


Abstract
Acute myeloid leukemia (AML) with t(8;21)(q22;q22.1);RUNX1-RUNX1T1, one of the core-binding factor leukemias, is one of the most common subtypes of AML with recurrent genetic abnormalities and is associated with a favorable outcome. The translocation leads to the formation of a pathological RUNX1-RUNX1T1 fusion that leads to the disruption of the normal function of the core-binding factor, namely, its role in hematopoietic differentiation and maturation. The consequences of this alteration include the recruitment of repressors of transcription, thus blocking the expression of genes involved in hematopoiesis, and impaired apoptosis. A number of concurrent and cooperating mutations clearly play a role in modulating the proliferative potential of cells, including mutations in KIT, FLT3, and possibly JAK2. RUNX1-RUNX1T1 also appears to interact with microRNAs during leukemogenesis. Epigenetic factors also play a role, especially with the recruitment of histone deacetylases. A better understanding of the concurrent mutations, activated pathways, and epigenetic modulation of the cellular processes paves the way for exploring a number of approaches to achieve cure. Potential approaches include the development of small molecules targeting the RUNX1-RUNX1T1 protein, the use of tyrosine kinase inhibitors such as dasatinib and FLT3 inhibitors to target mutations that lead to a proliferative advantage of the leukemic cells, and experimentation with epigenetic therapies. In this review, we unravel some of the recently described molecular pathways and explore potential therapeutic strategies.
Introduction
Acute myeloid leukemia (AML) with t(8;21)(q22;q22.1) constitutes ∼4% to 8% of AML. This subtype is associated with the M2 subtype of the FAB classification, younger age at presentation, a characteristic immunophenotype, frequent coexpression of CD19, and additional cytogenetic abnormalities, including loss of sex chromosome, del(9q), and trisomy 8, and a favorable outcome.1
Much has been learned in recent decades of the role of this translocation in leukemogenesis, of the concurrent mutations, and the downstream pathways that may be involved. In this review, we present an up-to-date overview of the molecular features of this subtype of AML, focusing on the disruption caused by RUNX1-RUNX1T1 fusion, the cooperating mutations that have been uncovered, the activation of oncogenes and signaling pathways, and the role of microRNAs (miRs). A greater understanding of the mechanisms of disease paves the way for exploring novel targets for therapy.
The role of RUNX1 and RUNX1T1 in normal cells
The RUNX1T1 gene is a transcriptional corepressor belonging to the human RUNX1T1 homolog family, that includes MTG16 (Myeloid Translocation Gene on chromosome 16) and MTGR1 (Myeloid translocation Gene-Related protein 1). The gene comprises 4 Nervy homology domains (Nervy homology regions [NHR] 1 to 4) (Figure 1). RUNX1T1 is expressed in many normal tissues, especially brain and heart, and is implicated in angiogenesis.2 Hematopoietic expression of RUNX1 is limited to erythropoiesis, albeit transiently, where its expression has been shown to be regulated by GATA-1.3 RUNX1T1 does not interact directly with DNA, but is recruited by transcription factors, including GFI1 and BCL6, forming multiprotein complexes. RUNX1T1 in turn recruits a number of corepressors to facilitate transcriptional repression.

Schematic model of RUNX1, RUNX1T1, and RUNX1-RUNX1T1 domains. The translocation t(8;21)(q22;q22) generates a fusion gene consisting of the RUNX1 gene from chromosome 21 and the RUNX1T1 gene from chromosome 8. AD, activation domain; ID, inhibitory domain; RHD, runt homology domain; TAD, transactivation domain.
Schematic model of RUNX1, RUNX1T1, and RUNX1-RUNX1T1 domains. The translocation t(8;21)(q22;q22) generates a fusion gene consisting of the RUNX1 gene from chromosome 21 and the RUNX1T1 gene from chromosome 8. AD, activation domain; ID, inhibitory domain; RHD, runt homology domain; TAD, transactivation domain.
The RUNX1 gene comprises 12 exons, including RHD and the transactivation domain, and encodes the α subunit of the core-binding factor that binds DNA (Figure 1). RUNX1 is a key transcriptional factor crucial for hematopoietic differentiation and myeloid development.4 Its role is evidenced by the absence of fetal liver hematopoiesis and lethality in knockout animal models, by the germline mutations predisposing to familial platelet disorder, and by the recently defined World Health Organization entity of myeloid neoplasms with RUNX1 mutations.5,6
Structure and function of RUNX1-RUNX1T1 fusion protein
The t(8;21)(q22;q22.1) translocation is a leukemogenic alteration that leads to a novel chimeric gene RUNX1-RUNX1T1, generated on the derivative chromosome 8. This translocation generates a fusion gene, which consists of the RUNX1 gene from chromosome 21 and the RUNX1T1 gene from chromosome 8. The breakpoint in RUNX1 occurs between exons 5 and 6; the translocation juxtaposes the 5′ end of the RUNX1 gene, including the RHD domain, with the 3′ end of the RUNX1T1 gene, with its 4 NHR domains (Figure 1). The generated fusion protein consists of 752 amino acids; the first 177 amino acids are derived from RUNX1, whereas the 575 amino acids are from RUNX1T1.7 Structurally, RUNX1-RUNX1T1 therefore has 5 domains: the RHD from RUNX1 and NHR domains 1 to 4 from RUNX1T1.4
The RUNX1T1 segment mediates the recruitment of corepressors complex histone deacetylase (HDAC), SMRT, N-CoR, and mSin3 to the binding domain of RUNX1, which results in the blocking of RUNX1 target genes.8,9 This prevents cell differentiation, increases cell survival, and ultimately, promotes leukemogenesis (Figure 2A-B).8,10 Although the expression of RUNX1T1 is low in normal hematopoietic cells, t(8;21) leads to high levels of RUNX1T1 expression.11 The RUNX1-RUNX1T1 fusion transcript may be detected in the blood and bone marrow of t(8;21) patients with AML and is a marker for minimal residual disease detection.12
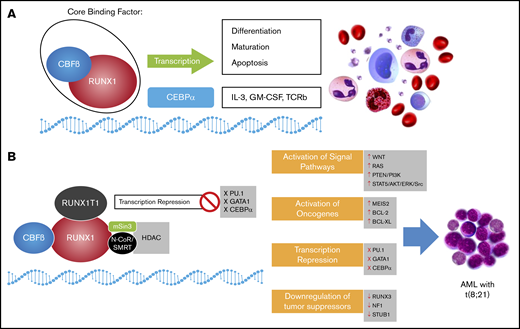
A schematic representation of the role of normal core binding factor and RUNX1-RUNX1T1 fusion protein in hematopoiesis and leukemogenesis, respectively. (A) Core binding factor (CBF) and its role in transcription: The CBF comprises 2 subunits (α and β) that are involved in transcription of genes involved in hematopoiesis. (B) The RUNX1-RUNX1T1 rearrangement leads to recruitment of ETO (encoded by RUNX1T1) that recruits transcriptional repressors, including N-Cor/SMRT and HDAC, that inhibit transcription of hematopoietic genes. GM-CSF, granulocyte-macrophage colony-stimulating factor; IL-3, interleukin-3; TCRb, T-cell receptor beta.
A schematic representation of the role of normal core binding factor and RUNX1-RUNX1T1 fusion protein in hematopoiesis and leukemogenesis, respectively. (A) Core binding factor (CBF) and its role in transcription: The CBF comprises 2 subunits (α and β) that are involved in transcription of genes involved in hematopoiesis. (B) The RUNX1-RUNX1T1 rearrangement leads to recruitment of ETO (encoded by RUNX1T1) that recruits transcriptional repressors, including N-Cor/SMRT and HDAC, that inhibit transcription of hematopoietic genes. GM-CSF, granulocyte-macrophage colony-stimulating factor; IL-3, interleukin-3; TCRb, T-cell receptor beta.
Molecular pathogenesis of RUNX1-RUNX1T1 fusion protein and role in leukemogenesis
RUNX1-RUNX1T1 directly regulates transcription and blocks differentiation to promote leukemia progression. It has been proposed that the interaction between the Runt domain of the RUNX1-RUNX1T1 protein and the non-DNA binding partner of the RUNX protein CBFβ is essential for leukemogenesis.13 In fact, RUNX1-RUNX1T1 acts as a dominant repressor for all RUNX1-mediated hematopoietic genes to disrupt normal hematopoietic differentiation and promote a preleukemic state (Figure 2).14 In addition, RUNX1-RUNX1T1 blocks critical hematopoietic transcription factors by interfering with their transcriptional activity. These transcription factors include PU.1, GATA1, and CEBPA, which are key regulators for myeloid differentiation.15-17 RUNX1-RUNX1T1 with the N-CoR corepressor also blocks differentiation by colocalizing primarily at promoter regions enriched with motifs for hematopoietic regulator and myeloid differentiation factors.18 The repressor function of RUNX1-RUNX1T1 is not limited to differentiation genes but also extends to inhibit some tumor suppressor genes, including RUNX3 and neurofibromatosis-1 (NF1)19,20 (Figure 3).

Summary of the various molecular mechanisms that may interact in the pathogenesis of AML t(8;21); RUNX1-RUNX1T1, and potential therapies. Fusion RUNX1-RUNX1T1 leads to aberrations in a number of intracellular pathways including downregulation of tumor suppressors, activation of oncogenes, abnormal activation of specific signaling pathways, and block of transcription factors, and often occurs with concurrent mutations. These may be amenable to therapeutic targeting. PARP, poly(ADP-ribose) polymerase.
Summary of the various molecular mechanisms that may interact in the pathogenesis of AML t(8;21); RUNX1-RUNX1T1, and potential therapies. Fusion RUNX1-RUNX1T1 leads to aberrations in a number of intracellular pathways including downregulation of tumor suppressors, activation of oncogenes, abnormal activation of specific signaling pathways, and block of transcription factors, and often occurs with concurrent mutations. These may be amenable to therapeutic targeting. PARP, poly(ADP-ribose) polymerase.
RUNX1-RUNX1T1 contributes to increased proliferation through its interaction with a number of factors. Hypoxia-inducible factor 1α (HIF1α) is a transcription factor that is overexpressed in RUNX1-RUNX1T1–positive patients with AML. HIF1α/RUNX1-RUNX1T1 interactions are essential to drive a high proliferation rate both in in vitro and in mice models. HIF1α is overexpressed in RUNX1-RUNX1T1–positive patients with AML and drives proliferation through increasing DNMT3a activity, causing DNA hypermethylation.21 RUNX1-RUNX1T1 fusion also mediates upregulation of Pontin, a highly conserved AAA+ ATPase involved in multiple cellular functions, to drive cell cycle progression and proliferation necessary for the oncogenic growth of t(8;21) cells.22 WNT and RAS signaling are also constitutively active to drive proliferation in patients with AML with t(8;21).23,24 In addition, Myeloid Ecotropic Viral Integration Site (MEIS2) expression levels are high in patients with AML t(8;21) and found to drive growth of RUNX1-RUNX1T1 cells by binding strongly to the DNA binding domain of RUNX1.25
RUNX1-RUNX1T1 expression in hematopoietic progenitors leads to enhanced self-renewal and a block in differentiation, and the forkhead box protein 01 (FOXO1) transcription factor has been implicated in this role. FOXO1 is aberrantly activated to drive self-renewal pathways of preleukemic stem cells (LSCs) in t(8;21) AML.26 Informative binding studies have shown that the DNA binding motif of FOXO1 binds and activates a stem cell molecular signature in RUNX1-RUNX1T1 cells. High FOXO1 expression thus plays an oncogenic role in mediating self-renewal and impaired differentiation of human CD34+RUNX1-RUNX1T1 hematopoietic stem cells (HSCs).
Recently, a new role for RUNX1-RUNX1T1 in promoting leukemogenesis has been proposed. In general, normal hematopoietic cells express physiological levels of aminoterminal enhancer of split and small nucleolar RNAs, but in contrast, RUNX1-RUNX1T1 cells induce high aminoterminal enhancer of split expression and small nucleolar RNAs formation, in what may facilitate LSC proliferation.27
Epigenetically, RUNX1-RUNX1T1 cells have been found to express unique epigenetic signatures to promote leukemogenesis. RUNX1-RUNX1T1 affects histone deacetylation and DNA methylation through mediating HDACs and DNMT1 recruitments, which lead to transcriptional silencing. Moreover, JMJD1C, a histone demethylase, is reported to function as a transcriptional coactivator for RUNX1-RUNX1T1 to increase proliferation in leukemia cells.28
A role for RUNX1-RUNX1T1 in inhibiting apoptosis has also been proposed. Antiapoptotic proteins BCL-2 and BCL-XL are frequently overexpressed in RUNX1-RUNX1T1 rearranged cells.29,30 Depletion of RUNX1-RUNX1T1 has been shown to increase apoptosis and cell cycle arrest. Mitogen-activated protein kinase 1 mediates resistance to RUNX1-RUNX1T1 suppression.31 In addition, RUNX1-RUNX1T1 has been reported to impair the activity of genes involved in the DNA repair machinery. Furthermore, POLE and OGG1 genes have been found to be downregulated in RUNX1-RUNX1T1 cells, causing an increase in DNA damage.32,33 Finally, the E3 ubiquitin ligase STUB1 has been identified as a negative regulator of the RUNX1-RUNX1T1 fusion protein. STUB1 binds to the RUNX1-RUNX1T1 complex to promote its ubiquitination and degradation. STUB1 overexpression causes dramatic growth inhibition in RUNX1-RUNX1T1 cells but not in other leukemia cells or normal blood cells (Figure 3).34
Is the RUNX1-RUNX1T1 fusion an early or late aberration in AML with t(8;21)? A number of lines of evidence, both preclinical and clinical, point to t(8;21) being an early event in leukemogenesis. RUNX1-RUNX1T1 transcripts can be detected in Guthrie cards from newborns who develop AML later on in life.35 Analysis of clonal composition of a series of AML cases by polymerase chain reaction and targeted sequencing has revealed that RUNX1-RUNX1T1 aberrations carry the features of a preleukemic lesion, namely, that they are present early in the preleukemic clone, are also found in clones when patients relapse, and lead to nonleukemic hematopoiesis in the marrows of mice in xenotransplantation models.36
Studies of RUNX1-RUNX1T1 rearranged mouse models
Mouse models have been crucial to our understanding, in vivo, of the mechanisms of leukemogenesis of AML t(8;21). Mouse studies have demonstrated that RUNX1 knockout during embryonic development leads to complete failure of HSC production, whereas targeting RUNX1 in hematopoietic cells promotes a preleukemic state.37 To prevent embryonic lethality, a conditional RUNX1-RUNX1T1 knock-in led to expansion of myeloid progenitor cells but did not block their differentiation nor initiate leukemia.38 RUNX1-RUNX1T1 alone is not thought to be sufficient to induce leukemogenesis, and additional mutations are needed to develop leukemia in mice.38-40 However, 2 RUNX1-RUNX1T1 truncation variants within the carboxyterminal have been reported to directly induce leukemia in vivo.39,41 RUNX1-ETO9a is a RUNX1-RUNX1T1 splice variant that lacks the NHR3/NHR4 domains and was able to induce AML after a few months when transplanted into mice.9,42 This splice variant was also detected in t(8;21)-positive patients with AML and found to correlate with short survival. A further study suggested that expressing RUNX1-RUNX1T1 fusion protein in mice lead to increased cell migration and impaired cell adhesion. This provides an explanation for an additional feature for RUNX1-RUNX1T1–containing cells that mediate migration across the bone marrow barrier to accelerate tumor progression and may help explain why this subtype is associated with extramedullary disease more often.43
Cooperating mutations in AML with RUNX1-RUNX1T1
As highlighted above, the occurrence of the RUNX1-RUNX1T1 rearrangement is not thought to be sufficient to initiate leukemia, and secondary genetic events are needed. High expression of RUNX1-RUNX1T1 in progenitor HSCs promotes differentiation and cell self-renewal but does not appear to be sufficient to induce AML.27,44,45 A number of cooperative mutations may contribute to leukemogenesis.38,39 By carrying out high-throughput screening on 106 patient samples, Duployez and colleagues reported that 96% of t(8;21) AML cases carried additional cytogenetic or mutational anomalies. In addition to confirming mutations in KIT, FLT3-ITD, and RAS (40%, 26%, and 10%, respectively), 42% had mutations in epigenetic regulators (ASXL1/2 and EZH2) and 18% of samples had cohesin complex mutations, notably of RAD21, STAG2, SMC1a, and SMC3. Concurrent tyrosine kinase, chromatin modifier, and cohesin mutations were associated with the highest risk of relapse, indicating the multiple pathways, including chromatin interactions with RUNX1, that may effect leukemogenesis.46 These findings were further confirmed by Christen and colleagues, who, in addition to reporting frequent class I mutations that rendered a proliferative advantage affecting the RAS/RTK pathway, including KIT, JAK2, FLT3, NRAS/KRAS, CBL, and PTPN11, also reported on hitherto unreported mutations in GIGYF2, DHX15, and G2E3, and were able to present a chronology of leukemogenesis whereby epigenetic mutations occurred before tyrosine kinase mutations.47
The aforementioned studies also confirmed the poor prognostic impact of high burden KIT mutations.46 KIT mutations are reported in up to 46% of patients with AML t(8;21), and these are associated with worse outcomes.48 Several reports indicate that the RUNX1-RUNX1T1 fusion protein and c-KIT play a fundamental role in tumorigenesis of AML. KIT mutations were found necessary in transforming HSCs into LSCs in RUNX1-RUNX1T1 cells.49,50 Mechanistically, RUNX1-RUNX1T1 transactivates KIT expression by direct binding to KIT promoter through the RUNX1 motif.51 This finding is supported by significantly high c-KIT expression levels in t(8;21) patients with AML. In fact, high c-KIT expression predicts poor prognosis in these patients.52 A possible mechanism for the worse outcome may be the finding that active c-KIT along with REtr, a C-terminal truncated variant of RUNX1T1, has been reported to expand proliferation of CD34+ progenitor cells and augment DNA repair machinery to drive chemoresistance.50 RUNX1-RUNX1T1 along with KIT mutations has been shown to transform murine bone marrow cells to AML.53 Moreover, it has been reported that amyloid precursor protein (APP) in cooperating with KIT mutations promotes apoptosis but not proliferation through the PI3K/AKT signaling pathway.54 Knockdown of APP arrests both colony formation and migration but not differentiation, cell cycle, or apoptosis in Kasumi-1 cells. In RUNX1-RUNX1T1–rearranged patients with AML, APP was significantly correlated with extramedullary leukemia, bone marrow cellularity, white blood cell count, and KIT mutations. This study also suggested that RUNX1-RUNX1T1 might adversely affect the prognosis of patients with AML with t(8;21) by upregulating APP expression.55
FLT3 mutations have been reported in up to 16% of t(8;21) patients, although evidence for their impact on prognosis has been conflicting. FLT3-ITD mutations with a high allelic burden appear to portend inferior survival. In contrast, FLT3-TKD mutations, especially with a high mutant level, have been associated with a better outcome.47 RAS mutations are reported in 20% to 30% of cases, with NRAS being more frequent than KRAS mutations. These mutations do not, however, appear to have an impact on relapse or survival. CBL mutations are frequently mutated in t(8;21) AML and have been found to play a critical factor in expanding CD34+ cells. CBL mutations in coordination with RUNX1-RUNX1T1 can cause both myeloid proliferation and expansion of human CD34+ cells through STAT5/AKT/ERK/Src signaling.56,57 JAK2 mutations have also been shown to impact survival. The PTPN11 gene, coding for a phosphatase acting downstream of tyrosine kinases, is infrequently mutated, and mutations in this gene lead to constitutive autophosphorylation of KIT kinase activity, although this does not appear to impact outcome.58,59 GIGYF2, an RTK signaling regulator, was also recently reported to be mutated in 2% of cases.47
Mutations of epigenetic regulators have been reported in AML with t(8;21). Mutations in DNA methylation regulator TET2 with the presence of RUNX1-RUNX1T1 have been shown to induce leukemia in mice models.60 The additional sex combslike (ASXL2) gene is frequently mutated in patients with AML with t(8;21) but not in patients with other cytogenetic abnormalities or RUNX1-mutated AML.61,62 In at least 1 series, patients carrying ASXL2 mutations were found to have a higher incidence of relapse.61 Unlike age-related clonal hematopoiesis, ASXL1 and ASXL2 mutations appear to be late events in AML t(8;21).47
Recently, mutations in the transcription regulator ZBTB7A that mediates hematopoietic development have been associated with t(8;21) in patients with AML, as have mutations in DHX15 and those involving the cohesin complex; the role of these mutations in leukemogenesis and clinical outcome remains to be determined.63,64
RUNX1-RUNX1T1 and miR
miRs are small noncoding RNA molecules involved in the regulation of gene expression.65,66 It has been proposed that RUNX1-RUNX1T1 mediates the expression of some miRs that are known to function as tumor suppressor genes or oncogenes.67 The miR miR-130a acts as an oncogene in t(8;21) AML cells by mediating leukemogenesis and a more aggressive disease course. RUNX1-RUNX1T1 was found to directly upregulate miR-130a expression and drive resistance to etoposide.68 MiR-126 is another miR that is mediated by RUNX1-RUNX1T1. Overexpression of miR-126 is correlated with unfavorable survival in patients with AML with t(8;21). MiR-126 also mediates leukemogenesis by activating genes that are highly expressed in LSCs and progenitor cells.69 MiR-383 expression has been reported to suppress expression of one of the thanatos-associated proteins (THAPs), namely, THAP10, that regulate chromatin modification, apoptosis, cell proliferation, and transcriptional regulation.70 THAP10 acts as a tumor suppressor, and RUNX1-RUNX1T1 transcriptionally induces the expression of miR-383 that in turn suppresses THAP10 function.71
RUNX1-RUNX1T1 also mediates downregulation of some miRs involved in cell differentiation and tumor suppression, further implicating miRs in leukemogenesis. MiR-223 and miR-222/221 are known to regulate differentiation and have been found to be downregulated in t(8;21) patients with AML as compared with normal bone marrow cells.72 The expression of miR-9 has been negatively correlated with expression levels of silent mating type information regulation homolog-1 that is a class III HDAC that mediates differentiation, cell proliferation, and gene expression. In addition, overexpression of miR-9 decreases silent mating type information regulation homolog-1 expression and blocks proliferation in RUNX1-RUNX1T1–positive AML cell lines.73 On the other hand, epigenetic silencing of tumor suppressor gene miR-193a was found to accelerate the oncogenic activity of RUNX1-RUNX1T1 cells by activating the PTEN/PI3K signal pathway.74
Therapeutic targeting of AML with t(8;21); RUNX1-RUNX1T1
Therapy of AML with t(8;21), especially in the upfront setting, relies on standard anthracycline and cytarabine, followed by 2 to 4 courses of cytarabine consolidation. The increased chemosensitivity compared with other AML subtypes may to some extent be explained by the demonstration of an activated p53 pathway, leading to increased apoptosis and DNA damage.33 Addition of gemtuzumab ozogamicin (GO), an anti-CD33 antibody, further improves outcomes, and the beneficial effect is seen despite the expression of CD33 often being weak in AML t(8;21) and regardless of the level of expression, possibly due to its action on CD33+ hematopoietic progenitors. It is thought that the efficacy of GO is attenuated by the expression of p-glycoprotien that contributes to multidrug resistance; p-glycoprotein is reduced in AML t(8;21) compared with other subtypes, and this may further offer an explanation of why these patients have better outcomes with GO.75-77 Despite this relative chemosensitivity, treatment failure, especially in relapse, remains a challenge. Strategies to search for a cure can therefore target one or more of a number of steps in leukemogenesis.
Can RUNX1/RUNX1T1 be targeted at the level of the gene? RUNX1 deletion in RUNX1-RUNX1T1–transformed human CD34+ cells induced apoptosis and inhibited tumor growth. In these studies, targeting the Runt domain of RUNX1 proteins blocked the RUNX1/CBFβ interaction and decreased survival and proliferation of leukemic cells.78,79 These experiments provide proof of principle of such an approach in a preclinical setting.
The most attractive prospect would be the development of a small molecule that can block the activity of the abnormal RUNX1-RUNX1T1 protein. A novel compound that binds specifically to the Runt domain of RUNX proteins has been shown to disrupt the interaction of RUNX1-RUNX1T1 with CBFβ and has shown high antitumor activity in RUNX1-RUNX1T1 leukemia cell lines.80 Comprehensive screening of more than two thousand US Food and Drug Administration–approved drugs identified 2 common agents, namely, methylprednisolone and methotrexate, as highly effective in Kasumi-1 cells.81 Virtual screening of an array of molecules identified a compound, named Compound 7.44, that was able to disrupt RUNX1/RUNX1T1 tetramerization and thus block its oncogenic potential, while allowing differentiation and promoting prolonged survival in mouse models.82
Some natural agents have been also reported to be effective in targeting the RUNX1-RUNX1T1 fusion protein. Celastrol is a natural compound extracted from the Chinese herb Tripterygium wilfordii Hook. Celastrol has been shown to block cell proliferation and induce apoptosis by targeting the RUNX1-RUNX1T1 protein.83 Oroxylin A also shows antitumor activity in RUNX1-RUNX1T1 cells. Oroxylin A reduces the expression of RUNX1-RUNX1T1 protein, promotes the expression of differentiation-related proteins C/EBPα and P21, and prolongs the survival of AML-bearing mice.84 A further compound, Oridonin, was shown by investigators in China to cleave RUNX1-RUNX1T1 oncoprotein and selectively promote apoptosis in t(8;21) leukemia cells.85 Esculetin is another natural agent that was shown to therapeutically target RUNX1-RUNX1T1 cells and was shown to dramatically decrease the half-life of RUNX1-RUNX1T1 messenger RNA.86
Because RUNX1-RUX1T1 leads to recruitment of HDAC to RUNX1 binding domains, epigenetic approaches have been studied in RUNX1-RUNX1T1 models, and there are a number of hypomethylating agents and HDAC inhibitors used in therapy that would make them attractive agents. RUNX1T1 has been reported to interact with HDACs to induce histone acetylation levels in RUNX1-RUNX1T1 cells. The pan-HDAC inhibitor panobinostat depletes APP levels and inhibits proliferation.87 In addition, treatment of mice bearing t(8;21) AML with panobinostat leads to dramatic responses that were independent of functional p53 or activation of apoptotic markers.88 Moreover, a panobinostat and arsenic trioxide combination degraded the RUNX1-RUNX1T1 protein and caused cell cycle arrest, increased differentiation of AML blasts, and lead to complete responses in a mouse model.89 HDAC inhibitors also block leukemogenesis in RUNX1-RUNX1T1 cells.90 An HDAC inhibitor phenylbutyrate has been found to promote differentiation and apoptosis by targeting the RUNX1-RUNX1T1 protein.91 Cell lines with RUNX1-RUNX1T1 have been shown to be some of the most sensitive AML cell lines to Lysine-specific demethylase 1 inhibition; targeting Lysine-specific demethylase 1 completely eradicated tumor growth in a RUNX1-RUNX1T1 xenograft model.92
Approaches that target antiapoptotic pathways have been investigated with positive results in preclinical models. The proteosome inhibitor bortezomib and the BCL-2 inhibitor ABT-737 induce apoptosis and block proliferation of RUNX1-RUNX1T1 cells both in vitro and in vivo.93,94 A novel oxazine derivative, ZGDHu, was found to block the growth of Kasumi 1 cells via decreasing nuclear factor κB activity, depleting RUNX1-RUNX1T1 fusion protein and causing cell cycle arrest. A further drug that has been found to be highly effective in inducing apoptosis of RUNX1-RUNX1T1 cells is fingolimod. Fingolimod is a sphingosine analog and a US Food and Drug Administration–approved drug for treating patients with multiple sclerosis.95 Fingolimod has shown high antitumor activity in mice models and primary leukemic cells isolated from patients with AML t(8;21).96 A further mechanism that may be targeted to enhance leukemic cell death is poly(ADP-ribose) polymerase inhibition, and RUNX1-RUNX1T1 AML cells were shown to be highly sensitive to poly(ADP-ribose) polymerase inhibitors.97
Finally, given the occurrence of secondary mutations, including KIT, FLT3, and JAK2, as uncovered by high-throughput screening studies in AML t(8;21) and their impact on survival, these may be the most amenable targets with currently available tyrosine kinase inhibitors.23,24 Dasatinib, a tyrosine kinase inhibitor with activity against KIT mutations, has been trialed both in patients with minimal residual disease and in the upfront setting, and although initial reports suggest a modest impact, results of randomized trials are awaited.98 Modest antileukemic responses have been reported with the JAK2 inhibitor ruxolitinib in refractory AML.99 The recently approved FLT3 inhibitor midostaurin was demonstrated to result in improved survival in all FLT3 mutated patients and patients with t(8;21), and an FLT3 mutation would be eligible for this agent, although the proportion of patients with favorable risk in the trial was relatively small.100 A further surface protein that may be targeted is protease activated receptor-1, one of a family of integral membrane G protein–coupled receptors that are upregulated on some AML cells, and targeting protease activated receptor-1 has been shown to inhibit leukemogenesis in RUNX1-RUNX1T1 cells.101
Conclusions and future directions
Great strides have been made in understanding the pathobiology of AML with t(8;21), especially with the aid of molecular technologies that have revealed the mutational landscape, downstream pathways, and cooperating mutation and their role in leukemogenesis. However, a number of questions remain: why do patients with this subtype of AML have better survival than others with chemotherapy, and how can this relative chemosensitivity be further exploited? As the translocation is considered an early lesion in leukemogenesis, can the fusion protein be better targeted with small molecules, to recapitulate the CML success story? Also, while deep sequencing technologies have further revealed a pattern of concurrent mutations, their role in leukemogenesis remains to be elucidated.
Despite progress in the understanding of the biology of AML t(8;21), there is a relative paucity, partly due to this subtype of AML comprising a minor subset of patients with AML, of an adequate number of dedicated clinical trials of agents targeting the pathways involved in leukemogenesis. This will require prospective phase 1 to 2 multinational trials involving a large number of centers, which may not always be feasible or supported. Although a disease-specific targeted therapy is a modality that will continue to be pursued, in the absence of such a drug, combinations of standard chemotherapeutic agents with or without anti-CD33 immunoconjugates are likely to be trialed with pathway-specific agents and tyrosine kinase inhibitors. Greater implementation in clinical practice of myeloid next-generation sequencing panels will mean more of the recently discovered cooperating mutations may be identified and potentially targeted. Given the recent reports of the occurrence of JAK2 mutations, albeit in a small proportion, further trials of JAK2 inhibitors would be warranted. Potential pathways that warrant further studies as potential targets include the hedgehog signaling pathway, for which a drug (Glasdegib) has recently been approved in AML, newer BCL-2 inhibitors, and targeting GLI1. Targeting FOXO1 by genetic and pharmacological approaches inhibits proliferation of t(8;21) AML cell lines and provides a further potential approach for targeting cancer stem cells.
Although for relapsing patients an allogenic transplant will salvage a significant proportion, rapid advancements in the field of immunotherapy, bispecific T-cell engagers, and chimeric antigen receptor T-cell and NK cells hold promise in adding to the armamentarium of available therapies for those who fail current lines of therapy.
Authorship
Contribution: S.A.-H. and S.O.A.A. wrote the manuscript; S.O.A.A., M.A., M.M., and F.A. conceptualized the review, edited the manuscript, and provided input; and all authors reviewed and approved the draft individually.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Syed Osman Ali Ahmed, Adult Hematology/Bone Marrow Transplantation, Oncology Center, MBC-64, King Faisal Specialist Hospital and Research Center, POB 3354, Riyadh 11211, Saudi Arabia; e-mail: syedahmed@kfshrc.edu.sa.

