0.50 ± 0.04 compared with 2.1 ± 0.2, respectively; P < .0003), and there was no difference between the Lman1−/− and Lman1gt1/gt1 mice. The hypomorphic Lman1cgt/cgt mice exhibit an intermediate ratio of post-ER A1AT:ER-retained A1AT, which is significantly lower than that in wild-type mice (0.73 ± 0.04 vs 2.1 ± 0.2; P < .0007) and significantly higher than that in Lman1−/− null mice (P < .02); this finding, however, was not significantly different from that in Lman1gt1/gt1 mice, consistent with an intermediate level of intracellular A1AT accumulation.
Although A1AT (and presumably other LMAN1 cargoes) accumulates in the ER in LMAN1-deficient mice, there is no difference in the expression level of multiple unfolded protein response genes in Lman1−/− mice compared with wild-type control mice, consistent with a prior report (supplemental Figure 3).28
LMAN1 deficiency results in perinatal lethality with incomplete penetrance
Confirming our previous results,28 intercrosses of Lman1gt1/+ mice on a C57BL/6J genetic background resulted in reduced numbers of homozygous Lman1gt1/gt1 mice at 3 weeks of age (17% observed vs 25% expected; P < .003) (Table 2). Similar incompletely penetrant lethality was observed for Lman1−/− mice (from an Lman1+/− intercross) (P < .007). Genotyping of pups dying spontaneously on the first day of life revealed an excess of Lman1−/− pups (11 of 22; P < .007), consistent with perinatal lethality. No obvious histologic abnormalities were observed on necropsy of Lman1−/− pups and wild-type littermates euthanized at E18.5 to suggest a cause of death; histologic examination of adult Lman1−/− mice was also unremarkable (supplemental Figure 4). In contrast, the expected number of Lman1cgt/cgt mice at weaning was observed from an Lman1cgt/+ intercross (P > .19). Similar to previously reported results for the Lman1gt1 allele,28 no perinatal lethality was observed for Lman1−/− pups on a mixed C57BL/6J-129S1/SvImJ genetic background (P > .79). Intercrosses between Lman1+/−/Mcfd2+/− mice confirmed the partial perinatal lethality for the Lman1−/− but not the Mcfd2−/− genotype (Table 3).32
Discussion
We report the characterization of a series of gene-targeted Lman1 alleles directing gene expression levels ranging from 0% to 7% of wild type, as confirmed by qRT-PCR and western blot expression studies in multiple tissues. The observation of similar plasma FV and FVIII levels in Lman1gt1/gt1 and Lman1−/− mice excludes trace residual LMAN1 expression from the Lman1gt1 allele as the explanation for more modest reductions in FV and FVIII in LMAN1-deficient mice compared with humans.28 These findings also suggest that, at least in mice, an LMAN1-independent mechanism for FV and FVIII secretion may account for the residual ∼50% levels in LMAN1 null animals. This alternative mechanism could be mediated by another cargo receptor, potentially a paralog of LMAN1 (eg, ERGL). Such an alternative cargo receptor could be expressed constitutively, or only induced in the absence of LMAN1. The confirmation of the previously reported perinatal, strain-specific lethality28 with an independent Lman1– allele also excludes a passenger gene effect40 not due to LMAN1 deficiency as the cause of lethality in Lman1gt1/gt1 mice. In contrast, mice homozygous for the hypomorphic Lman1cgt allele are not significantly reduced in number. Because fetal loss would not be expected to result from moderate FV and FVIII deficiency, these data suggest the existence of another, as yet unknown, critical LMAN1-dependent cargo(s). The lack of evidence for perinatal loss in human F5F8D patients despite much greater reductions in FV and FVIII,16 together with normal survival of Lman1−/− mice on a mixed genetic background, are consistent with enhanced susceptibility of inbred mice to reduced ER exit of a key LMAN1 cargo. However, differences in the repertoire of LMAN1 cargoes between mouse and human cannot be excluded.
We report a dose–response relationship between the levels of LMAN1 expression and the steady-state plasma levels of FV and FVIII. These results are surprising given the apparent enrichment for null mutations among LMAN1 alleles identified in F5F8D patients,41 which suggested that even low levels of residual LMAN1 function are sufficient to support normal FV and FVIII secretion. Taken together, these observations raise the possibility that human subjects with hypomorphic LMAN1 mutations and residual LMAN1 activity might present with more modest reductions in FV and FVIII, potentially resulting in only mild bleeding. Such mild disease could potentially have eluded clinical detection or a specific genetic diagnosis to date. Our results thus suggest considering genetic testing in patients with more modestly reduced FV and FVIII levels than those typically associated with F5F8D. These findings also suggest that therapeutic targeting of LMAN1 to reduce FV and FVIII levels as an anticoagulant strategy may only require partial inhibition of LMAN1 function.
Original data or protocols may be requested by contacting the corresponding author (David Ginsburg; e-mail: ginsburg@umich.edu).
Acknowledgments
The authors thank Mark Hoenerhoff from the University of Michigan Unit for Laboratory Animal Medicine for performing the histologic analyses.
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R35 HL135793 [D.G.]; R01 HL094505 [B.Z.]; K08 HL128795 and R01 HL148333 [R.N.K.]). This work was also supported by Mcubed, a research seed-funding program for faculty at the University of Michigan (R.N.K.) and by the University of Michigan Rogel Cancer Center (R.N.K.). R.N.K. was a recipient of an American Society of Hematology Scholar Award. D.G. is an investigator of the Howard Hughes Medical Institute.
Authorship
Contribution: L.A.E., R.N.K., and D.G. designed the research; L.A.E. and R.N.K. performed the experiments; L.A.E., R.N.K., B.Z., and D.G. analyzed the data; L.A.E., R.N.K, and D.G. wrote the manuscript; and all authors reviewed and commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David Ginsburg, Life Sciences Institute, University of Michigan, 210 Washtenaw Ave, Ann Arbor, MI 48109; e-mail: ginsburg@umich.edu.

