

Key Points
Transient suppression of immune responses to LVs and LV-transduced cells increased and prolonged transgene expression.
IO delivery of FVIII variant LVs effectively improved therapeutic effect of FVIII-containing platelets for hemophilia treatment.

Abstract
Our previous studies demonstrated that intraosseous (IO) infusion of lentiviral vectors (LVs) carrying a modified B domain–deleted factor VIII (FVIII) transgene driven by a megakaryocyte-specific promoter (GP1Bα promoter; G-F8/N6-LV) successfully transduced hematopoietic stem cells (HSCs) to produce FVIII stored in the platelet α-granules. Platelet FVIII corrected the bleeding phenotype with limited efficacy in hemophilia A (HemA) mice with and without preexisting anti-FVIII inhibitors. The present study sought to further enhance the therapeutic efficacy of this treatment protocol by increasing both the efficiency of LV transduction and the functional activity of platelet FVIII. A combined drug regimen of dexamethasone and anti-CD8α monoclonal antibody enhanced the percentage of transduced bone marrow and HSCs over time. In G-F8/N6-LV–treated HemA mice, significant improvement in phenotypic correction was observed on day 84. To improve platelet FVIII functionality, genes encoding FVIII variant F8X10K12 with increased expression or F8N6K12RH with increased functional activity compared with F8/N6 were incorporated into LVs. Treatment with G-F8X10K12-LV in HemA mice produced a higher level of platelet FVIII but induced anti-FVIII inhibitors. After treatment with combined drugs and IO infusion of G-F8/N6K12RH-LV, HemA mice showed significant phenotypic correction without anti-FVIII inhibitor formation. These results indicate that new human FVIII variant F8/N6K12RH combined with immune suppression could significantly enhance the therapeutic efficacy of in vivo platelet-targeted gene therapy for murine HemA via IO delivery. This protocol provides a safe and effective treatment for hemophilia that may be translatable to and particularly beneficial for patients with preexisting inhibitory antibodies to FVIII.
Introduction
Inhibitory antibody formation against human factor VIII (hFVIII) is a significant complication in the treatment of a patient with hemophilia A (HemA). Ectopic expression of FVIII in platelets could be an effective approach for treating HemA. Unlike circulating plasma FVIII, FVIII stored in α-granules of platelets is protected from being processed by antigen-presenting cells, thereby significantly reducing the chance of inducing anti-FVIII immune responses.1,2 Moreover, platelet FVIII is not neutralized by preexisting anti-FVIII inhibitory antibodies (inhibitors), which will benefit HemA patients who have already developed inhibitors.3 During bleeding, the activated platelets locally release FVIII that can directly participate in the coagulation cascade and promote clot formation. It has recently been demonstrated that even low levels of platelet FVIII can partially correct the HemA phenotype in animal models.4-6
Our approach to direct long-term expression of FVIII in platelets is intraosseous (IO) infusion of lentiviral vectors (LVs) carrying an FVIII transgene controlled by megakaryocyte-specific promoter GP1Bα (G-F8-LV).4 In this in vivo gene therapy protocol for treating HemA, hematopoietic stem cells (HSCs) were efficiently transduced by LVs in situ, resulting in FVIII expression in megakaryocytes and storage in platelet α-granules. A single IO infusion of G-F8-LV resulted in >3% of the platelets containing hFVIII, leading to partial phenotypic correction in HemA mice with and without preexisting inhibitors.
In vivo delivery of LVs can avoid many difficulties and potential toxicities encountered by ex vivo gene therapy, including low engraftment potential and preconditioning of the patient. Furthermore, both myeloablative and nonmyeloablative preconditioning regimens used by ex vivo gene therapy induce thrombocytopenia. Because bypassing agents often achieved low efficacy compared with FVIII replacement therapy,7 the induction of concurrent thrombocytopenia in severe HemA inhibitor patients may pose a major hemostatic risk.5 In vivo gene therapy protocols can bypass this significant hemostatic risk. Additionally, recent clinical testing using a humanized bispecific antibody (emicizumab; ACE910)8 showed impressive results in treating hemophilia patients with and without inhibitory antibodies. However, frequent infusions of costly reagents are required, and potential long-term adverse effects still need to be evaluated. Compared with drug or protein therapy, gene therapy can achieve a prolonged therapeutic effect with only 1 or several treatments over a patient’s lifetime.
However, a potential limitation of in vivo gene therapy protocols is the induction of LV-specific immune responses, which may decrease LV transduction efficiency and eliminate LV-transduced cells.9,10 It has been shown that dexamethasone (Dex) can suppress inflammatory responses after IV delivery of LVs into immunocompetent mice and increase LV transduction efficiency.11,12 We have also shown that rapamycin can enhance LV transduction efficiency for both in vivo and ex vivo gene therapies.13
An ideal transgene for effective in vivo gene therapy for hemophilia would confer a higher gene expression level and a transgene product with enhanced biological activity. For more efficient packaging, shorter complementary DNAs (cDNAs) coding for B domain–deleted (BDD) FVIII variants that have similar or higher functional activity than full-length FVIII14 were used in gene therapy preclinical research.15,16 Additional mutations were incorporated into the gene coding for BDD FVIII to increase its expression/secretion, such as F8/N6 with a 226–amino acid B domain variant sequence17 and others.18-20 Some FVIII variants, like FVIII-RH20 and furin cleavage site–deleted BDD FVIII variants,21,22 exhibit an increase in biological activity compared with BDD FVIII.
In this study, we aimed to improve the therapeutic efficacy of HemA gene therapy with IO infusion of LVs targeting FVIII expression in platelets. The immunocompetent mice were pretreated with Dex to suppress inflammatory responses and anti-CD8α monoclonal antibody (mAb) to inhibit cytotoxicity by transient depletion of CD8+ T cells. The pharmacological intervention with combined Dex and anti-CD8α mAb treatment improved LV transduction efficiency and increased long-term transgene expression levels in mice. Furthermore, 2 new hFVIII variants with higher expression and enhanced biological activity were also tested in immunocompetent HemA mice. The results demonstrated that combined drug treatment plus IO infusion of LVs containing the FVIII variant gene with enhanced biological activity significantly improved hemophilia phenotypic correction.
Materials and methods
Animals
All mice were kept in a specific pathogen-free environment at Seattle Children’s Research Institute (SCRI) according to National Institutes of Health guidelines for animal care and the guidelines of SCRI. The protocols were approved by the Institutional Animal Care and Use Committee at SCRI. HemA mice (F8 exon 16 knockout) with a C57BL/6 (BL/6) genetic background were generated by crossing the mixed-background HemA mice (SV129/BL6) with BL/6 mice for 8 generations.23 BL/6 mice and Rag2−/− mice with hematopoietic and immune system defects, including arrested B- and T-cell development,24 were purchased from the Jackson Laboratory. Only male HemA mice were used in this study.
LV constructs carrying GFP and human F8 cDNA variants
The self-inactivating LV construct encoding green fluorescent protein (GFP) was driven by an MND promoter and LV constructs carrying various BDD hFVIII variants, including hF8/N625 (a control vector used in previous studies4 ), hF8X10K12,26-28 and hF8/N6K12RH,20,26 were under the control of a ubiquitous EF1α promoter or platelet-specific GP1Bα promoter (Figure 1A). F8X10K12 encodes a novel hFVIII variant with a deleted B domain and mutations in the A1 domain to enhance expression/secretion (X10; V86I, F105Y, S108A, E115D, H117Q, L129F, K132G, Q134H, T147M, and P152L) and in the C1 and C2 domains to increase functional activity (K12; V1857I, H1859R, M1907K, M1926K, L1975V, A1993V, H2007Q, D2066E, K2085M, Q2113H, S2157N, and R2159H) to enhance FVIII activity. RH in F8/N6K12RH denotes an R1645H mutation20 for generating a more stable FVIII single-chain molecule.
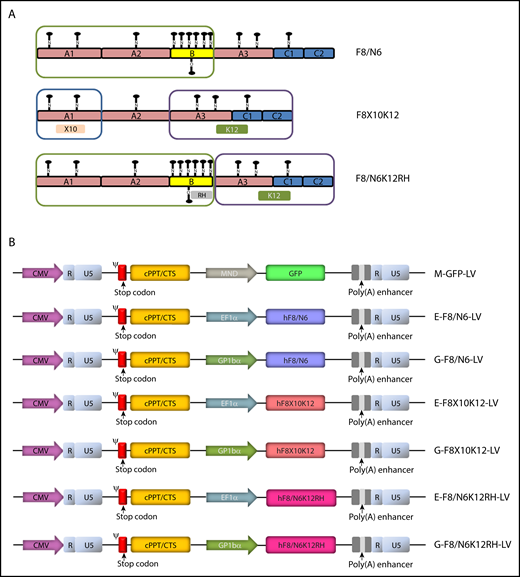
Schematics of new hFVIII variants and LVs incoporating cDNAs encoding GFP or hFVIII varants. (A) hFVIII variants. Compared with F8/N6, F8X10K12 had a deleted B domain, a 10–amino acid change in the A1 domain, and a 12–amino acid change in the light chain, and F8/N6K12RH had a 12–amino acid change in the light chain and an amino acid change at the furin cleavage site within the B domain (position R1645H). (B) Schematics of self-inactivating LV constructs encoding GFP under the control of an MND promoter or various hFVIII variants, including F8/N6, F8X10K12, and F8/N6K12RH, under the control of a ubiquitous EF1α promoter or platelet-specific GP1Bα promoter. CMV, cytomegalovirus; cPPT, central polypurine tract; CTS, central termination sequence.
Schematics of new hFVIII variants and LVs incoporating cDNAs encoding GFP or hFVIII varants. (A) hFVIII variants. Compared with F8/N6, F8X10K12 had a deleted B domain, a 10–amino acid change in the A1 domain, and a 12–amino acid change in the light chain, and F8/N6K12RH had a 12–amino acid change in the light chain and an amino acid change at the furin cleavage site within the B domain (position R1645H). (B) Schematics of self-inactivating LV constructs encoding GFP under the control of an MND promoter or various hFVIII variants, including F8/N6, F8X10K12, and F8/N6K12RH, under the control of a ubiquitous EF1α promoter or platelet-specific GP1Bα promoter. CMV, cytomegalovirus; cPPT, central polypurine tract; CTS, central termination sequence.
IO infusion of LVs
Immunosuppression treatment
Dex (MWIVet) and anti-CD8α mAb (BioXcell; clone YTS169.4) were used to transiently deplete innate immune responses and cytotoxicity, respectively. LVs were infused into the mice on day 0. Single drug–pretreated mice were given Dex (5 mg/kg at −24, −4, 4, and 24 hours) or anti-CD8α mAb (4 mg/kg on days −1, 4, and 11) by intraperitoneal injection. Combined drug–pretreated mice were administrated Dex (5 mg/kg at −24, −4, 4, and 24 hours) and anti-CD8α mAb (4 mg/kg on days −1, 4, 11, 16, and 21).
Characterization of transgene (GFP or hFVIII) expression
GFP expression in bone marrow cells of BL6 mice treated with M-GFP-LV4 (LV carrying a GFP gene driven by the modified myeloid proliferative sarcoma virus promoter30,31 ; Figure 1B) and hFVIII expression in F8-LV–transduced 293T cells were detected by flow cytometry. hFVIII expression in platelets of F8-LV–treated HemA mice was measured by enzyme-linked immunosorbent assay as previously described.4 Detailed information about the process of isolating bone marrow cells, white blood cells, and platelets and about the antibodies used in this study is provided in the supplemental Data.
Assays for measuring hFVIII activity and anti-hFVIII antibodies
Plasma samples were isolated from peripheral blood in experimental mice collected by retroorbital bleeding. hFVIII activity was analyzed using a modified activated partial thromboplastin time assay, and anti-hFVIII antibodies were measured by hFVIII Bethesda inhibitor assay as previously described.23,32
Assays to characterize phenotypic correction in HemA mice
Correction of the bleeding phenotype of LV-treated HemA mice was initially examined using a modified tail clip assay as previously described.4 For more sensitive evaluation of functional platelet FVIII, the blood flow rate of the right carotid artery in LV-treated HemA mice after local damage was measured in a ferric chloride (FeCl3)–induced thrombosis model. In addition, platelet-stored hFVIII function in LV-treated HemA mice was evaluated by a rotational thromboelastometry (ROTEM) assay using freshly collected whole blood. Detailed information about the ROTEM procedure and the FeCl3 injury model is provided in the supplemental Data.
Statistical analyses
Data are expressed as mean ± standard deviation of the mean. All statistical analyses were performed using GraphPad Prism 7 software. One-way analysis of variance (ANOVA) was used to evaluate the significance of single–time point experiments, and repeated measures ANOVA (2-way ANOVA followed by post hoc Bonferroni’s multiple comparison test) was used to determine statistical significance in experiments involving measurements over an extended time course. Differences were considered significant at P < .05.
Results
Comparison of GFP expression in immunocompetent and immunodeficient mice over time after IO delivery of M-GFP-LV
To evaluate if immune responses affected transgene expression, M-GFP-LV (1.1 × 108 infectious units [ifu] per animal; Figure 1B) was delivered via IO infusion into immunodeficient Rag2−/− mice and immunocompetent BL6 mice. Seven days after infusion, we observed that GFP expression levels in total bone marrow cells and HSCs in Rag2−/− mice were comparable to those in BL6 mice (Figure 2A). Subsequently, GFP levels were increased in Rag2−/− mice but decreased in BL6 mice on day 28. At 84 days after infusion, Rag2−/− mice had higher GFP expression in total bone marrow cells and HSCs than BL6 mice (Figure 2A). Overall, after M-GFP-LV transduction in bone marrow, GFP expression in total bone marrow and HSCs, respectively, was maintained at relatively stable high levels in Rag2−/− mice for up to 240 days (experimental duration; 5% to 12% in bone marrow and 20% to 40% in HSCs at day 240), whereas GFP expression levels gradually decreased over time in immunocompetent BL6 mice (Figure 2B-C). These results suggest that immune responses elicited by LVs, likely including cytotoxic lymphocytes, may reduce the number of transduced cells over time.
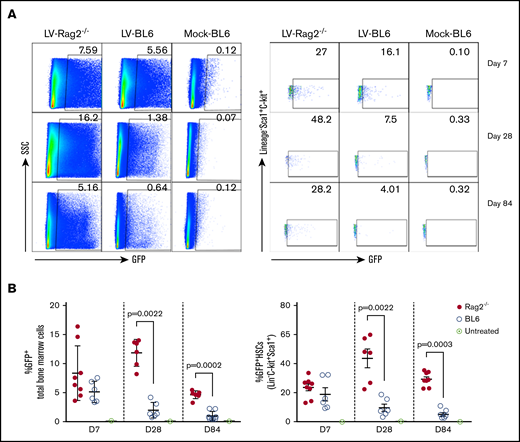
GFP expression in bone marrow cells after IO infusion of M-GFP-LV in immunocompetent and immunodeficient mice. Both C57BL/6J (BL6) and B6(Cg)-Rag2tm1.1Cgn/J (Rag2−/−) mice received IO infusion with self-inactivating LVs encoding GFP under the control of an MND promoter (M-GFP-LV; 1.1 × 108 ifu per animal) or sterile phosphate-buffered saline (20 μL per animal; mock) on day 0. The experimental mice were euthanized on day 7, 28, or 84. The bone marrow cells were isolated, and GFP expression levels in total bone marrow cells (A-B, left) and HSCs (Lin−C-Kit+Sca1+; A-B, right) were examined by flow cytometry. Data are shown as representative flow images (A) and summary plot over time (B). Data shown are expressed as mean ± standard deviation of the mean. Each symbol represents an individual animal. SSC, side scatter.
GFP expression in bone marrow cells after IO infusion of M-GFP-LV in immunocompetent and immunodeficient mice. Both C57BL/6J (BL6) and B6(Cg)-Rag2tm1.1Cgn/J (Rag2−/−) mice received IO infusion with self-inactivating LVs encoding GFP under the control of an MND promoter (M-GFP-LV; 1.1 × 108 ifu per animal) or sterile phosphate-buffered saline (20 μL per animal; mock) on day 0. The experimental mice were euthanized on day 7, 28, or 84. The bone marrow cells were isolated, and GFP expression levels in total bone marrow cells (A-B, left) and HSCs (Lin−C-Kit+Sca1+; A-B, right) were examined by flow cytometry. Data are shown as representative flow images (A) and summary plot over time (B). Data shown are expressed as mean ± standard deviation of the mean. Each symbol represents an individual animal. SSC, side scatter.
Pharmacological approach to enhance GFP expression in immunocompetent mice after IO delivery of M-GFP-LV
Based on the results obtained from LV-treated immunocompetent and immunodeficient mice, we explored whether pharmacological agents designed to nonspecifically or specifically block these immune responses would limit clearance of transduced cells, leading to more persistent and higher levels of sustained transgene expression. First, Dex was given to the BL6 mice with IO infusion of M-GFP-LV (8.8 × 108 ifu per animal on day 0; Figure 3A). Seven days after LV infusion, the Dex plus LV–treated mice produced significantly higher GFP expression levels in both total bone marrow cells and HSCs than those in mice treated with LVs only (Figure 3B), indicating that Dex could efficiently inhibit early immune responses and enhance GFP expression.
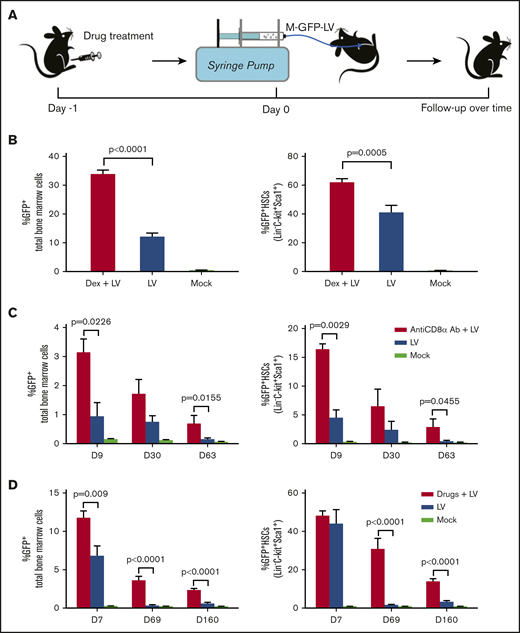
Transient immune suppression to enhance in situ transduction efficiency of bone marrow cells after IO infusion of M-GFP-LVs. (A) Schematic of IO infusion of M-GFP-LV into BL6 mice pretreated with intraperitoneal injection of Dex (5 mg/kg, 4 times: −24, −4, 4, and 24 hours) or anti-CD8α mAb (4 mg/kg, 3 times: days −1, 4, and 11 or 5 times: days −1, 4, 11, 16, and 21) or combined drugs (Dex 4 times + anti-CD8α mAb 5 times). GFP expression in total bone marrow cells and HSCs (Lin−C-Kit+Sca1+) was measured by flow cytometry. (B) BL6 mice were pretreated with Dex after IO infusion of M-GFP-LV (8.8 × 108 ifu per animal; n = 8) or treated with LVs only (n = 6) or sterile phosphate-buffered saline (PBS; 20 μL per animal; mock; n = 3). GFP expression in total bone marrow cells (left) and HSCs (right) was detected on day 7. (C) BL6 mice were pretreated with anti-CD8α mAb (3 times) after IO infusion of GFP-LV (8.8 × 107 ifu per animal; n = 4) or treated with LV only (n = 4) or sterile PBS (20 μL per animal; mock; n = 3). GFP expression in total bone marrow cells (left) and HSCs (right) was detected on days 9, 30, and 63. (D) BL6 mice were pretreated with combined drugs after IO infusion of GFP-LV (3.6 × 108 ifu per animal; n = 6) or treated with LVs only (n = 6) or sterile PBS (20 μL per animal; mock; n = 3). GFP expression in total bone marrow cells (left) and HSCs (right) was detected on days 7, 69, and 160. Data are expressed as mean ± standard deviation of the mean.
Transient immune suppression to enhance in situ transduction efficiency of bone marrow cells after IO infusion of M-GFP-LVs. (A) Schematic of IO infusion of M-GFP-LV into BL6 mice pretreated with intraperitoneal injection of Dex (5 mg/kg, 4 times: −24, −4, 4, and 24 hours) or anti-CD8α mAb (4 mg/kg, 3 times: days −1, 4, and 11 or 5 times: days −1, 4, 11, 16, and 21) or combined drugs (Dex 4 times + anti-CD8α mAb 5 times). GFP expression in total bone marrow cells and HSCs (Lin−C-Kit+Sca1+) was measured by flow cytometry. (B) BL6 mice were pretreated with Dex after IO infusion of M-GFP-LV (8.8 × 108 ifu per animal; n = 8) or treated with LVs only (n = 6) or sterile phosphate-buffered saline (PBS; 20 μL per animal; mock; n = 3). GFP expression in total bone marrow cells (left) and HSCs (right) was detected on day 7. (C) BL6 mice were pretreated with anti-CD8α mAb (3 times) after IO infusion of GFP-LV (8.8 × 107 ifu per animal; n = 4) or treated with LV only (n = 4) or sterile PBS (20 μL per animal; mock; n = 3). GFP expression in total bone marrow cells (left) and HSCs (right) was detected on days 9, 30, and 63. (D) BL6 mice were pretreated with combined drugs after IO infusion of GFP-LV (3.6 × 108 ifu per animal; n = 6) or treated with LVs only (n = 6) or sterile PBS (20 μL per animal; mock; n = 3). GFP expression in total bone marrow cells (left) and HSCs (right) was detected on days 7, 69, and 160. Data are expressed as mean ± standard deviation of the mean.
Second, we transiently depleted CD8+CD3+ T cells by intraperitoneal injection of anti-CD8α mAb to suppress cytotoxic responses (supplemental Figure 1A). The mice infused with M-GFP-LV (1.1 × 108 ifu per animal) were treated with anti-CD8α mAb (Figure 3A). CD8α+CD3ε+ T cells were depleted in blood and bone marrow in treated mice 4 days after anti-CD8α mAb treatment (day 3 after IO infusion) and then recovered gradually to normal levels (supplemental Figure 1B). Although GFP expression levels decreased over time, such levels in both total bone marrow cells and HSCs of anti-CD8α mAb plus LV–treated mice were higher than those in mice treated with LVs only on days 9, 30, and 63 after LV infusion (Figure 3C; supplemental Figure 2), indicating that anti-CD8α mAb treatment partially rescued GFP expression, likely by transient depletion of CD8α+CD3ε+ T cells.
To increase LV transduction and preserve survival of transduced cells, we administered a short course of combination drug treatment to the BL6 mice along with M-GFP-LV (3.6 × 108 ifu per animal on day 0; Figure 3A). Seven days after IO infusion, GFP expression levels in total bone marrow cells in drug plus LV–treated mice were significantly higher than those in mice treated with LV only (Figure 3D left). No significant difference in GFP expression levels was observed in HSCs at this early time point (Figure 3D right). Nevertheless, in the long term, GFP expression levels in both total bone marrow cells and HSCs in drug plus LV–treated mice were significantly higher than those in mice treated with LVs only (Figure 3D; supplemental Figure 3), and >10% of GFP+ HSCs were seen at the end of 5 months. Furthermore, a trend toward higher lentiviral copy numbers were detected in blood cells and total bone marrow cells in drug plus LV–treated mice than in LV-treated mice on day 160 (supplemental Figure 4).
Pharmacological approach to improve phenotypic correction in immunocompetent HemA mice with IO delivery of G-F8/N6-LV
Next, we applied the same pharmacological approach to improve the therapeutic effects of IO infusion of G-F8/N6-LV in HemA mice. HemA mice were given combined drug treatment (Dex plus anti-CD8α mAb) plus an IO infusion of G-F8/N6-LV (2.2 × 106 ifu per animal). Ten weeks after IO infusion, hemophilia phenotypic correction was evaluated with 2 injury mouse models: tail clipping and FeCl3-induced thrombosis. In the tail clipping assay, the mice treated with LVs only or drugs plus LVs had significantly reduced blood loss than HemA control mice, with blood loss in the drugs plus LVs group lower than in the LV-only group (Figure 4A). Consistent with that, in the FeCl3-induced carotid artery injury model, the average blood flow rates in the mice treated with LVs only or drugs plus LVs were significantly slower than those in HemA control mice, with the flow rates in the drugs plus LVs group lower than in the LV-only group (Figure 4B). These results confirm that IO infusion of G-F8/N6-LV into HemA mice can enhance their blood clotting function. Furthermore, combined drug treatment with Dex and anti-CD8α Ab significantly enhanced the therapeutic efficiency of IO infusion of G-F8/N6-LV in HemA mice and achieved persistent FVIII expression for >1 year in treated mice (experimental follow-up duration). In addition, there was neither detectable plasma FVIII activity nor anti-FVIII inhibitors in blood (Figure 4C).

Transient immune suppression to enhance phenotypic correction in HemA mice after IO infusion of G-F8/N6-LV. HemA mice were pretreated with combined drugs (Dex 4 times + anti-CD8α mAb 5 times) and then given an IO infusion of self-inactivating LVs encoding hFVIII variant with the proximal 226–amino acid region of the B domain (F8/N6) under the control of GP1Bα promoter (G-F8/N6-LV; 2.2 × 106 ifu per animal) on day 0. (A) HemA phenotypic correction in mice treated with G-F8/N6-LV or G-F8/N6-LV plus drugs was evaluated by tail clip assay on day 70 (n = 6-8 per group). The average blood loss of untreated HemA mice was set as 100%. Wild-type C57BL/6 mice were used as positive controls. (B) HemA phenotypic correction in mice treated with G-F8/N6-LV plus drugs or G-F8/N6-LV was also evaluated by measuring carotid artery blood flow rate on day 84. (C) Plasma samples were collected from mice treated with G-F8/N6-LV plus drugs or G-F8/N6-LV on day 84. hFVIII activity and anti-FVIII antibodies were measured by activated partial thromboplastin time and Bethesda assays, respectively. Each symbol represented an individual animal. Data were expressed as mean ± standard deviation of the mean.
Transient immune suppression to enhance phenotypic correction in HemA mice after IO infusion of G-F8/N6-LV. HemA mice were pretreated with combined drugs (Dex 4 times + anti-CD8α mAb 5 times) and then given an IO infusion of self-inactivating LVs encoding hFVIII variant with the proximal 226–amino acid region of the B domain (F8/N6) under the control of GP1Bα promoter (G-F8/N6-LV; 2.2 × 106 ifu per animal) on day 0. (A) HemA phenotypic correction in mice treated with G-F8/N6-LV or G-F8/N6-LV plus drugs was evaluated by tail clip assay on day 70 (n = 6-8 per group). The average blood loss of untreated HemA mice was set as 100%. Wild-type C57BL/6 mice were used as positive controls. (B) HemA phenotypic correction in mice treated with G-F8/N6-LV plus drugs or G-F8/N6-LV was also evaluated by measuring carotid artery blood flow rate on day 84. (C) Plasma samples were collected from mice treated with G-F8/N6-LV plus drugs or G-F8/N6-LV on day 84. hFVIII activity and anti-FVIII antibodies were measured by activated partial thromboplastin time and Bethesda assays, respectively. Each symbol represented an individual animal. Data were expressed as mean ± standard deviation of the mean.
G-F8X10K12-LV generated higher expression, secretion, and functional activity of FVIII but induced anti-FVIII antibody formation
To improve the therapeutic effects of in vivo gene therapy of HemA mice, we next incorporated cDNAs encoding FVIII variants with high expression and functional activity. F8X10K12 encodes a novel hFVIII variant with a deleted B domain and mutations in the A1 domain to enhance expression/secretion (×10) and mutations in C1 and C2 domains to increase functional activity (K12; Figure 1A). We first tested its expression by hydrodynamic injection of the plasmid, pEF1α-F8X10K12 driven by a ubiquitous EF1α promoter, into HemA mice. Compared with pEF1α-F8/N6, pEF1α-F8X10K12 produced dramatically higher levels of FVIII in treated mouse plasma on day 4 (Figure 5A left). We also generated E-F8X10K12-LV (Figure 1B), driven by the EF1α promoter to transduce 293T cells (multiplicity of infection, 100). Higher percentage and mean fluorescence intensity of F8 gene expression were detected in E-F8X10K12-LV–transduced cells compared with E-F8/N6-LV–transduced cells on day 4 (Figure 5A right), confirming that F8X10K12 enhanced FVIII production after LV transduction.

IO infusion of G-F8X10K12-LV induced formation of inhibitory antibodies in HemA mice. (A) FVIII expression from new FVIII variant F8X10K12 in mice and cell culture. Both BDDF8X10K12 and F8/N6 were cloned into a lentiviral transgene backbone plasmid controlled under a ubiquitous promoter EF1α (pEF1α-F8X10K12 and pEF1α-F8/N6, respectively). HemA mice were hydrodynamically injected with pEF1α-F8X10K12 (n = 3) or pEF1α-F8/N6 (n = 9; 50 μg per animal) or sterile phosphate-buffered saline (PBS; mock; 2 mL per animal). Plasma samples were collected on day 4 postinjection, and hFVIII activity was measured by activated partial thromboplastin time assay (left). E-F8X10K12-LV and E-F8/N6-LV were generated to transduce 293T cells (multiplicity of infection, 100) on day 0. On day 5, hFVIII expression levels in 293T cells were detected by flow cytometry (right). (B) HemA mice were treated with IO infusion of G-F8X10K12-LV (2.2 × 106 ifu per animal) or G-F8/N6-LV (2.2 × 106 ifu per animal) or sterile PBS (mock; 20 μL per animal) on day 0. hFVIII levels in platelet lysates in G-F8X10K12-LV– or G-F8/N6-LV–treated or mock mice were measured by enzyme-linked immunosorbent assay on day 90. (C) Anti-FVIII antibodies in the plasma samples collected from the treated and mock mice were measured by Bethesda assay on day 120. Each symbol represents an individual animal. Data are expressed as mean ± standard deviation of the mean.
IO infusion of G-F8X10K12-LV induced formation of inhibitory antibodies in HemA mice. (A) FVIII expression from new FVIII variant F8X10K12 in mice and cell culture. Both BDDF8X10K12 and F8/N6 were cloned into a lentiviral transgene backbone plasmid controlled under a ubiquitous promoter EF1α (pEF1α-F8X10K12 and pEF1α-F8/N6, respectively). HemA mice were hydrodynamically injected with pEF1α-F8X10K12 (n = 3) or pEF1α-F8/N6 (n = 9; 50 μg per animal) or sterile phosphate-buffered saline (PBS; mock; 2 mL per animal). Plasma samples were collected on day 4 postinjection, and hFVIII activity was measured by activated partial thromboplastin time assay (left). E-F8X10K12-LV and E-F8/N6-LV were generated to transduce 293T cells (multiplicity of infection, 100) on day 0. On day 5, hFVIII expression levels in 293T cells were detected by flow cytometry (right). (B) HemA mice were treated with IO infusion of G-F8X10K12-LV (2.2 × 106 ifu per animal) or G-F8/N6-LV (2.2 × 106 ifu per animal) or sterile PBS (mock; 20 μL per animal) on day 0. hFVIII levels in platelet lysates in G-F8X10K12-LV– or G-F8/N6-LV–treated or mock mice were measured by enzyme-linked immunosorbent assay on day 90. (C) Anti-FVIII antibodies in the plasma samples collected from the treated and mock mice were measured by Bethesda assay on day 120. Each symbol represents an individual animal. Data are expressed as mean ± standard deviation of the mean.
Next, we delivered G-F8X10K12-LV and control G-F8/N6-LV in 2 groups of HemA mice, respectively. The average platelet FVIII antigen levels in mice treated with G-F8X10K12-LV only were significantly higher than those in mice treated with G-F8/N6-LV only on day 90 (Figure 5B), suggesting that F8X10K12 produced higher platelet FVIII levels. However, we found that all mice treated with G-F8X10K12-LV only (n = 9) generated anti-hFVIII antibodies, whereas no anti-FVIII antibody was detected in G-F8/N6-LV–treated mice (Figure 5C). It was suspected that significant portions of FVIII produced in G-F8X10K12-LV–treated mice were secreted into the plasma as a result of the enhanced expression and secretion of F8X10K12 protein, thus facilitating the generation of anti-FVIII antibodies.
G-F8/N6K12RH-LV combined with drug treatment produced FVIII with higher functional activity and significantly enhanced HemA phenotypic correction
To avoid the induction of high-titer anti-FVIII antibody production, we cloned F8/N6K12RH (Figure 1A) into the LV in an effort to increase synthesis and functional activity of FVIII without enhancing FVIII secretion. As shown in earlier sections, F8/N6 is a BDD FVIII variant that can enhance FVIII gene expression without induction of inhibitory anti-hFVIII antibodies (Figures 4 and 6). RH in F8/N6K12RH denotes an R1645H mutation20 for generating a more stable FVIII single-chain molecule and together with K12 mutations can significantly enhance the biological activity of FVIII compared with BDD FVIII (supplemental Figure 5). As expected, the mice treated with hydrodynamic injection of pEF1α-F8/N6K12RH had significantly higher plasma FVIII activity than mice treated with pEF1α-F8/N6 (Figure 6A).
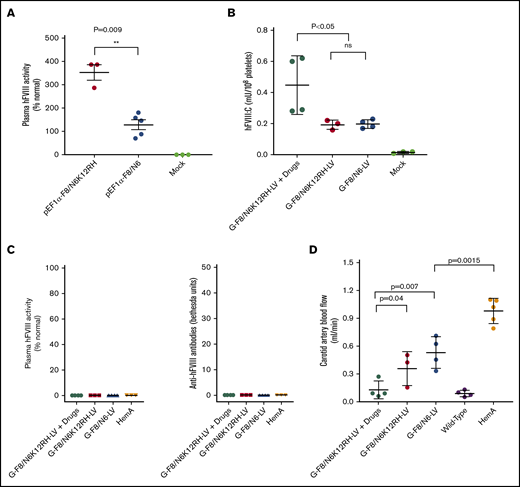
Phenotypic correction was achieved in transiently immunosuppressed HemA mice after IO infusion of G-F8/N6K12RH-LV. (A) F8/N6K12RH was cloned into a lentiviral transgene backbone plasmid controlled under a ubiquitous promoter EF1α (pEF1α-F8/N6K12RH). HemA mice were hydrodynamically injected with pEF1α-F8/N6K12RH (n = 3; 50 μg per animal) or pEF1α-F8/N6 (n = 5; 50 μg per animal) or sterile phosphate-buffered saline (PBS; n = 3; mock; 2 mL per animal). Plasma samples were collected on day 4 postinjection, and hFVIII activity was measured by activated partial thromboplastin time (aPTT) assay. (B) HemA mice were pretreated with drugs (Dex 4 times + anti-CD8α mAb 5 times) after IO infusion of G-F8/N6K12RH-LV (2.2 × 106 ifu per animal) or G-F8/N6-LV (2.2 × 106 ifu per animal) or sterile PBS (mock; 20 μL per animal) on day 0. hFVIII levels in platelet lysates in mice treated with G-F8/N6K12RH-LV plus drugs, G-F8/N6K12RH-LV only, or G-F8/N6-LV only and mock mice were measured by enzyme-linked immunosorbent assay on day 84. (C) Plasma samples were collected from the treated or mock mice on day 84. hFVIII activity and anti-FVIII antibodies were measured by aPTT and Bethesda assays, respectively. (D) HemA phenotypic correction in mice treated with G-F8/N6-LV only or G-F8/N6K12RH-LV plus drugs was also evaluated by measuring carotid artery blood flow rate on day 84. Each symbol represents an individual animal. Data are expressed as mean ± standard deviation of the mean. **P < .01. ns, not significant.
Phenotypic correction was achieved in transiently immunosuppressed HemA mice after IO infusion of G-F8/N6K12RH-LV. (A) F8/N6K12RH was cloned into a lentiviral transgene backbone plasmid controlled under a ubiquitous promoter EF1α (pEF1α-F8/N6K12RH). HemA mice were hydrodynamically injected with pEF1α-F8/N6K12RH (n = 3; 50 μg per animal) or pEF1α-F8/N6 (n = 5; 50 μg per animal) or sterile phosphate-buffered saline (PBS; n = 3; mock; 2 mL per animal). Plasma samples were collected on day 4 postinjection, and hFVIII activity was measured by activated partial thromboplastin time (aPTT) assay. (B) HemA mice were pretreated with drugs (Dex 4 times + anti-CD8α mAb 5 times) after IO infusion of G-F8/N6K12RH-LV (2.2 × 106 ifu per animal) or G-F8/N6-LV (2.2 × 106 ifu per animal) or sterile PBS (mock; 20 μL per animal) on day 0. hFVIII levels in platelet lysates in mice treated with G-F8/N6K12RH-LV plus drugs, G-F8/N6K12RH-LV only, or G-F8/N6-LV only and mock mice were measured by enzyme-linked immunosorbent assay on day 84. (C) Plasma samples were collected from the treated or mock mice on day 84. hFVIII activity and anti-FVIII antibodies were measured by aPTT and Bethesda assays, respectively. (D) HemA phenotypic correction in mice treated with G-F8/N6-LV only or G-F8/N6K12RH-LV plus drugs was also evaluated by measuring carotid artery blood flow rate on day 84. Each symbol represents an individual animal. Data are expressed as mean ± standard deviation of the mean. **P < .01. ns, not significant.
Next, we generated and delivered G-F8/N6K12RH-LV (Figure 1B) to HemA mice with combined drug treatment. As expected, the average platelet FVIII antigen levels in mice treated with G-F8/N6K12RH-LV only and those treated with G-F8/N6-LV only were similar. However, the platelet FVIII levels in mice treated with G-F8/N6K12RH-LV plus drugs were significantly higher than those in mice treated with G-F8/N6K12RH-LV only and G-F8/N6-LV only on day 84 (Figure 6B), suggesting that the combined drug treatment enhanced the production and maintenance of higher platelet FVIII levels. There was neither detectable plasma FVIII activity nor anti-FVIII inhibitors in blood of the treated mice on day 84 (Figure 6C). Phenotypic correction in the treated mice was next evaluated by the blood flow rate change in the FeCl3-induced carotid artery injury mouse model. The average flow rates were compared among 5 mouse groups; rates in untreated wild-type mice were the same or lower than those in mice treated with G-F8/N6K12RH-LV plus drugs, followed by mice treated with G-F8/N6K12RH-LV only, then mice treated with G-F8/N6-LV only, and finally untreated HemA mice on day 84 (Figure 6D), with several G-F8/N6K12RH-LV plus drug–treated mice exhibiting the same flow rates as those observed in wild-type mice. These results correlate well with the phenotypic correction data obtained from the tail bleeding assay (data not shown). The function of platelet FVIII in these 5 groups of mice was also evaluated using an ROTEM assay. The results were consistent with the flow rate data (Table 1). Interestingly, we saw that the maximum clot firmness was easily corrected by platelet FVIII, but it was more difficult to correct the clotting time. Our ROTEM assay results are comparable to results obtained from a previous ex vivo platelet-targeted gene therapy33 and a therapy using infused FVIII-expressing platelets34 for hemophilia.
ROTEM assay results using whole blood samples collected on day 140 after IO infusion of LVs to evaluate platelet FVIII function in HemA mice
| Mice | Mean ± standard deviation of mean | |||
|---|---|---|---|---|
| CT, s | CFT, s | α, degree | MCF, mm | |
| G-F8/N6K12RH-LV + drugs | 146 ± 4* | 104 ± 21 | 70 ± 4** | 66 ± 4 |
| G-F8/N6K12RH-LV | 388 ± 193 | 165 ± 123 | 62 ± 15* | 61 ± 7 |
| G-F8/N6-LV | 564 ± 367 | 261 ± 182 | 43 ± 13 | 62 ± 6 |
| Wild type | 164 ± 49* | 57 ± 10* | 79 ± 2** | 63 ± 1 |
| HemA | 956 ± 185* | 2370 ± 1150* | 4 ± 6*** | 26 ± 19* |
| Mice | Mean ± standard deviation of mean | |||
|---|---|---|---|---|
| CT, s | CFT, s | α, degree | MCF, mm | |
| G-F8/N6K12RH-LV + drugs | 146 ± 4* | 104 ± 21 | 70 ± 4** | 66 ± 4 |
| G-F8/N6K12RH-LV | 388 ± 193 | 165 ± 123 | 62 ± 15* | 61 ± 7 |
| G-F8/N6-LV | 564 ± 367 | 261 ± 182 | 43 ± 13 | 62 ± 6 |
| Wild type | 164 ± 49* | 57 ± 10* | 79 ± 2** | 63 ± 1 |
| HemA | 956 ± 185* | 2370 ± 1150* | 4 ± 6*** | 26 ± 19* |
Each assay was carried out for 1 h to obtain values of clotting time (CT), clot formation time (CFT), α angle, and maximum clot firmness (MCF). Statistical significance was analyzed based on relevance to G-F8/N6-LV–treated mouse group. Mice were treated with G-F8/N6K12RH-LV + drugs, G-F8/N6K12RH-LV, or G-F8/N6-LV. Untreated wild-type normal mice and HemA mice were used as positive and negative controls, respectively.
P < .05, **P < .01, ***P < .001.
Discussion
Recent clinical trials for HemA35,36 or HemB36-38 gene therapy using recombinant adeno-associated viral (AAV) vectors have shown promising results. However, lentiviral gene therapy can be superior in several aspects.39,40 First, AAV vectors persists as episomal, concatemerized vectors after in vivo gene transfer. Over time, transgene expression may decrease, and repeated dosing will be required. It remains to be shown whether repeated dosing is feasible or effective.41 In contrast, LVs integrate into the genome of transduced cells, so a single treatment with LV-mediated gene therapy may be sufficient. In addition, compared with AAV vectors, which have a transgene capacity of ∼4.5 kb, LVs accommodate much larger cDNAs, such as F8. Furthermore, LVs can efficiently transduce both dividing and nondividing cells, leading to the efficient transduction of primitive HSCs. Clinical trials of ex vivo HSC gene therapy recently showed remarkable therapeutic benefits and safety records39,42 in patients with severe genetic diseases, including X-linked severe combined immunodeficiency,43 Wiskott-Aldrich syndrome,44 spinal muscular atrophy,45 and sickle cell disease.46 Most significantly, genotoxicity by clonal dominance or significant clonal expansions from vector integration were not observed in these patients.
In the current study, our novel gene therapy protocol for HemA patients involved IO infusion of LVs expressing hFVIII driven by a megakaryocyte-specific promoter, leading to transgene FVIII expression in platelets. Compared with ex vivo gene therapy,47,48 IO infusion could efficiently transduce HSCs4 in situ to avoid the lengthy and complicated procedures of cell manipulation and transplantation.42 Most importantly, our approach avoids the requirement of myeloablative conditioning regimens that may result in concurrent thrombocytopenia in the posttransplantation period. Compared with other in vivo LV-mediated gene therapy approaches, such as IV delivery of LV-targeting gene expression in hepatocytes,49,50 IO delivery is unique. Slow injection of LVs into the bone marrow cavity avoids the systemic toxicity caused by IV delivery of LVs,12 AAV vectors,51,52 and other candidate gene therapy vectors.53,54 In addition, compared with hepatocyte-targeted LV gene transfer, which involves a high risk of antibody formation after FVIII is secreted into circulation, our platelet-targeted FVIII gene therapy generated FVIII stored in platelets and protected from uptake by antigen-presenting cells and neutralization by anti-FVIII inhibitors.4,55 During bleeding, FVIII was released by activated platelets on site to efficiently enhance clot formation. The bleeding phenotype is corrected in HemA mice with or without preexisting anti-FVIII inhibitors.4,56,57 Additionally, targeting FVIII expression to platelets for HemA gene therapy did not increase thrombotic risks58 but promoted immune tolerance in HemA mice.2 Therefore, our strategy of IO LV delivery targeting FVIII expression in platelets is highly promising for treatment of HemA patients, especially those with inhibitors.
However, immune responses associated with in vivo delivery of LVs may decrease LV transduction efficiency and eliminate transduced cells.9,10,59 We administered Dex to suppress the inflammatory cytokine production from IO injection of LVs and followed by treating the mice with anti-CD8α to transiently deplete the potential infiltration of cytotoxic CD8+ T cells, thus yielding higher LV transduction efficiency with higher and more persistent expression of both GFP reporter and FVIII genes. These agents in combination synergistically augmented LV transduction. Dex has also previously been shown to improve LV transduction by decreasing phagocytosis.60 Furthermore, whether anti-CD8α can help reduce a cytotoxic lymphocyte response or decrease interferon production and phagocytosis, thus enhancing LV transduction, will need to be investigated in future studies. However, we still observed a gradual decrease of GFP+ HSCs in combined drug–treated mice over time, suggesting that other immunomodulation regimens could potentially further enhance the persistence of transgene expression after IO delivery of LVs.
A modified FVIII with a higher expression level, enhanced bioactivity, and longer half-life would be desirable for successful gene therapy in HemA. Compared with human FVIII, canine FVIII61 is functionally more active, whereas porcine FVIII62 is expressed at higher levels. Novel recombinant hFVIII is therefore designed with mutations at specific sites, replacing amino acids with porcine- or canine-corresponding sequences to increase expression and functionality.20,61,63-65 Compared with mice treated with IO delivery of G-F8/N6-LV directed by a platelet-specific promoter, G-F8X10K12-LV–treated mice produced higher FVIII expression levels in platelets; however, anti-FVIII antibodies were induced, likely as a result of FVIII secretion into the circulation. The F8X10 variant27,28 with significantly enhanced secretion capability may not increase the storage of FVIII in platelets; therefore, it is not suitable for this particular application. It was also reported by Greene et al66 that high levels of FVIII expression in platelets could induce platelet apoptosis. Therefore, we next focused our platelet-targeted gene therapy for HemA on enhancing bioactivity only (F8/N6K12RH with an R1645H mutation20 and K12 mutations26 in the light chain) rather than expression levels. We demonstrated that hemophilia phenotypic correction in mice treated with G-F8/N6K12RH-LV plus combined drugs was significantly improved compared with G-F8/N6-LV–treated mice without the generation of anti-FVIII inhibitors.
As mentioned earlier, hFVIII ectopic expression in platelets provides an effective pathway for HemA gene therapy. FVIII stored in platelets is protected from neutralizing antibodies and also improves the bleeding phenotype, with as few as 1% to 5% of peripheral blood platelets containing FVIII.4,39,40,48 The therapeutic effect of platelet FVIII was characterized by several FVIII function assays. However, results from the chromogenic assay67,68 could not accurately predict therapeutic efficacy because of the very low levels of platelet FVIII, even in transgenic mice.68 Therefore, in addition to the chromogenic assay, we used an ROTEM assay to confirm the functional activity of FVIII-containing platelets. It is particularly interesting that by using an ROTEM assay, we found that maximum clot firmness could be easily corrected by G-F8/N6K12RH-LV plus drugs, G-F8/N6K12RH-LV only, and G-F8/N6-LV only. However, only mice treated with G-F8/N6K12RH-LV plus drugs achieved more complete correction of clotting time. This could be because platelet FVIII can only be released and functional after the platelet is activated. To examine platelet FVIII function in vivo, the tail clip assay4,69 was used to show that IO infusion of LVs significantly corrected bleeding phenotype compared with HemA control mice. However, therapeutic effects between a combined drug regimen plus LV treatment and LV-only treatment could not be clearly distinguished by this assay because of the high variance in the results. To more accurately evaluate the functional activity of platelet FVIII, the FeCl3 carotid artery thrombosis assay,70,71 where blood flow rate was monitored after local damage to the carotid artery,71 was used. Compared with the modified tail clipping assay, FeCl3-induced carotid thrombosis seemed to be more sensitive to monitor the role of platelet FVIII in enhancing local blood clot formation in arteries71,72 and capable of distinguishing therapeutic differences in the treatment groups. It was shown that platelet FVIII was effective in enhancing blood clotting and that a combination drug regimen with the new FVIII variant F8/N6K12RH achieved significant phenotypic correction of HemA in mice. Our results are consistent with a previous report demonstrating that platelet FVIII function is more potent in the arterial thrombosis model.71 Platelets are localized in the core of a thrombus with little flow. During platelet degranulation, FVIII is released to directly participate in the coagulation cascade. In this setting, the FVIII variant with enhanced activity is particularly efficacious in promoting blood clotting.
In conclusion, persistent FVIII gene expression in platelets was achieved by IO infusion of G-F8/N6K12RH-LV into HemA mice pretreated with a combination of Dex and anti-CD8α mAb. Transient suppression of immune responses to LVs and LV-transduced cells significantly increased LV transduction efficiency and produced long-term, stable transgene expression. In addition, F8/N6K12RH, with enhanced bioactivity, effectively improved the therapeutic effect of FVIII-containing platelets in a phenotypic correction assay. The function of platelet FVIII isolated from LV-treated mice was confirmed using an ROTEM assay. Phenotypic correction of hemophilia was clearly demonstrated in a carotid artery injury model. Taken together, these results indicate that this new approach may provide a readily translatable treatment for hemophilia in large animals and clinical trials.
Requests for data sharing should be e-mailed to the corresponding author, Carol H. Miao (e-mail: miao@u.washington.edu).
Acknowledgments
This work was supported by grants from the National Heart, Lung, and Blood Institute, National Institutes of Health (R01 HL134321 [C.H.M.], R01 HL123326 [C.H.M.], and R01 HL132557 [M.P.]).
Authorship
Contribution: X.W. designed and performed experiments, analyzed data, and participated in the writing of the paper; R.Y.F., C.L., C.-Y.C., J.F., and J.Z. performed research; B.A.K., W.X., and L.L. helped with the revision of the paper; M.P. provided research advice and revised the paper; and C.H.M. designed the project, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: W.X. holds equity in Ivygen Corporation. The remaining authors declare no competing financial interests.
Correspondence: Carol H. Miao, Center for Immunity and Immunotherapies, Seattle Children’s Research Institute, 1900 Ninth Ave, C9S-7, Seattle, WA 98101; e-mail: miao@u.washington.edu.

