

Key Points
KIT exon 17 mutation is a poor prognostic factor in AML patients with RUNX1-RUNX1T1, but not in those with CBFB-MYH11.
NRAS mutation is a poor prognostic factor in AML patients with CBFB-MYH11.

Abstract
The prognostic impact of KIT mutation on core-binding factor acute myeloid leukemia (CBF-AML) remains controversial. We registered 199 newly diagnosed de novo CBF-AML patients, aged 16 to 64 years, who achieved complete remission. They received 3 courses of high-dose cytarabine therapy and no further treatment until hematological relapse. Mutations in exons 8, 10-11, and 17 of the KIT gene were analyzed. Furthermore, we analyzed mutations in 56 genes that are frequently identified in myeloid malignancies and evaluated minimal residual disease (MRD). The primary end point was relapse-free survival (RFS) according to KIT mutations. The RFS in KIT-mutated patients was inferior to that in unmutated patients (hazard ratio, 1.92; 95% confidence interval, 1.23-3.00; P = .003). Based on subgroup analysis, KIT mutations had a prognostic impact in patients with RUNX1-RUNX1T1, but not in those with CBFB-MYH11, and only exon 17 mutation had a significant prognostic impact. Multivariate Cox regression analysis with stepwise selection revealed that the KIT exon 17 mutation and the presence of extramedullary tumors in patients with RUNX1-RUNX1T1, and loss of chromosome X or Y and NRAS mutation in patients with CBFB-MYH11 were poor prognostic factors for RFS. MRD was evaluated in 112 patients, and it was associated with a poorer RFS in the patients with CBFB-MYH11, but not in those with RUNX1-RUNX1T1. These results suggested that it is necessary to separately evaluate AML with RUNX1-RUNX1T1 or CBFB-MYH11 according to appropriate prognostic factors. This study was registered at www.umin.ac.jp/ctr/ as #UMIN000003434.
Introduction
Acute myeloid leukemia (AML) with RUNX1-RUNX1T1 or CBFB-MYH11 is categorized into a favorable cytogenetic risk group, and allogeneic hematopoietic stem cell transplantation (HSCT) is not generally recommended during the first complete remission (CR).1 However, several prognostic factors, including genetic alterations, have been demonstrated.2-8 In particular, KIT mutation has been suggested to be associated with a poor prognosis in AML patients with RUNX1-RUNX1T1 or CBFB-MYH11.3,5,9-12 Many types of KIT mutations have been identified in cancer cells, but there are 3 mutation hot-spots (exon 8, exon 10-11, and exon 17) in AML.3,10,12-14 Several groups previously reported that KIT mutation was a poor prognostic factor for overall survival (OS), event-free-survival, and/or relapse-free-survival (RFS) in AML with RUNX1-RUNX1T1 or CBFB-MYH11.3,10,12,15,16 On the other, some groups reported that KIT mutation was not associated with the long-term prognosis.1,17-19 This controversy may be caused by several study limitations such as the prognostic relevance being mostly evaluated retrospectively, not all types of KIT mutations being evaluated, and the analyzed patient number being insufficient for statistical power. Furthermore, the prognostic impact of recently identified recurrent mutations, such as ASXL1, ASXL2, and ZBTB7A, on AML with RUNX1-RUNX1T1 or CBFB-MYH11 remains unclear. We therefore conducted a prospective, multicenter cooperative study (Japan Adult Leukemia Study Group [JALSG] core-binding factor [CBF]-AML209-KIT) to evaluate the prognostic impact of KIT mutation in AML patients with RUNX1-RUNX1T1 or CBFB-MYH11 who were treated using the same high-dose cytarabine (HiDAC) regimen. Furthermore, we evaluated the frequency and clinical relevance of other gene mutations and prognostic impact of minimal residual disease (MRD).
Methods
Patients
Patients aged 16 to 64 years old with newly diagnosed de novo AML according to the World Health Organization 2008 classification, and an Eastern Cooperative Oncology Group performance status of 2 or lower were eligible for enrollment if they had a RUNX1-RUNX1T1 or CBFB-MYH11 chimeric transcript and achieved CR within 2 courses of standard induction therapy. All patients in this study were registered in the JALSG registration study after being diagnosed with AML and were treated using a standard dose of idarubicin + cytarabine or daunorubicin + cytarabine for induction therapy, as shown in supplemental Table 1. Other inclusion criteria were: serum alanine aminotransferase or serum aspartate aminotransferase level up to 2.5 times the institutional upper limit of normal; serum bilirubin up to 2.0 mg/dL; serum creatinine level up to 1.5 times the institutional upper limit of normal; left ventricular ejection fraction greater than 50% on ultrasound echocardiography; or PaO2 greater than 60 Torr or SpO2 greater than 90% under room air. We excluded patients with secondary AML, those with a history of hematological abnormalities before registration, those with other types of malignant tumors, those with a history of craniotomy, and those with a history of receiving whole brain radiation to have a more uniform patient background and to exclude safety concerns associated with previous treatments.
We also excluded patients with cardiac dysfunction corresponding to either of the following: Patients who need to use cardiac pacemakers, those with a complete left bundle branch block, those with 2 branch blocks, those with ventricular or atrial tachyarrhythmia requiring treatment, those with a digestive tract ulcer of A2 stage or higher, those with uncontrolled diabetes mellitus, those with a fasting blood glucose level ≤200 that could not be maintained by insulin administration, and those with active uncontrolled infections. Written informed consent was received from all patients. The protocol was approved by the ethics committees of all participating institutions. This study was registered in the UMIN Clinical Trials Registry (UMIN000003434, http://www.umin.ac.jp/ctr/).
Treatments
All patients received 3 courses of HiDAC therapy (2 g/m2 by 3-hour infusion every 12 hours for 5 days), as previously reported.20,21 For patients older than 60 years of age, 1 dose of cytarabine was reduced to 1.5 g/m2. We recommended that patients be hospitalized in the lower than NASA Class 10 000 clean room during treatment. Best supportive care, including administration of antibiotics and platelet transfusion, was performed if indicated. When patients had life-threatening documented infections during neutropenia, the use of granulocyte colony-stimulating factor was permitted. Bone marrow (BM) examination was performed to confirm CR before each course and at the end of the last course. After the completion of 3 courses of HiDAC therapy, patients did not receive further chemotherapy, immunotherapy, or HSCT until hematological relapse was observed. If patients developed hematological relapse, the best treatment, including HSCT, was applied at each institute.
Cytogenetic and molecular analyses
Cytogenetic G-banding analysis was performed using standard methods at each institute. Chimeric gene transcripts of RUNX1-RUNX1T1 and CBFB-MYH11 were centrally quantified by the real-time quantitative polymerase chain reaction (RT-qPCR) method using BM or peripheral blood samples at diagnosis according to a previous report.22 FLT3-ITD mutation was centrally examined by the PCR method; genomic PCR was performed and the amplified products were subjected to agarose gel electrophoresis as previously reported.23 These results were immediately reported to each institute. Residual DNA and RNA samples were preserved at the JALSG sample storage center. Mutations in exons 8, 10, 11, and 17 in the KIT gene were analyzed using the preserved DNA extracted from AML cells at diagnosis, as previously reported.24 We also analyzed mutations in FLT3, NPM1, CEBPA, NRAS, TP53, WT1, and IDH1 genes, and partial tandem duplication of the KMT2A gene (KMT2A-PTD) in 198 patients (supplemental Table 2). In addition, we analyzed mutations in 49 other genes in 170 patients using the TruSight Myeloid Sequencing Panel (Illumina, San Diego, CA), as previously reported (supplemental Table 2).8,25
Assessment of MRD
The chimeric transcript level of RUNX1-RUNX1T1 or CBFB-MYH11 using BM samples was evaluated after the hematological recovery from the third course of HiDAC therapy by RT-qPCR, as previously reported.22 Because the lower detection limit of RUNX1-RUNX1T1 and CBFB-MYH11 transcripts was 50 copies/μg RNA in our system, we defined MRD as positive if each transcript was ≥50 copies/μg RNA. To avoid interfering with protocol treatment, the results of gene mutations and MRD levels were not disclosed to institutes until 2 years after registration.
Definitions and study end points
Relapse after CR was defined as the presence of at least 1 of the following: reappearance of leukemic blasts in the peripheral blood, recurrence of more than 5% blasts in BM not attributable to any other cause, such as BM regeneration after chemotherapy, and development of extramedullary leukemia. We did not include chimeric transcript levels of RUNX1-RUNX1T1 or CBFB-MYH11 in the definition of CR or relapse. OS was defined as the time from the start date of induction therapy to death from any cause or last follow-up. RFS was defined as the time from the date of CR to relapse or death of any cause or the last follow-up.
The primary end point was RFS in AML patients with RUNX1-RUNX1T1 or CBFB-MYH11 according to KIT mutations. Secondary end points were OS according to KIT mutations, clinical relevance of the genetic alterations, prognostic impact of known prognostic factors, and MRD levels after the completion of therapy.
Statistical analysis
This study was prospectively powered to demonstrate a lower RFS of AML patients with RUNX1-RUNX1T1 or CBFB-MYH11 harboring KIT mutations than in those without them. With a sample size of 175 patients, the study had a power >90% at a 5% level of significance by the log-rank test if the incidence of KIT mutation in AML with RUNX1-RUNX1T1 or CBFB-MYH11 was 25%, and the 2-year RFS of patients with and without KIT mutations was 35% and 60%, respectively, according to previous reports.9,10,15,26 The RFS and OS were estimated by the Kaplan-Meier method; differences in survival distributions were evaluated using the log-rank test. Differences in continuous variables were analyzed by the Mann-Whitney U test for distribution between 2 groups. Analysis of frequencies was performed using Fisher's exact test for 2 × 2 tables or Pearson's χ2 test for larger tables. The prognostic significance of the clinical variables was assessed using the Cox proportional hazards model. We also attempted to construct a prognostic factor model in CBF-AML by adding a molecular type that stratifies the prognosis of CBF-AML. Factors that independently affect disease-free survival were narrowed down using the stepwise method and multivariate analysis using the Cox proportional hazard model. Factors that were candidates for model construction were factors whose P < .1 in the univariate analysis. Two-sided P < .05 was considered significant. Analyses were performed using Stata version 13·1 (StataCorp, College Station, TX).
One planned interim analysis for the primary end point was to be performed 1 year after the 100th patient was enrolled; this analysis took place independently of the study secretariat in March 2014, and the JALSG data and safety monitoring board made the decision to continue this study. Significance for the primary end point followed the O’Brien-Fleming method to maintain a 5% level of significance. Allogeneic transplantation was performed during the first remission period for 3 patients, and they were treated as deviations and censored at the time of transplantation in RFS analysis, including the multivariate analysis.
Results
Enrollment
Between May 2010 and September 2014, 203 patients from 85 institutes were enrolled. Four patients were excluded: 3 did not fulfill the eligibility criteria and 1 received other treatment (Figure 1). Thus, 199 patients consisting of 132 (66.3%) patients with RUNX1-RUNX1T1 and 67 (33.7%) with CBFB-MYH11 were included in the primary analysis.
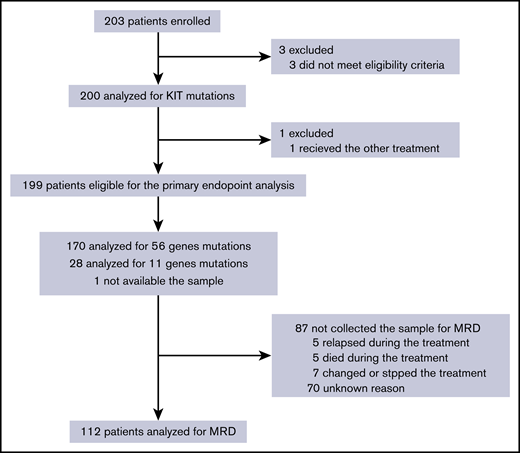
CONSORT flow diagram. The primary end point, relapse-free survival, was evaluated in 199 eligible patients. Prognostic analysis of MRD was performed on 112 patients whose samples were collected.
CONSORT flow diagram. The primary end point, relapse-free survival, was evaluated in 199 eligible patients. Prognostic analysis of MRD was performed on 112 patients whose samples were collected.
KIT mutations and patient characteristics
KIT mutations were identified in 63 of the 199 patients (31.7%): 42 of 132 (31.8%) and 21 of 67 (31.3%) patients with RUNX1-RUNX1T1 and CBFB-MYH11, respectively (Table 1; supplemental Table 3). A total of 68 mutations were identified in the 63 patients with KIT mutations and mutation in exon 17 was the most frequently identified (50/68, 73.5%), followed by that in exon 8 (14/68, 20.6%) and in exons 10-11 (4/68, 5.9%) (supplemental Table 4). KIT mutation in exon 8 was more frequent in AML with CBFB-MYH11 (9/24, 37.5%) than in that with RUNX1-RUNX1T1 (5/44, 11.4%) (P = .014). Although mutation at the N822 residue in exon 17 was identified in 13 of 44 (29.5%) KIT mutations of the patients with RUNX1-RUNX1T1, no patient with CBFB-MYH11 had this mutation (P = .008); however, mutation at the D816 residue was equally identified in patients with RUNX1-RUNX1T1 (21/44, 47.7%) and CBFB-MYH11 (13/24, 54.1%). Patient characteristics according to KIT mutation are presented in Table 1 and supplemental Table 4. The median BM blast percentage and white blood cell (WBC) index27 in the patients with RUNX1-RUNX1T1 were higher in the KIT-mutated patients than in the unmutated patients. G-banding karyotype analysis was performed on 197 patients. An additional cytogenetic abnormality was observed in 126 patients. However, there was no significant difference in additional cytogenetic abnormalities between KIT-mutated and unmutated patients (supplemental Table 5).
Patient characteristics according to KIT mutation
| Characteristic | All patients (n = 199) | KIT unmutated (n = 136) | KIT mutated (n = 63) | P |
|---|---|---|---|---|
| Chimera transcript type, n (%) | 1.00 | |||
| RUNX1-RUNX1T1 | 132 (66) | 90 (66) | 42 (67) | |
| CBFB-MYH11 | 67 (34) | 46 (34) | 21 (33) | |
| Sex, n (%) | .53 | |||
| Male | 125 (63) | 81 (61) | 42 (67) | |
| Female | 74 (37) | 55 (39) | 21 (33) | |
| Age, y | .72 | |||
| Median | 41 | 41 | 41 | |
| Range | 16-64 | 17-64 | 16-64 | |
| WBC, ×109/L | .12 | |||
| Median | 9.6 | 8.5 | 12.1 | |
| Range | 0.80-287.0 | 0.80-287.0 | 1.84-192.4 | |
| BM blasts, % | <.001 | |||
| Median | 61.2 | 53.8 | 73.5 | |
| Range | 10.8-97.0 | 10.8-96.5 | 26.1-97.0 | |
| WBC index* | .002 | |||
| Median | 4.73 | 3.35 | 6.11 | |
| Range | 0.35-41.3 | 0.35-41.3 | 0.93-29.6 | |
| Extramedullary tumor, n (%) | 20 (10) | 13 (10) | 7 (11) | .80 |
| CD19 expression, n/N (%) | 68/192 (35) | 49/129 (38) | 19/63 (30) | .34 |
| CD56 expression, n/N (%) | 91/193 (47) | 56/130 (43) | 35/63 (56) | .13 |
| Induction therapy, n (%) | .27 | |||
| Daunorubicin base | 68 (34) | 43 (32) | 25 (40) | |
| Idarubicin base | 131 (66) | 93 (68) | 38 (60) | |
| Induction cycle, n (%) | 1.00 | |||
| 1 course | 187 (94) | 128 (94) | 59 (93) | |
| 2 courses | 12 (6) | 8 (6) | 4 (6) | |
| Additional cytogenetic abnormalities (n = 197), n/N (%) | ||||
| Loss of X/Y | 78/197 (40) | 55/135 (40) | 23/62 (38) | .64 |
| Trisomy 8 | 7/197 (4) | 4/135 (3) | 3/62 (5) | .68 |
| Trisomy 22 | 18/197 (9) | 11/135 (8) | 7/62 (11) | .60 |
| del(9q) | 14/197 (7) | 12/135 (9) | 2/62 (3) | .23 |
| del(7q)/-7 | 4/197 (2) | 3/135 (2) | 1/62 (2) | 1.00 |
| Complex | 14/197 (7) | 7/135 (5) | 7/62 (11) | .14 |
| Characteristic | All patients (n = 199) | KIT unmutated (n = 136) | KIT mutated (n = 63) | P |
|---|---|---|---|---|
| Chimera transcript type, n (%) | 1.00 | |||
| RUNX1-RUNX1T1 | 132 (66) | 90 (66) | 42 (67) | |
| CBFB-MYH11 | 67 (34) | 46 (34) | 21 (33) | |
| Sex, n (%) | .53 | |||
| Male | 125 (63) | 81 (61) | 42 (67) | |
| Female | 74 (37) | 55 (39) | 21 (33) | |
| Age, y | .72 | |||
| Median | 41 | 41 | 41 | |
| Range | 16-64 | 17-64 | 16-64 | |
| WBC, ×109/L | .12 | |||
| Median | 9.6 | 8.5 | 12.1 | |
| Range | 0.80-287.0 | 0.80-287.0 | 1.84-192.4 | |
| BM blasts, % | <.001 | |||
| Median | 61.2 | 53.8 | 73.5 | |
| Range | 10.8-97.0 | 10.8-96.5 | 26.1-97.0 | |
| WBC index* | .002 | |||
| Median | 4.73 | 3.35 | 6.11 | |
| Range | 0.35-41.3 | 0.35-41.3 | 0.93-29.6 | |
| Extramedullary tumor, n (%) | 20 (10) | 13 (10) | 7 (11) | .80 |
| CD19 expression, n/N (%) | 68/192 (35) | 49/129 (38) | 19/63 (30) | .34 |
| CD56 expression, n/N (%) | 91/193 (47) | 56/130 (43) | 35/63 (56) | .13 |
| Induction therapy, n (%) | .27 | |||
| Daunorubicin base | 68 (34) | 43 (32) | 25 (40) | |
| Idarubicin base | 131 (66) | 93 (68) | 38 (60) | |
| Induction cycle, n (%) | 1.00 | |||
| 1 course | 187 (94) | 128 (94) | 59 (93) | |
| 2 courses | 12 (6) | 8 (6) | 4 (6) | |
| Additional cytogenetic abnormalities (n = 197), n/N (%) | ||||
| Loss of X/Y | 78/197 (40) | 55/135 (40) | 23/62 (38) | .64 |
| Trisomy 8 | 7/197 (4) | 4/135 (3) | 3/62 (5) | .68 |
| Trisomy 22 | 18/197 (9) | 11/135 (8) | 7/62 (11) | .60 |
| del(9q) | 14/197 (7) | 12/135 (9) | 2/62 (3) | .23 |
| del(7q)/-7 | 4/197 (2) | 3/135 (2) | 1/62 (2) | 1.00 |
| Complex | 14/197 (7) | 7/135 (5) | 7/62 (11) | .14 |
WBC index calculated in patients with RUNX1-RUNX1T1.
Landscape of gene mutations in CBF-AML
Identified gene mutations in analyzed AML patients are shown in Figure 2A. KIT mutation (31.7%) was the most frequently identified, followed by NRAS (21.7%), FLT3 (12.1%), and ASXL2 (11.8%) mutations in AML with RUNX1-RUNX1T1 or CBFB-MYH11; however, the mutation status was different between AML with RUNX1-RUNX1T1 and CBFB-MYH11 (Figure 2B). ASXL2, ASXL1, RAD21, and ZBTB7A mutations were more frequent in AML with RUNX1-RUNX1T1 than in that with CBFB-MYH11. In contrast, NRAS, KRAS, and FLT3-TKD mutations were more frequent in AML with CBFB-MYH11 than in that with RUNX1-RUNX1T1 (Figure 2B). Significantly overlapping mutations were observed between KIT and ASXL2, NRAS and KRAS, and CSF3R and ASXL1. Mutually exclusive mutations were observed between KIT and NRAS and KIT and ZBTB7A (Figure 2C-E; supplemental Figure 1).
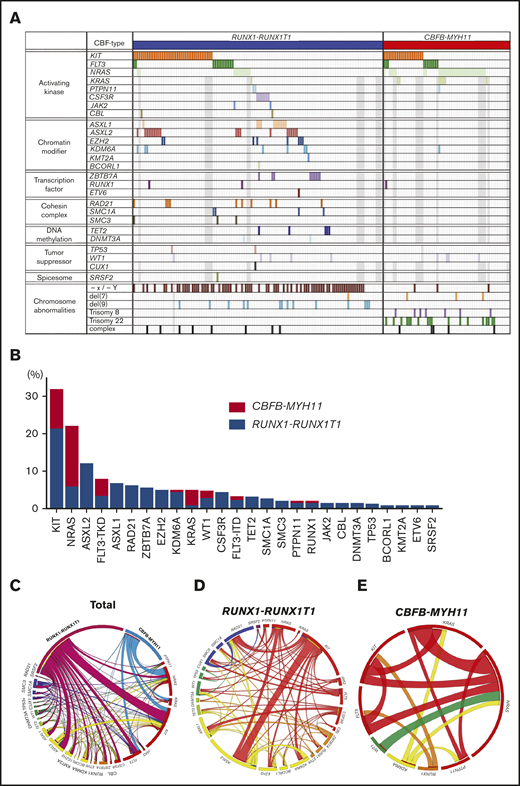
Mutation landscape of AML with RUNX1-RUNX1T1 or CBFB-MYH11. (A) Identified mutations in analyzed patients are shown. Gray boxes indicate the patients whose samples were not analyzed. (B) The frequency of recurrently mutated genes by CBF-AML fusion type is shown. (C) Circos plots illustrate the association of mutated genes in AML with RUNX1-RUNX1T1 or CBFB-MYH11. (D-E) Circos plots illustrate the association of mutated genes in AML with RUNX1-RUNX1T1 and AML with CBFB-MYH11. The width of the arches indicates the percentage of mutations.
Mutation landscape of AML with RUNX1-RUNX1T1 or CBFB-MYH11. (A) Identified mutations in analyzed patients are shown. Gray boxes indicate the patients whose samples were not analyzed. (B) The frequency of recurrently mutated genes by CBF-AML fusion type is shown. (C) Circos plots illustrate the association of mutated genes in AML with RUNX1-RUNX1T1 or CBFB-MYH11. (D-E) Circos plots illustrate the association of mutated genes in AML with RUNX1-RUNX1T1 and AML with CBFB-MYH11. The width of the arches indicates the percentage of mutations.
Prognostic impact of KIT mutation
The median follow-up period was 1566 days (range, 356-2453), and the 2-year RFS and OS in the entire cohort were 61.31% (95% confidence interval [95% CI]: 54.11-67.72) and 85.79% (95% CI: 80.09-89.97), respectively. By chimeric transcripts, the RFS and OS of patients with RUNX1-RUNX1T1 were not significantly different from those of patients with CBFB-MYH11: the 2-year RFS rates were 62.3% (95% CI, 53.3-70.0) and 59.6% (95% CI, 46.8-70.2) for RUNX1-RUNX1T1 and CBFB-MYH11, respectively (P = .88) (supplemental Figure 4).
The 2-year RFS rates were 48.6% (95% CI, 35.7-60.3) and 67.1% (95% CI, 58.5-74.4) in KIT-mutated and unmutated patients, respectively (hazard ratio [HR], 1.92; 95% CI, 1.23-3.00; P = .003 by log-rank test) (Figure 3A). Among the 3 types of KIT mutations, only the mutation in exon 17 had a lower prognostic impact on the RFS of CBF-AML patients (HR, 2.30; 95% CI, 1.45-3.64; P < .001) (Figure 3B). Furthermore, mutations at D816 and N822 residues had a significant prognostic impact, whereas the prognostic impact of other mutations in exon 17 was unclear because of the small number of patients (supplemental Figure 2).

RFS according to KIT mutation. (A) Kaplan-Meier estimates of RFS according to KIT mutation in 199 CBF-AML patients. Kaplan-Meier estimates of RFS in patients with (C) RUNX1-RUNX1T1 or (E) CBFB-MYH11. (B,D,F) Kaplan-Meier estimates of RFS according to the KIT mutation type.
RFS according to KIT mutation. (A) Kaplan-Meier estimates of RFS according to KIT mutation in 199 CBF-AML patients. Kaplan-Meier estimates of RFS in patients with (C) RUNX1-RUNX1T1 or (E) CBFB-MYH11. (B,D,F) Kaplan-Meier estimates of RFS according to the KIT mutation type.
Although there was no significant difference in RFS between the patients with RUNX1-RUNX1T1 and CBFB-MYH11 (supplemental Figure 3), based on subgroup analysis, KIT mutations had a prognostic impact on RFS only in patients with RUNX1-RUNX1T1: the 2-year RFS rates were 39.5% (95% CI, 24.7-53.9) and 72.8% (95% CI, 62.2-80.9) in KIT-mutated and unmutated patients, respectively (HR, 3.27; 95% CI, 1.90-5.64; P < .001) (Figure 3C). Furthermore, only the KIT exon 17 mutation had a lower prognostic impact on the RFS of AML patients with RUNX1-RUNX1T1 (HR, 3.82; 95% CI, 2.21-6.60; P < .001) (Figure 3D). In contrast, no KIT mutations affected the RFS of patients with CBFB-MYH11 (Figure 3E, F). KIT mutation was also associated with a poorer OS for AML with RUNX1-RUNX1T1, but not for that with CBFB-MYH11 (supplemental Figure 4), and the prognostic impact of each KIT mutation on OS was the same as that on RFS.
Prognostic factors in CBF-AML
We examined prognostic factors for RFS in 199 patients who were eligible for analysis of the primary end point. Multivariate Cox regression analysis with stepwise selection demonstrated that only the KIT exon 17 mutation was an independent poor prognostic factor for RFS in CBF-AML patients (HR, 2.42; 95% CI, 1.52-3.85; P < .001).
By CBF-subtype, KIT exon 17 mutation (HR, 4.17; 95% CI, 2.38-7.34; P < .001) and the presence of extramedullary tumors (HR, 3.85; 95% CI, 1.35-10.9; P = .011) in patients with RUNX1-RUNX1T1, and loss of chromosome X or Y (HR, 5.79; 95% CI, 1.21-27.6; P = .03) and NRAS mutation (HR, 2.38; 95% CI, 1.03-5.53; P = .04) in those with CBFB-MYH11 were identified as poor prognostic factors for RFS by multivariate analysis (Table 2).
Multivariate analysis for RFFS
| Variables | RUNX1-RUNX1T1 | CBFB-MYH11 | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| KIT exon17 mutation | 4.17 | 2.38-7.34 | <.001 | |||
| Extramedullary tumor | 3.85 | 1.35-10.93 | .011 | |||
| Loss of X/Y | 5.79 | 1.21-27.6 | .03 | |||
| NRAS mutation | 2.38 | 1.03-5.53 | .04 | |||
| Variables | RUNX1-RUNX1T1 | CBFB-MYH11 | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| KIT exon17 mutation | 4.17 | 2.38-7.34 | <.001 | |||
| Extramedullary tumor | 3.85 | 1.35-10.93 | .011 | |||
| Loss of X/Y | 5.79 | 1.21-27.6 | .03 | |||
| NRAS mutation | 2.38 | 1.03-5.53 | .04 | |||
We also analyzed the prognostic impact of gene mutation in 170 patients in whom 56 gene mutations were examined. By multivariate analysis, KIT mutation (HR, 3.56; 95% CI, 1.97-6.44; P < .001) and TET2 mutation (HR, 2.53; 95% CI, 1.37-11.5; P = .01) in patients with RUNX1-RUNX1T1, and NRAS mutation (HR, 2.36; 95% CI, 1.00-5.58; P = .05) in patients with CBFB-MYH11 were found to be poor prognostic factors for RFS (supplemental Table 5).
MRD analysis
We evaluated the MRD level after the completion of 3 courses of HiDAC therapy in 112 patients. MRD was positive in 32 of 75 (42.7%) and 16 of 37 (43.2%) patients with RUNX1-RUNX1T1 and CBFB-MYH11, respectively (Figure 4A). The RFS of patients with MRD was lower than that of those without MRD (HR, 2.39; 95% CI, 1.24-4.61; P = .009) (Figure 4B). Of note, the presence of MRD was associated with a poorer RFS in patients with CBFB-MYH11 (HR, 4.55; 95% CI, 1.20-17.2; P = .03), but not in those with RUNX1-RUNX1T1 (P = .11) (Figure 4C-D). The presence of MRD was significantly associated with KIT exon 17 mutation in the patients with RUNX1-RUNX1T1 (supplemental Table 6). Multivariate analysis of 112 patients demonstrated the presence of MRD (HR, 5.49; 95% CI, 1.43-21.0; P = .01) and NRAS mutation (HR, 3.93; 95% CI, 1.03-15.0; P = .05) to be poor prognostic factors for RFS in patients with CBFB-MYH11. In contrast, WBC count (>50 × 109/L) (HR, 5.57; 95% CI, 1.24-15.0; P = .03), KIT exon17 mutation (HR, 3.71; 95% CI, 1.60-8.64; P = .002), and FLT3-TKD mutation (HR, 3.39; 95% CI, 1.13-10.2; P = .03) were poor prognostic factors in patients with RUNX1-RUNX1T1 (supplemental Table 7).
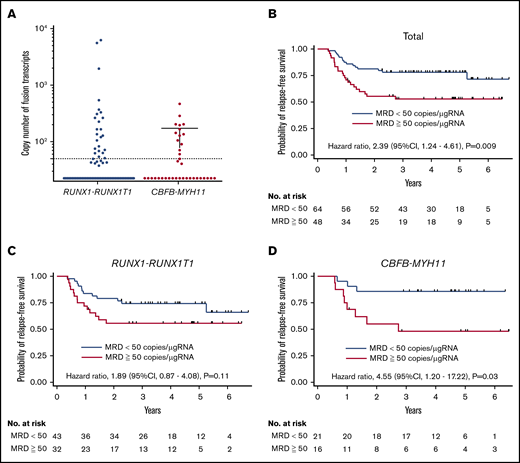
RFS according to the MRD level. (A) The RUNX1-RUNX1T1 or CBFB-MYH11 chimeric transcript level in each patient after the completion of 3 courses of HiDAC therapy is shown. (B) Kaplan-Meier estimates of RFS according to the MRD status in 112 CBF-AML patients. Kaplan-Meier estimates of RFS in patients with (C) RUNX1-RUNX1T1 or (D) CBFB-MYH11.
RFS according to the MRD level. (A) The RUNX1-RUNX1T1 or CBFB-MYH11 chimeric transcript level in each patient after the completion of 3 courses of HiDAC therapy is shown. (B) Kaplan-Meier estimates of RFS according to the MRD status in 112 CBF-AML patients. Kaplan-Meier estimates of RFS in patients with (C) RUNX1-RUNX1T1 or (D) CBFB-MYH11.
Discussion
The prognostic impact of KIT mutation is a major clinical concern in AML patients with RUNX1-RUNX1T1 and CBFB-MYH11 because controversial results were reported by several groups. In this large prospective study, we demonstrated that the adverse effects of KIT mutation were observed only in AML patients with RUNX1-RUNX1T1 and not in AML patients with CBFB-MYH11, although our study included a small number of patients with CBFB-MYH11 compared with the previous study.3 Furthermore, there was no significant difference in the RFS or OS between patients with RUNX1-RUNX1T1 and CBFB-MYH11 in this study. Because the results of mutation analysis were not reported to each institute until the completion of the protocol therapy and any further intervention was prohibited until hematological relapse, the present results are sufficient to evaluate the clinical relevance of KIT mutations and other molecular abnormalities in adult patients with CBF-AML treated using HiDAC. Moreover, in the patients with CBFB-MYH11, NRAS mutation was preferentially identified in KIT-unmutated patients, and had an adverse effect on RFS, whereas NRAS mutation did not affect the RFS of patients with RUNX1-RUNX1T1 (Table 2). The fusion transcripts RUNX1-RUNX1T1 and CBFB-MYH11 are not sufficient for leukemia development and additional driver mutations, such as KIT, FLT3, and RAS mutations, are required for its onset.28 However, the present study suggested that the prognostic impact of these driver mutations differs between patients with RUNX1-RUNX1T1 and CBFB-MYH11.
Several groups previously reported that the MRD level examined by the chimeric transcripts using RT-qPCR was useful for predicting the long-term prognosis of CBF-AML patients; however, there are several opinions regarding thresholds and time points for MRD assessment.1,19,29-32 Although we evaluated MRD after completing the 3-course consolidation therapy, MRD samples were not collected from 87 patients for several reasons, including disease progression (Figure 1). The RFS was significantly lower in the sample-uncollected patients than the collected patients among those with either RUNX1-RUNX1T1 or CBFB-MYH11. Therefore, the present study evaluated the clinical significance of MRD in patients who were able to maintain CR during consolidation therapy. Although further studies are required to clarify when MRD should be evaluated, our study demonstrated the importance of MRD for evaluating the prognosis of AML patients with CBFB-MYH11.
In conclusion, we clarified the prognostic impact of KIT mutation and the MRD status in adult AML patients with RUNX1-RUNX1T1 or CBFB-MYH11 who were treated using HiDAC, refining the concept of risk stratification of AML patients with RUNX1-RUNX1T1 and CBFB-MYH11. Other treatment strategies, including allogeneic HSCT during the first remission, or addition of gemtuzumab or ozogamicin to chemotherapy, should be considered for patients with a high risk of relapse identified by this study.33 The molecular risk groups presented in this study are amenable to routine diagnostic assessment, and provide a foundation for future clinical trials and research.
Acknowledgments
The authors thank the clinicians and leaders of the 85 institutions who entered their patients into the JALSG CBF-AML209-KIT study and provided the necessary data to make this study possible.
This study was supported by a grant from the Practical Research for Innovative Cancer Control from Japan Agency for Medical Research and Development, AMED (17ck0106251) (H. Kiyoi).
Authorship
Contribution: H. Kiyoi was the chief investigator of the trial; H. Kiyoi, Y.I., N.K., Y.A., I.M., Y. Miyazaki, and T.N. were involved in conception and study design; I.S., M.S., N. Dobashi, H.Y., N. Doki, A.T., T.K., S.K., H. Kanamori, N.I., A.K., Y. Moriuchi, N. Asada, D.H., K.T., T.S., M.H., T.T., Y.Y., and S.O. were involved in patient accrual and data acquisition; H. Kiyoi, Y.I., N.K., and N. Asou performed laboratory experiments and analysis; Y.I. and Y.A. performed the statistical analysis; H. Kiyoi, Y.I., N.K., S.O., Y. Miyazaki, and Y.A. were responsible for data analysis and interpretation; H. Kiyoi, Y.I., and N.K. were responsible for the preparation and writing of the manuscript; and all authors contributed to and approved the final manuscript.
Conflict-of interest disclosure: H. Kiyoi received research funding from Chugai Pharmaceutical Co. Ltd., Kyowa Hakko Kirin Co. Ltd., Zenyaku Kogyo Co. Ltd., FUJIFILM Corporation, Astellas Pharma Inc., Otsuka Pharmaceutical Co. Ltd., Nippon Shinyaku Co. Ltd., Eisai Co. Ltd., Pfizer Japan Inc., Takeda Pharmaceutical Co. Ltd., Novartis Pharma K.K., Sumitomo Dainippon Pharma Co. Ltd., Sanofi K.K., and Celgene Corporation; consulting fees from Astellas Pharma Inc., Amgen Astellas BioPharma K.K., and Daiichi Sankyo Co. Ltd.; honoraria from Bristol-Myers Squibb, Astellas Pharma Inc., and Novartis Pharma K.K. N. Dobashi received research funding from Pfizer Japan Inc., Chugai Pharmaceutical Co. Ltd., Astellas Pharma Inc., Kyowa Hakko Kirin Co. Ltd., Zenyaku Kogyo Co. Ltd., Eisai Co. Ltd., Otsuka Pharmaceutical Co. Ltd., Celgene Co., and Sysmex Co. N. Asou received research funding from Chugai Pharmaceutical Co. Ltd. and Toyama Chemical Co., Ltd., and consulting fees from SRL Inc. The remaining authors declare to competing financial interests.
A list of participating investigators and institutes of Japan Adult Leukemia Study Group, in addition to the authors, appears in the supplemental appendix.
Correspondence: Hitoshi Kiyoi, Department of Hematology and Oncology, Nagoya University Graduate School of Medicine, Tsurumai-cho 65, Showa-ku, Nagoya 466-8550, Japan; e-mail: kiyoi@med.nagoya-u.ac.jp.

