

Key Points
Undetectable MRD in PB or BM comparably predicts longer-term survival in patients with CLL receiving venetoclax.
The majority of patients with relapsed/refractory CLL who achieve undetectable MRD on venetoclax therapy do so within 24 months.

Abstract
The highly selective BCL2 inhibitor venetoclax achieves deep responses in patients with relapsed or refractory (R/R) chronic lymphocytic leukemia (CLL), including undetectable minimal residual disease (uMRD). We retrospectively reviewed 62 patients with CLL treated with venetoclax to investigate the performance of peripheral blood (PB) compared with bone marrow (BM) assessment of MRD; the kinetics, clinicopathological associations, and longer-term outcomes of uMRD attainment and recrudescence; and the ability of venetoclax dose escalation to deepen responses. Among 16 patients who achieved PB uMRD and had contemporaneous BM assessments, 13 (81%) had confirmed BM uMRD, and patients with PB uMRD had outcomes at least as favorable as those with BM uMRD for time to progression, overall survival, and MRD recrudescence. Excluding 2 patients lacking earlier assessment, the median time to PB uMRD was 18 (range, 5-26) months, with 90% of instances achieved by 24 months. There was no new PB uMRD attainment after 24 months without treatment intensification. The dominant association with earlier attainment of uMRD was concurrent rituximab (P = .012). Complex karyotype was associated with inferior uMRD attainment after 12 months of therapy (P = .015), and patients attaining uMRD whose disease harbored TP53 abnormalities demonstrated a trend toward earlier recrudescence (P = .089). Of patients who received venetoclax dose escalations, 4 (27%) of 15 achieved improvements in response. For patients with R/R CLL receiving venetoclax, PB uMRD commonly correlates with BM uMRD and is associated with a comparable longer-term prognosis. Concurrent rituximab augments uMRD attainment, but dose escalation and further treatment beyond 24 months infrequently deepen responses.
Introduction
Chronic lymphocytic leukemia (CLL) is the most prevalent leukemia in the Western world,1 and is characterized by constitutive overexpression of the prosurvival protein BCL2.2 Venetoclax (ABT-199/GC-0199) is an orally bioavailable, highly selective small-molecule inhibitor of BCL23 with significant efficacy in the treatment of CLL, including disease with adverse features, such as fludarabine (F)-refractoriness, bulky adenopathy, TP53 abnormalities, and unmutated IGHV.4-6
In patients with relapsed or refractory (R/R) CLL, venetoclax monotherapy achieves an overall response rate (ORR) of ∼80%, with a complete remission (CR) rate of 20%, including 5% with undetectable minimal residual disease (uMRD)4,7 measured by multiparameter flow cytometry.8 Similar responses are seen in patients with deletion 17p [del(17p)].5 Combination therapy using venetoclax plus rituximab is associated with an ORR of 86%, an improved CR rate of 51%, and uMRD achievement in 61% of patients, opening the possibility of time-limited therapy among deep responders.9,10 This observation, combined with more recently demonstrated improved progression-free survival (PFS) among patients treated with venetoclax-rituximab compared with bendamustine-rituximab,11 has led to paradigm shifts in CLL management in recent years. Two potential strategies for using targeted therapy in CLL have emerged: prolonging PFS without deep remission with Bruton tyrosine kinase inhibitors12 or pursuing deep remissions with associated prolonged PFS using venetoclax-based therapy.13 However, the longer-term implications of achieving uMRD among patients with R/R CLL treated with venetoclax remain unclear.
Unfortunately, there is a significant risk for secondary resistance with prolonged continuous exposure to venetoclax,14 as with other targeted therapies including ibrutinib,15 furthering the argument for time-limited treatment. To date, the best early predictor of durability of response is achievement of CR, and particularly uMRD.6 Suggested clinicopathological associations with inferior PFS in R/R CLL treated with venetoclax include TP53 dysfunction, bulky adenopathy, NOTCH1 mutations, prior B-cell receptor therapy failure,4,6 F-refractoriness, and complex karyotype (CK).16 Although clinical experience with BCL2 inhibitors continues to accumulate, many questions remain on how best to monitor and personalize therapy for individual patients based on their clinicopathological risk factors.
We have previously published an analysis of a cohort of patients with R/R CLL treated with continuous venetoclax in early-phase clinical trials.16 Many of these patients had regular peripheral blood (PB) and bone marrow (BM) MRD assessments while receiving venetoclax, using multiparameter flow cytometry as per European Research Initiative in CLL (ERIC) criteria.8 Using these data, we report here the performance of PB MRD monitoring compared with BM, the timing of uMRD attainment, the longer-term outcomes associated with uMRD attainment, the clinicopathological associations with uMRD attainment, the kinetics of MRD recrudescence, and the capacity for venetoclax dose escalation to deepen response.
Methods
Subjects
A retrospective analysis was performed on data from 62 patients with CLL treated with venetoclax who had objective responses at The Royal Melbourne Hospital and Peter MacCallum Cancer Centre from June 2011 to September 2018. All but 2 patients had been previously treated for CLL. Patients were enrolled on 1 of 3 venetoclax trials: M12-175 phase 1 study of venetoclax monotherapy (NCT01328626; 36 patients), M13-365 phase 1b study of venetoclax plus rituximab combination therapy (NCT01682616; 14 patients), or M13-982 phase 2 study of venetoclax monotherapy in del(17p) CLL (NCT01889186; 12 patients). Eligibility criteria and other details for each of these trials have been published.4,5,9 In all studies, patients received venetoclax 150 to 600 mg (mostly 400 mg) daily until disease progression or discontinuation for another reason. Patients on the M13-365 trial also received 6 doses of monthly rituximab (375 mg/m2 in month 1 and 500 mg/m2 in months 2-6) after completion of the dose ramp-up of venetoclax. All patients provided written informed consent, and study protocols were approved by local institutional review boards and conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines.
Clinical data
Baseline patient and disease characteristics were recorded at enrolment, including age, number of prior therapies, F-refractoriness (defined as primary failure to respond or disease progression within 6 months of F-based therapy17 ), presence of bulky adenopathy (defined as lymph nodes >5 cm on CT scan18 ), and genetic features, including del(17p), del(11q), CK (defined as ≥3 clonal chromosomal aberrations on conventional metaphase karyotype19,20 ), and TP53 mutation. TP53 variants were detected with a sensitivity of 5% variant allele frequency using targeted next-generation sequencing of all coding exons of TP53 (NM_000546.5). TP53 variants were assessed for pathogenicity by using a variety of curation sources, including the Genome Aggregation Database (https://gnomad.broadinstitute.org/), the Catalogue of Somatic Mutations in Cancer (https://cancer.sanger.ac.uk/cosmic), and the International Agency for Research on Cancer TP53 Database (http://p53.iarc.fr/). The best disease response to venetoclax, as assessed by clinical investigators in accordance with 2008 International Workshop in CLL (iwCLL) criteria,21 was documented. The details of all venetoclax dose escalations were collected. Disease progression on venetoclax was defined as per iwCLL guidelines.21
MRD assessment
Patients were serially monitored for MRD in PB and BM at clinician discretion, as no study protocol initially mandated MRD monitoring. MRD was quantified using multiparameter flow cytometry by ERIC methodology.8,22,23 Patients were recorded as having uMRD if there was less than 1 CLL cell detectable per 10 000 leukocytes (<0.01%) in either PB or BM (analyzing a minimum of 200 000 leukocytes), and patients were considered MRD positive if the MRD burden in all PB and BM assessments was at least 0.01%. Patients in whom no detectable CLL was found on flow cytometric assays with sensitivity inferior to less than 0.01% were deemed MRD indeterminate. In the analyses presented, patients whose deepest response was MRD indeterminate were conservatively characterized as MRD positive. Patients who had no MRD assessments because of early progression were considered MRD positive. Changes in MRD status during treatment were described using PB MRD as a result of the higher frequency of assessment. MRD recrudescence was defined as the first date of detection of MRD at least 0.01% in PB after prior achievement of uMRD in either PB or BM, or first date of iwCLL progression if this occurred without prior confirmation of MRD positivity. BM and PB samples were considered contemporaneous if their dates of collection were within 3 months.
Statistical analysis
The Kaplan-Meier method was used to estimate time to progression (TTP) and overall survival (OS) for patients receiving continuous venetoclax therapy, measured from date of trial entry to date of progression, death, drug cessation, or last follow-up on study. To describe outcomes on continuous therapy, TTP analysis considered iwCLL progression on drug as an event and censored patients who ceased drug (ceased drug in good response [n = 5]; ceased for other reasons: new diagnosis lung cancer and myelodysplastic syndrome [n = 1], withdrew consent [n = 1], ceased trial for allogeneic stem cell transplant [n = 1], or died in remission [n = 1], acute myocardial infarction in BM uMRD CR). OS analysis considered death from any cause as an event irrespective of treatment cessation. Landmark analyses were performed to compare TTP and OS for patients who did or did not achieve uMRD, using 24 months (at which time >90% of ultimate uMRD attainment had occurred) as the landmark. Patients without an event were censored at the date of last follow-up or the prespecified data cutoff date of 30 September 2018 if next follow-up occurred after this date and confirmed no intervening events. Estimates of proportional uMRD attainment were expressed as cumulative incidence. Associations between clinicopathological variables and time to uMRD achievement were analyzed using the Cox proportional hazard model to calculate cause-specific hazard ratios with an α level set at 0.05; progressive disease before dose escalation (PD) and death from other causes were considered competing risks and treated as censoring events. All data were analyzed using Stata 14.1 for Mac (StataCorp, College Station, TX) and GraphPad Prism version 6.0h for Mac (GraphPad Software, La Jolla, CA).
Results
Patient and treatment characteristics
Clinicopathological characteristics of the study cohort are shown in Table 1. The median age was 67 (range, 45-87) years. The median number of prior therapies was 3 (range, 0-12), with 2 previously untreated patients. At enrolment, the frequency of chemoimmunotherapy adverse disease risk factors were F-refractoriness (30/62; 48%), bulky adenopathy (28/62; 45%), TP53 abnormalities (34/58; 59%), and CK (15/42; 36%). Ninety percent of patients had 1 or more adverse characteristic before venetoclax therapy. No patients had received prior Bruton tyrosine kinase inhibitor or idelalisib therapy. Sixty-nine percent (43/62) of patients received an initial dose of at least 400 (median, 400; range, 100-600) mg/d. Concomitant rituximab was administered to the 14 (23%) of 62 patients within the M13-365 trial. Median survivor follow-up for the study population was 62 (range, 41-81+) months.
Factors associated with uMRD in the peripheral blood or bone marrow at study entry
| Variable | n | Association with PB/BM uMRD attainment | ||
|---|---|---|---|---|
| Cause-specific HR (95%CI) | P | |||
| Age ≥65 y | Y | 38 | 1.28 (0.57-2.87) | .558 |
| N | 24 | |||
| ≥4 prior lines of therapy | Y | 25 | 0.59 (0.24-1.44) | .240 |
| N | 37 | |||
| Fludarabine refractory | Y | 30 | 0.68 (0.29-1.57) | .359 |
| N | 32 | |||
| Bulky disease (>5 cm) | Y | 28 | 1.14 (0.50-2.61) | .752 |
| N | 34 | |||
| Rituximab combination therapy | Y | 14 | 2.86 (1.22-6.69) | .012 |
| N | 48 | |||
| Dose ≥400 mg | Y | 43 | 1.03 (0.43 – 2.47) | .947 |
| N | 19 | |||
| Del(17p) and/or TP53 mutation | Y | 34 | 0.58 (0.26-1.33) | .192 |
| N | 24 | |||
| Complex karyotype | Y | 15 | 0.58 (0.19-1.73) | .320 |
| N | 27 | |||
| Prior navitoclax | Y | 9 | 0.210 (0.03-1.59) | .095 |
| N | 53 | |||
| Variable | n | Association with PB/BM uMRD attainment | ||
|---|---|---|---|---|
| Cause-specific HR (95%CI) | P | |||
| Age ≥65 y | Y | 38 | 1.28 (0.57-2.87) | .558 |
| N | 24 | |||
| ≥4 prior lines of therapy | Y | 25 | 0.59 (0.24-1.44) | .240 |
| N | 37 | |||
| Fludarabine refractory | Y | 30 | 0.68 (0.29-1.57) | .359 |
| N | 32 | |||
| Bulky disease (>5 cm) | Y | 28 | 1.14 (0.50-2.61) | .752 |
| N | 34 | |||
| Rituximab combination therapy | Y | 14 | 2.86 (1.22-6.69) | .012 |
| N | 48 | |||
| Dose ≥400 mg | Y | 43 | 1.03 (0.43 – 2.47) | .947 |
| N | 19 | |||
| Del(17p) and/or TP53 mutation | Y | 34 | 0.58 (0.26-1.33) | .192 |
| N | 24 | |||
| Complex karyotype | Y | 15 | 0.58 (0.19-1.73) | .320 |
| N | 27 | |||
| Prior navitoclax | Y | 9 | 0.210 (0.03-1.59) | .095 |
| N | 53 | |||
CI, confidence interval; N, no; Y, yes.
Analytical correlation of PB and BM MRD assessment
MRD assessment was performed at least once for 59 (95%) of 62 patients, with 3 cases of early-onset Richter transformation not tested for MRD. With a total period of observation of 186 person-years, there were 684 MRD assays in PB (3.7 per person-year) and 199 (1 per person-year) in BM. For the 58 patients on study with more than 1 assessment, the median interval between PB MRD assessments was 3 (minimum, 1; mode, 2) months; the median interval of BM MRD assessments for the 49 repeatedly tested patients was 10 (minimum, 3; mode, 11) months. uMRD was confirmed in PB for 19 patients, and in BM for 21 patients. Twenty-six (42%) patients had confirmed uMRD in 1 or both compartments.
We interrogated the relationship between BM and PB MRD status for individual patients at the time of first achievement of PB uMRD. There were 7 patients who achieved uMRD in BM that could not be confirmed in PB because of the lack of a sensitive PB assessment to confirm disease below 10−4 (ie, MRD indeterminate in PB and confirmed uMRD in the BM because of the higher sensitivity of BM assay), such that patients were classified as PB MRD positive (n = 6), or detectable MRD (0.01%-1%) in the PB 6 months before the uMRD BM assessment without a contemporaneous PB assessment (n = 1). Of the 16 patients who achieved uMRD in the PB and who had contemporaneous BM assessments, 13 (81%) had uMRD confirmed in both compartments, with the 3 remaining cases having residual disease less than 0.1% in BM.
Timing of uMRD attainment
The temporal distribution of PB MRD status within the cohort during venetoclax therapy is shown in Figure 1A. The proportion of patients with deep responses (either MRD indeterminate or confirmed uMRD) was highest between 9 and 18 months receiving therapy. Excluding 2 cases of PB uMRD confirmed after ∼4 years without prior MRD assessment, the median time to PB uMRD was 18 (range, 5-26) months, with 90% of uMRD attainment occurring at or before 24 months (Figure 2B). Timing of BM uMRD was similar at a median of 18 (range, 4-31) months, excluding 3 late uMRD confirmations without prior assessment (94% attained ≤24 months). We next interrogated the trajectory of MRD burden beyond 24 months in the 30 patients continuing to receive venetoclax with subsequent informative PB MRD assessments. Ten (33%) had no change in MRD burden (7 stable uMRD; 3 stable positive 0.01%-1% MRD), whereas 18 (60%) had rising MRD and only 2 (7%) had falling MRD (supplemental Figure 1). The 2 cases of falling MRD with further venetoclax after 24 months both occurred after dose escalations (see lanes 1 and 4, Figure 5). Thus, in the absence of dose escalation, there were no instances of MRD clearance with additional venetoclax therapy after 24 months. Among the 36 patients who did not achieve PB or BM uMRD, 28 developed PD at a median time of 19 (range, 4-73) months, 4 remain on study with stable detectable MRD 1% or less (median follow-up, 52 [range, 41-63+] months), and 4 ceased drug for other reasons (withdrew consent [5 months], cease therapy in MRD-detectable CR [7 months], intercurrent lung cancer [11 months], and planned allogeneic stem cell transplant [30 months]). These data suggest that the majority of patients who ultimately achieve uMRD with venetoclax therapy do so within 24 months, and ongoing unaltered therapy beyond this time rarely eradicates persistent MRD.
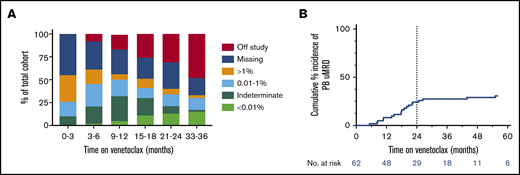
Kinetics of MRD during venetoclax therapy. (A) Serial peripheral blood MRD status during venetoclax therapy. (B) Cumulative incidence of attainment of uMRD in the peripheral blood. Dotted line shows 24 months on therapy, at which time 90% of ultimate peripheral blood uMRD was achieved, excluding 2 nonrepresentative late events (significant delay to MRD assessment).
Kinetics of MRD during venetoclax therapy. (A) Serial peripheral blood MRD status during venetoclax therapy. (B) Cumulative incidence of attainment of uMRD in the peripheral blood. Dotted line shows 24 months on therapy, at which time 90% of ultimate peripheral blood uMRD was achieved, excluding 2 nonrepresentative late events (significant delay to MRD assessment).

Outcomes after uMRD attainment in the peripheral blood and bone marrow. (A) Landmark analysis of time to iwCLL progression stratified by MRD status at 24 months in the peripheral blood and bone marrow. Patients who progressed or came off study before 24 months (at which time >90% of uMRD attainment had occurred) were excluded. (B) Landmark analysis of overall survival stratified by MRD status at 24 months in the peripheral blood and bone marrow. Patients who died or came off study before 24 months (at which time >90% of uMRD attainment had occurred) were excluded.
Outcomes after uMRD attainment in the peripheral blood and bone marrow. (A) Landmark analysis of time to iwCLL progression stratified by MRD status at 24 months in the peripheral blood and bone marrow. Patients who progressed or came off study before 24 months (at which time >90% of uMRD attainment had occurred) were excluded. (B) Landmark analysis of overall survival stratified by MRD status at 24 months in the peripheral blood and bone marrow. Patients who died or came off study before 24 months (at which time >90% of uMRD attainment had occurred) were excluded.
Outcomes after uMRD attainment
The longer-term outcomes of patients who achieved uMRD in PB and BM are shown in Figure 2A-B. In landmark analyses, achievement of uMRD in PB was associated with significantly prolonged TTP (median TTP PB uMRD vs no PB uMRD: not reached vs 55 months; P = .0043; hazard ratio [HR], 0.09; 95% CI, 0.01-0.73) and OS (median OS PB uMRD vs no PB uMRD: not reached vs 82 months; P = .033; HR undefined). Patients who attained uMRD in the BM demonstrated similarly prolonged TTP (median TTP BM uMRD vs no BM uMRD: not reached vs 58 months; P = .032; HR, 0.22; 95% CI, 0.05-1.00) and a trend toward prolonged OS (median OS BM uMRD vs no BM uMRD: not reached vs 82 months; P = .107; HR, 0.21; 95% CI, 0.03-1.70]; Figure 2A-B). Overall, these data demonstrate that the favorable outcomes associated with uMRD attainment are comparably predicted by PB or BM assessment.
Clinicopathological correlates with uMRD attainment
Considering PB and BM uMRD as equivalent, univariate analyses were performed to understand the patient and disease characteristics at enrolment associated with attainment of uMRD. The variables considered were age 65 years or older, 4 or more lines of prior therapy, F-refractoriness, bulky adenopathy, upfront administration of rituximab, venetoclax dose 400 mg/d or higher, del(17p), and/or TP53 mutation, CK, and prior exposure to BCL2 inhibitor therapy with navitoclax24 (Table 1). The dominant factor associated with likelihood to attain uMRD and earlier time to uMRD was concomitant rituximab (estimated cumulative incidence of uMRD at 24 months for rituximab vs no rituximab: 64% vs 23%; P = .012; HR, 2.86; 95% CI, 1.22-6.69; Figure 3A). For patients who continued receiving therapy at 12 months but had not yet attained uMRD, initial rituximab co-administration maintained a significant positive association with subsequent uMRD attainment (supplemental Table 1). Furthermore, there was an association with inferior uMRD attainment with ongoing therapy after 12 months with patients whose disease harbored CK (estimated cumulative incidence of uMRD at 24 months for non-CK vs CK: 37% vs 27%; P = .015; HR undefined; Figure 3B). There was a statistically nonsignificant inferior uMRD attainment after 12 months for patients whose disease harbored abnormalities in TP53 (estimated cumulative incidence of uMRD at 24 months for TP53 abnormality vs no TP53 abnormality: 50% vs 24%; P = .067; HR, 0.38; 95% CI, 0.13-1.11) or was F-refractory (estimated cumulative incidence of uMRD at 24 months for F-refractory vs non-F-refractory: 38% vs 27%; P = .061; HR, 0.30; 95% CI, 0.08-1.14; Figure 3C-D).

Clinicopathological associations with uMRD attainment. Five patients with late confirmation of uMRD (2 in PB, 3 in BM) without prior assessment were included. (A) Cumulative incidence of uMRD in the peripheral blood or bone marrow stratified by coadministration of rituximab or not. (B) Cumulative incidence of uMRD in the peripheral blood or bone marrow stratified by complex karyotype or not. (C) Cumulative incidence of uMRD in the peripheral blood or bone marrow stratified by TP53 abnormality or not. (D) Cumulative incidence of uMRD in the peripheral blood or bone marrow stratified by fludarabine refractoriness.
Clinicopathological associations with uMRD attainment. Five patients with late confirmation of uMRD (2 in PB, 3 in BM) without prior assessment were included. (A) Cumulative incidence of uMRD in the peripheral blood or bone marrow stratified by coadministration of rituximab or not. (B) Cumulative incidence of uMRD in the peripheral blood or bone marrow stratified by complex karyotype or not. (C) Cumulative incidence of uMRD in the peripheral blood or bone marrow stratified by TP53 abnormality or not. (D) Cumulative incidence of uMRD in the peripheral blood or bone marrow stratified by fludarabine refractoriness.
MRD recrudescence
MRD recrudescence in PB occurred at a median of 46 (range, 3-52) months, with no apparent plateau (Figure 4A). Similar to TTP and OS outcomes, time to MRD recrudescence was at least as favorable among patients achieving PB uMRD as those with uMRD confirmed in BM (median time to MRD recrudescence PB uMRD vs BM uMRD: 46 vs 38 months; P = .178) (Figure 4B). When stratified by uMRD attainment before and exactly at the cohort median (18 months) or later, there was no difference in time to recrudescence between early and late attainers (median time to recrudescence early vs late: 38 vs 52 months; P = .873; Figure 4C), although this analysis was limited by cohort size. Univariate analysis of the associations between the aforementioned clinicopathological variables and time to recrudescence was performed (supplemental Table 2). Although concurrent rituximab was associated with uMRD attainment, no significant difference was observed in time to recrudescence between patients who achieved uMRD with or without concurrent rituximab (median time to recrudescence, 46 vs 37 months, respectively; P = .638; HR, 0.71; 95% CI, 0.17-2.94). There was a trend toward earlier MRD recrudescence in patients whose disease harbored del(17p) and/or TP53 mutations (median time to recrudescence del(17p) and/or TP53 mutation vs no p53 abnormality: 37 vs 45 months; P = .089; HR, 3.48; 95% CI, 0.75-16.08; Figure 4D). Among the 10 patients with PB MRD recrudescence, iwCLL progression occurred in 5 (50%) cases at a median time from recrudescence of 17 (range, 14-27) months. Testing for the recently described BCL2 Gly101Val mutation14 was completed at progression in 4 (80%) of these 5 patients and was detected in all cases at a median variant allele frequency of 1.0% (range, 0.01%-12.02%).

Patterns of MRD recrudescence. (A) Time to MRD recrudescence (whole cohort). (B) Time to uMRD recrudescence stratified by time from achievement in the peripheral blood or time from achievement in the bone marrow. (C) Time to MRD recrudescence stratified by attainment of uMRD before or after the median time to uMRD attainment (18 months). (D) Time to MRD recrudescence stratified by TP53 abnormality or not.
Patterns of MRD recrudescence. (A) Time to MRD recrudescence (whole cohort). (B) Time to uMRD recrudescence stratified by time from achievement in the peripheral blood or time from achievement in the bone marrow. (C) Time to MRD recrudescence stratified by attainment of uMRD before or after the median time to uMRD attainment (18 months). (D) Time to MRD recrudescence stratified by TP53 abnormality or not.
Responses after venetoclax dose escalation
We interrogated the capacity of venetoclax dose escalation to deepen responses and attain uMRD. Fifteen patients underwent dose escalations for iwCLL progression (n = 2), nodal enlargement not meeting iwCLL criteria (n = 1), and dose escalation within phase 1/2 trials (n = 12; Figure 5). The final dose to which patients were escalated was 600 mg/d in 6 cases, 400 mg/d in 4 cases, and less than 400 mg/d in 5 cases. No patient was escalated to a dose above 600 mg/d.

Outcomes after treatment escalation Swimmers plot for patients who received escalated doses of venetoclax. CR, complete response; PR, partial response; SD, stable disease.
Outcomes after treatment escalation Swimmers plot for patients who received escalated doses of venetoclax. CR, complete response; PR, partial response; SD, stable disease.
The 2 patients with disease progression who received dose escalation were documented to have rising MRD despite escalation, and came off study within 6 months (lanes 9 and 11). The BCL2 Gly101Val mutation was identified in 1 of these poor responders at a low variant allele frequency (0.07%) at progression (lane 9). For patients receiving dose intensification for reasons other than disease progression, objective improvement in response occurred in 4 (31%) of 13 patients (lanes 1-3 and 6). There were 3 instances of new uMRD attainment. Among the 6 patients who underwent dose escalation above 400 mg/d, there were 2 instances of new uMRD attainment (lanes 2 and 3; escalations occurred at 14 and 23 months from trial entry, respectively), 1 instance of deepening of iwCLL response from stable disease to partial response followed by PD within 6 months (lane 6), 1 instance of stable partial response (lane 4), and 2 instances of rising MRD shortly followed by iwCLL progression (lanes 9 and 15). One patient maintained PB uMRD status attained before dose escalation, and thus does not represent deepening response (lane 5). These data argue that dose escalation can deepen responses and achieve uMRD in a minority of cases.
Discussion
The maturing clinical trial data with venetoclax invites reconsideration of the goals of treatment with targeted agents in CLL. For the first time, clinicians have available a small-molecule inhibitor capable of reliably eliminating evidence of detectable MRD at conventional levels (10−4). Analogous to long-term follow-up data with chemoimmunotherapy in CLL25 and imatinib in chronic myeloid leukemia,26 it is apparent that uMRD achievement predicts a better clinical outcome in the longer term and should be the goal of venetoclax therapy. However, open questions remain about the compartment in which MRD is assessed most informatively, the kinetics of MRD attainment, and the clinicopathological correlates of uMRD achievement and recrudescence. In this analysis, we have taken steps toward understanding the role of uMRD in the longer-term outcomes of patients receiving venetoclax for R/R CLL.
Although our data are based on retrospective analysis, the comprehensive nature of MRD monitoring during such a prolonged period in this setting is unique and informative. A limitation of the work is the clinician-determined regimen of MRD testing, as this was neither protocol-specified nor standardized at the time. This is partially mitigated by the frequency with which testing was performed and our conservative definition of uMRD, such that patients with undetectable CLL who were assessed using insufficiently sensitive flow cytometric methods were deemed MRD positive for the purposes of analysis. The important prognostic implications of uMRD we report are consistent with other recently published trials.6,9,10
Debate has persisted over the role of BM biopsy in the monitoring of CLL.27 Our data show that MRD status in PB not only closely correlates to BM MRD status analytically but also has similar implications for longer-term outcomes. Although prospective trials of larger cohorts may definitively clarify the frequency and implications of PB/BM MRD discordance, these data argue that PB MRD monitoring could be used as a screen within clinical trials, with BM assessment reserved for patients who attain uMRD in the PB. Furthermore, the analytical and prognostic concordance we describe suggests PB MRD monitoring may be sufficient for clinical practice outside of clinical trials. Although alternative measures of MRD status such as polymerase chain reaction may offer greater sensitivity, our data support the assessment of MRD in PB using the more readily accessible method of flow cytometry in current routine clinical practice.
Although it is recognized that deeper responses are seen with greater duration of venetoclax therapy, we have now demonstrated that the majority of patients destined to achieve uMRD will have done so within 2 years of treatment. Moreover, patients who fail to achieve deep responses by that time are unlikely to deepen their response with further monotherapy, and dose escalation achieves clearance of MRD in only a minority of cases. Our observation that concurrent rituximab augments uMRD attainment is consistent with preclinical evidence of synergy28 and the higher rates of uMRD reported in venetoclax-rituximab combination trials compared with venetoclax monotherapy,4,9,11 although the study populations differed in several important prognostic factors. Although combination with rituximab may deepen responses when used upfront, the optimal strategy to achieve uMRD in patients whose disease demonstrates suboptimal responses to venetoclax remains unclear.
With our long follow-up, the lack of plateau in the MRD recrudescence curve of this cohort of heavily pretreated patients suggests there may be an inevitable rate of disease relapse even among those with the deepest responses, or if there is any enduring uMRD subset, it is small. It is not yet known whether this is also the case in the setting of front-line therapy. We observed a median delay of 17 months between detection of MRD recrudescence in PB and progression meeting iwCLL criteria. The finding that patients whose disease harbors TP53 abnormalities may have inferior attainment of uMRD and earlier MRD recrudescence may reflect the known accelerated proliferative rate of this biological subset,29 although longer-term follow-up of larger cohorts is required to clarify this association. Our data suggest the goal of uMRD in R/R CLL is predominantly achieved in the first 2 years of venetoclax therapy and is augmented by concurrent rituximab, with limited deepening of responses with dose escalation and extended therapy beyond 2 years for patients with suboptimal responses. Systematic analysis in prospective clinical trials is warranted to examine the optimal analytical technique for MRD assessment and the most effective strategies for intensifying therapy for patients who do not achieve uMRD on venetoclax.
Acknowledgments
The authors thank all the patients, family members, and staff who participated in the studies.
Authorship
Contribution: T.E.L., M.A.A., and J.F.S. designed the study; T.E.L., V.S.L., and S.M.H. collected the data; T.E.L. performed the data analysis and generated the figures; T.E.L., M.A.A., V.S.L., and S.M.H. made the primary contributions to manuscript preparation; and all authors reviewed, revised, and approved the final manuscript.
Conflict-of-interest disclosure: T.E.L., M.A.A., V.S.L., and A.W.R. are employees of the Walter and Eliza Hall Institute of Medical Research, which receives milestone and royalty payments related to venetoclax. T.E.L., M.A.A., and A.W.R. are recipients of a share in royalty payments paid to the Walter and Eliza Hall Institute of Medical Research. S.M.H. has received honoraria from Gilead and nonfinancial assistance from AbbVie. C.S.T. has received honoraria and research funding from AbbVie and Janssen and honoraria from BeiGene. A.W.R. receives research funding from AbbVie, Genentech, Servier, Janssen, and BeiGene. J.F.S. receives research funding from AbbVie, Genentech, Celgene, and Janssen and is an advisory board member and has received honoraria from AbbVie, Acerta, Celgene, Genentech, Janssen, Roche, Sunesis, and Takeda. M.A.A. has received honoraria from AbbVie and CSL. T.E.L. has received honoraria from AbbVie. The remaining authors declare no competing financial interests.
Correspondence: John F. Seymour, Department of Haematology, Peter MacCallum Cancer Centre & Royal Melbourne Hospital, 305 Grattan St, Melbourne, VIC 3000, Australia; e-mail: john.seymour@petermac.org.

