

Key Points
INCB053914 and ruxolitinib synergize to induce apoptosis of JAK2V617F-driven cells and to inhibit neoplastic growth of primary MPN cells.
INCB053914 antagonizes ruxolitinib persistence in an in vivo MPN model.

Abstract
Aberrant JAK2 tyrosine kinase signaling drives the development of Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs), including polycythemia vera, essential thrombocythemia, and primary myelofibrosis. However, JAK2 kinase inhibitors have failed to significantly reduce allele burden in MPN patients, underscoring the need for improved therapeutic strategies. Members of the PIM family of serine/threonine kinases promote cellular proliferation by regulating a variety of cellular processes, including protein synthesis and the balance of signaling that regulates apoptosis. Overexpression of PIM family members is oncogenic, exemplified by their ability to induce lymphomas in collaboration with c-Myc. Thus, PIM kinases are potential therapeutic targets for several malignancies such as solid tumors and blood cancers. We and others have shown that PIM inhibitors augment the efficacy of JAK2 inhibitors by using in vitro models of MPNs. Here we report that the recently developed pan-PIM inhibitor INCB053914 augments the efficacy of the US Food and Drug Administration–approved JAK1/2 inhibitor ruxolitinib in both in vitro and in vivo MPN models. INCB053914 synergizes with ruxolitinib to inhibit cell growth in JAK2-driven MPN models and induce apoptosis. Significantly, low nanomolar INCB053914 enhances the efficacy of ruxolitinib to inhibit the neoplastic growth of primary MPN patient cells, and INCB053914 antagonizes ruxolitinib persistent myeloproliferation in vivo. These findings support the notion that INCB053914, which is currently in clinical trials in patients with advanced hematologic malignancies, in combination with ruxolitinib may be effective in MPN patients, and they support the clinical testing of this combination in MPN patients.
Introduction
The identification of aberrant JAK2 tyrosine kinase activity (eg, JAK2V617F) as a driver of the Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs) polycythemia vera, essential thrombocythemia, and primary myelofibrosis led to the rapid assessment of JAK2 kinase inhibitors as targeted therapies for personalized medicine for these MPNs. Although numerous JAK2 inhibitors have been assessed in clinical trials, ruxolitinib is the only one approved by the US Food and Drug Administration for certain MPN patients.1,2 However, clinically tested JAK inhibitors generally improve the symptomology of MPN patients but fail to significantly decrease allele burden or induce disease remission. Recent data from long-term studies suggest that ruxolitinib can improve the natural course of disease by reversing myelofibrosis.3 This suggests that improved JAK2 inhibitors or improving the efficacy of ruxolitinib may provide therapeutic options that could lead to long-term remission. Although long-term ruxolitinib treatment may improve survival for patients with myelofibrosis,4-8 only a fraction of patients remains on therapy, further demonstrating the need for improved targeted MPN therapies.
The 3 members of the PIM family of serine/threonine kinases were initially identified as proto-oncogenes that cooperate with MYC to induce lymphomagenesis.9 PIM kinases have a variety of target substrates. For example, PIM activity augments mTORC1 activity via phosphorylation and inhibition of PRAS4010,11 and inhibits apoptosis by phosphorylating BAD.9,12,13 Thus, by regulating mTORC1, PIM activity can impinge on the control of a variety of additional cellular processes, including protein synthesis and metabolism, among others.14,15 Given the growth promoting and oncogenic potential of PIM kinases, PIM kinase inhibitors are being developed as targeted cancer therapies for numerous indications. For example, PIM inhibitors have been shown to be effective in models of solid cancer,16-20 as well as in blood cancers such as acute leukemia and myeloma, among others.21-24 However, only a small number of PIM kinase inhibitors have been successfully developed to the point of clinical testing for some of these indications. PIM kinases have also been shown to contribute to drug resistance in solid tumors as well as in hematopoietic cancers.17,25,26 Thus, PIM kinase inhibitors may play future roles in combination therapies aimed at improving the upfront efficacy of current targeted therapies, preventing the development of resistance to targeted therapies, and/or as second-line treatments to antagonize drug resistance.
Members of the PIM family play roles in hematopoiesis. For example, PIM1 has known roles in murine hematopoietic stem cell (HSC) function, including regulating the number and functionality of HSCs.27 Hematopoietic cells lacking all PIM kinases have reduced responses to certain cytokines,28 and mice lacking all 3 PIMs have lower numbers of platelets and hematopoietic progenitor colony-forming cells.29 However, mice deficient in all 3 PIM family proteins are viable and fertile,28 suggesting that therapeutic targeting with a pan-PIM inhibitor would be tolerated. PIM kinases are constitutively active and are thus regulated at the level of protein expression,9,30 including the transcription of PIM family members being induced via JAK/STAT signaling.9,31-36 Thus, PIM signaling is a downstream effector of JAK2 signaling and, given the progrowth and antiapoptotic nature of PIM activity, is a potential target for therapy in JAK2-driven neoplasms. Accordingly, PIM inhibitors have been assessed as potential therapeutic targets for MPNs.36-39 Here, we report the impressive efficacy of a recently described pan-PIM inhibitor INCB05391440 in combination with the JAK1/2 inhibitor ruxolitinib (approved by the US Food and Drug Administration) in preclinical MPN animal and human cell models.
Methods
Cell growth and apoptosis assays
UKE1 and SET2 cells were cultured as described.36 BaF3 cells transformed by expression of EpoR and JAK2V617F (BaF3-JAK2VF) were previously described.41 Relative viable cells were determined using CellTiter-Glo (Promega) and a Spectramax i3x reader (Molecular Devices). Synergy calculation is described in the supplemental Data. Total cell numbers and viability were determined by trypan blue exclusion. Drugs were refreshed at every time point during long-term growth studies. Apoptotic cells were determined by annexin V binding (BD Pharmingen), as described.36 Immunoblotting and antibody details are provided in the supplemental Data.
Ex vivo colony-forming assays
Peripheral blood was obtained from patients who provided consent through the Institutional Review Board–approved Moffitt Cancer Center Total Cancer Care protocol (MCC 14690) with approval by the Moffitt Cancer Center Scientific Review Committee. Primary MPN cell growth was assessed in colony-forming assays as described36 and as detailed in the supplemental Data.
Animal studies
Animal studies were conducted under the supervision of a veterinarian and in compliance with Incyte Corporation’s Animal Use Protocols, established and approved by the Incyte Institutional Animal Care and Use Committee.
Xenograft studies.
CB17 SCID mice (∼8 weeks old) (Charles River Laboratories) were subcutaneously injected into a single flank with 10 million JAK2V617F-driven patient-derived MPN model SET2 cells in matrigel. Tumor-bearing mice were randomly assigned 13 days after cell injection to generate cohorts of equal tumor sizes for treatment twice per day orally with drug vehicle (5% dimethylacetamide/95% of 0.5% methylcellulose), INCB053914 (100 mg/kg; n = 9), ruxolitinib (30 mg/kg; n = 7), or a combination of the 2 drugs (n = 10). Tumors were measured by using digital calipers. Tumor volumes were calculated by using the following formula: volume = (length × width2)/2, where the width was the smaller of the 2 dimensions.
MPL-W515L MPN model.
Drug effects on the development of MPN in vivo were determined using Balb/c mice containing transplanted MPL-W515L–expressing bone marrow cells.42 Briefly, bone marrow was ablated in 8-week-old Balb/c mice using 50 mg/kg 5-fluorouracil 5 days before transplantation with Balb/c bone marrow expressing MPL-W515L via retroviral infection. Blood counts were performed 7 days after transplantation, and 4 cohorts of mice (n = 9 mice in each cohort) with equal mean platelet numbers were generated for treatment with vehicle, INCB053914 (100 mg/kg), ruxolitinib (60 mg/kg), or a combination of INCB053914 and ruxolitinib twice per day orally. Blood counts were performed at 7, 14, 21, and 35 days of treatment, which corresponded to 14, 21, 28, and 42 days after transplantation. All mice treated with vehicle and INCB053914 died or were moribund by 21 days posttransplantation. Spleen weights of animals treated with vehicle (n = 4) and INCB053914 monotherapy (n = 1) were obtained when the mice became moribund on day 15 after transplantation (day 8 of treatment). The remaining animals treated with vehicle and INCB053914 monotherapy were moribund on day 21 after transplantation (day 14 of treatment). Three mice each from the ruxolitinib monotherapy and the combination therapy cohorts were euthanized to assess disease progression (spleen weight) on day 21 of treatment. Treatment continued for 35 days (day 42 after transplantation) at which point the experiment was stopped and all animals were euthanized.
Statistical analysis
Prism 8 (GraphPad Software, Inc., San Diego, CA) was used for graphical representation and statistical analyses of data.
Results
INCB053914 inhibits MPN model cell proliferation and synergizes with ruxolitinib to inhibit cell proliferation and induce apoptosis
INCB053914 is a recently described pan-PIM kinase inhibitor that exhibits potent activity against PIM1, PIM2, and PIM3 (biochemical 50% inhibitory concentration [IC50] values of 0.24, 30, and 0.12 nM, respectively).40 To assess the effects of INCB053914 on JAK2V617F-driven cell growth, we determined the concentration for 50% of maximal inhibition of cell proliferation (GI50) values of INCB053914 against 2 patient-derived JAK2V617F-expressing MPN model cell lines (UKE1 and SET2) and BaF3-JAK2V617F cells,41 which are cells transformed to cytokine independence by expression of EpoR and JAK2V617F. The GI50 values for INCB053914 against these cells were 1.3, 0.98, and 0.035 μM, respectively (Figure 1A). The ruxolitinib GI50 values on the same lines were 0.59, 0.083, and 0.055 μM, respectively (Figure 1B). We next assessed the effect of combining INCB053914 and ruxolitinib on the growth of these JAK2V617F-driven cells. In long-term growth studies, 0.5 μM INCB053914 as a single agent had only a slight effect in UKE1 cells but significantly antagonized the growth of these cells in the presence of ruxolitinib (Figure 2A). Similar results were obtained using SET2 cells (with 20 nM INCB053914) (Figure 2B) as well as BaF3-JAK2V617F cells (with 10 nM INCB053914) (Figure 2C). The combination of INCB053914 and ruxolitinib significantly reduced UKE1 and BaF3-JAK2V617F cell viability more than treatment with the single agents (supplemental Figure 1A). Cotreatment with INCB053914 reduced the ruxolitinib GI50 against UKE1 cells by more than 10-fold (Figure 2D) and, as calculated by the Bliss model of independence, this combination was synergistic across a wide range of ruxolitinib concentrations (Figure 2D). Similar findings of INCB053914 and ruxolitinib synergy were observed in SET2 cells (supplemental Figure 1B).
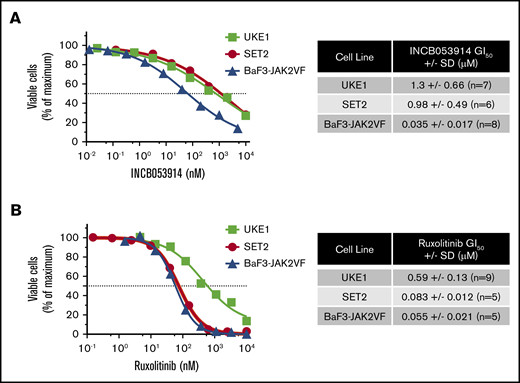
The pan-PIM inhibitor INCB053914 inhibits viable MPN model cell numbers in a concentration-dependent manner. (A) UKE1, SET2, and BaF3-JAK2VF cells were incubated with the indicated range of concentrations of INCB053914 (A) and ruxolitinib (B), and relative viable cells were determined after 72 hours (UKE1 and SET2) or 48 hours (BaF3-JAK2VF) by using CellTiter-Glo. Relative viable cell numbers are plotted as a percent of maximum for each drug concentration. Representative concentration-response curves are shown, and the average GI50’s of INCB053914 and ruxolitinib in each drug in each cell line are shown to the right of each graph.
The pan-PIM inhibitor INCB053914 inhibits viable MPN model cell numbers in a concentration-dependent manner. (A) UKE1, SET2, and BaF3-JAK2VF cells were incubated with the indicated range of concentrations of INCB053914 (A) and ruxolitinib (B), and relative viable cells were determined after 72 hours (UKE1 and SET2) or 48 hours (BaF3-JAK2VF) by using CellTiter-Glo. Relative viable cell numbers are plotted as a percent of maximum for each drug concentration. Representative concentration-response curves are shown, and the average GI50’s of INCB053914 and ruxolitinib in each drug in each cell line are shown to the right of each graph.
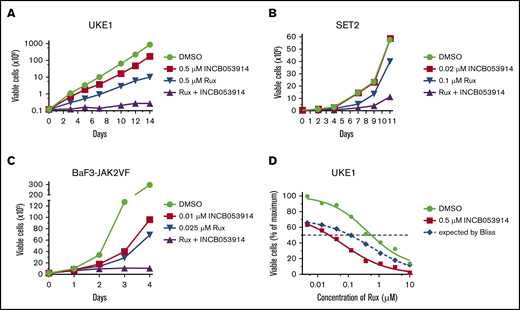
INCB053914 synergizes with ruxolitinib to inhibit the growth of MPN model cell lines. UKE1 cells were cultured with dimethyl sulfoxide (DMSO; 0.1%), 0.5 μM INCB053914, 0.5 μM ruxolitinib (Rux), and both drugs together (A); SET2 cells were cultured with DMSO (0.1%), 0.02 μM INCB053914, 0.1 μM ruxolitinib, and both drugs together (B); and BaF3-JAK2VF cells were cultured with DMSO (0.1%), 0.01 μM INCB053914, 0.025 μM ruxolitinib, and both drugs together, and viable cell numbers were determined by trypan blue exclusion over time (C). For panels A-B, drug and media were replenished at each time point. For panel C, drugs were added once and not replenished. (D) UKE1 cells were incubated with the indicated range of ruxolitinib in the absence or presence of 0.5 μM INCB053914, and relative viable cells were determined after 72 hours using CellTiter-Glo. The expected additive percent of relative viable cells at each ruxolitinib concentration in the presence of INCB053914 was determined by the Bliss model of independence and plotted as the blue dashed line. The reduced observed percent of relative viable cells (red line below the dashed line) demonstrates synergy between ruxolitinib and INCB053914.
INCB053914 synergizes with ruxolitinib to inhibit the growth of MPN model cell lines. UKE1 cells were cultured with dimethyl sulfoxide (DMSO; 0.1%), 0.5 μM INCB053914, 0.5 μM ruxolitinib (Rux), and both drugs together (A); SET2 cells were cultured with DMSO (0.1%), 0.02 μM INCB053914, 0.1 μM ruxolitinib, and both drugs together (B); and BaF3-JAK2VF cells were cultured with DMSO (0.1%), 0.01 μM INCB053914, 0.025 μM ruxolitinib, and both drugs together, and viable cell numbers were determined by trypan blue exclusion over time (C). For panels A-B, drug and media were replenished at each time point. For panel C, drugs were added once and not replenished. (D) UKE1 cells were incubated with the indicated range of ruxolitinib in the absence or presence of 0.5 μM INCB053914, and relative viable cells were determined after 72 hours using CellTiter-Glo. The expected additive percent of relative viable cells at each ruxolitinib concentration in the presence of INCB053914 was determined by the Bliss model of independence and plotted as the blue dashed line. The reduced observed percent of relative viable cells (red line below the dashed line) demonstrates synergy between ruxolitinib and INCB053914.
Ruxolitinib treatment induces apoptosis in MPN model cell lines.43 Combining INCB053914 with ruxolitinib led to synergistic induction of annexin V+ cells in UKE1, SET2, and BaF3-JAK2V617F cells (Figure 3A) when using concentrations of individual drugs that produced little to no induction of annexin V+ cells. This suggests that the decrease in cell growth and viability observed with the combination of INCB053914 and ruxolitinib was partly the result of an increase in apoptosis. The combination of INCB053914 and ruxolitinib did not block the growth or compromise the survival of non–JAK2V617F-driven Jurkat and K562 hematopoietic cells (supplemental Figure 1C).
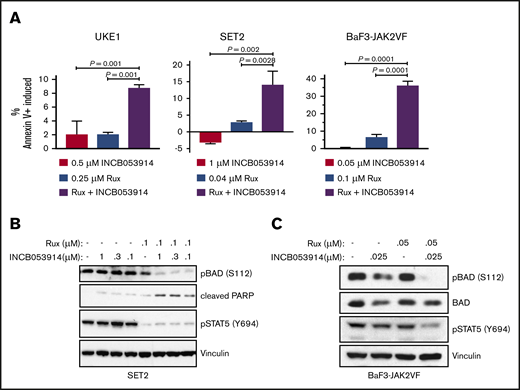
INCB053914 synergizes with ruxolitinib to induce apoptosis and inhibit phosphorylation of BAD. (A) UKE1, SET2, and BaF3-JAK2VF cells were incubated with DMSO (0.1%), INCB053914, ruxolitinib, or the combination of the 2 drugs for 72 hours, and annexin V+ cells were detected by flow cytometry. The percent of annexin V+ cells induced over DMSO-treated cells is plotted for each treatment. Error bars indicate standard deviation (SD) of biological triplicates, and P values were determined using one-way analysis of variance with multiple comparisons. Drug concentrations used were 0.5 μM INCB053914 and 0.25 μM ruxolitinib for UKE1 cells, 1 μM INCB053914 and 0.04 μM ruxolitinib for SET2 cells, and 0.05 μM INCB053914 and 0.1 μM ruxolitinib for BaF3-JAK2VF cells. Cell lysates of SET2 cells (B) and BaF3-JAK2VF cells (C) incubated with INCB053914 and ruxolitinib, alone and in combination as indicated for 4 hours (B) or 8 hours (C) were immunoblotted to assess effects on the levels of the indicated proteins. PARP, poly adenosine diphosphate (ADP)–ribose polymerase.
INCB053914 synergizes with ruxolitinib to induce apoptosis and inhibit phosphorylation of BAD. (A) UKE1, SET2, and BaF3-JAK2VF cells were incubated with DMSO (0.1%), INCB053914, ruxolitinib, or the combination of the 2 drugs for 72 hours, and annexin V+ cells were detected by flow cytometry. The percent of annexin V+ cells induced over DMSO-treated cells is plotted for each treatment. Error bars indicate standard deviation (SD) of biological triplicates, and P values were determined using one-way analysis of variance with multiple comparisons. Drug concentrations used were 0.5 μM INCB053914 and 0.25 μM ruxolitinib for UKE1 cells, 1 μM INCB053914 and 0.04 μM ruxolitinib for SET2 cells, and 0.05 μM INCB053914 and 0.1 μM ruxolitinib for BaF3-JAK2VF cells. Cell lysates of SET2 cells (B) and BaF3-JAK2VF cells (C) incubated with INCB053914 and ruxolitinib, alone and in combination as indicated for 4 hours (B) or 8 hours (C) were immunoblotted to assess effects on the levels of the indicated proteins. PARP, poly adenosine diphosphate (ADP)–ribose polymerase.
INCB053914 and ruxolitinib synergistically suppress cell signaling pathways that regulate BAD and mTORC1 activity
Exposure of cells to PIM inhibitors increases PIM protein levels36,37,40 and impairs phosphorylation of the PIM substrate PRAS40 at threonine 246.10,19,44 INCB053914 treatment of MPN model cells triggered increases in PIM1, PIM2, and PIM3 protein levels (supplemental Figure 2A) as well as inhibition of pPRAS40(T246) (supplemental Figure 2B), indicating on-target activity of INCB053914. The enhanced annexin V+ cells induced by the combination of INCB053914 and ruxolitinib suggested elevated signaling toward apoptosis. INCB053914 treatment induced low but detectable cleavage of poly adenosine diphosphate (ADP)–ribose polymerase (PARP), a marker of apoptosis, in SET2 cells, but had no apparent effect on levels of BAD phosphorylated at serine 112 (pBAD[S112]), which is a target of PIM kinase activity (Figure 3B).12 However, co-treatment of INCB053914 with ruxolitinib led to a significant reduction in levels of pBAD(S112) and to enhanced cleavage of PARP (Figure 3B). This is congruent with an increase in annexin V+ cells with the combination of drugs (Figure 3A) and suggests a synergistic effect on reversing the inhibition of the proapoptotic BAD protein that is mediated by phosphorylation of S112. INCB053914 treatment alone did not affect levels of pSTAT5(Y694) or enhance ruxolitinib-mediated inhibition of pSTAT5(Y694) in SET2 cells (Figure 3B) or levels of activated AKT (supplemental Figure 3). Similar synergistic suppression of pBAD(S112) was observed in BaF3-JAK2VF cells, in which the combination more modestly suppressed pSTAT5(Y694) (Figure 3C). Treatment of MPN model cells with INCB053914 also led to concentration-dependent decreases in biomarkers of mTORC1 activity. This includes decreased levels of pp70S6K(T389), pS6(S235/S236), and p4EBP1(T37/T46), the latter correlating with an increase in the faster sodium dodecyl sulfate-polyacrylamide gel electrophoresis–migrating hypophosphorylated form of 4EBP1 in SET2 (Figure 4A) and UKE1 cells (supplemental Figure 4). Importantly, the combination of INCB053914 and ruxolitinib synergistically suppressed these biomarkers compared with the individual effects of the drugs. Similar observations were made in BaF3-JAK2VF cells in which enhanced suppression of pPRAS40(T246) was also observed with the drug combination (Figure 4B).
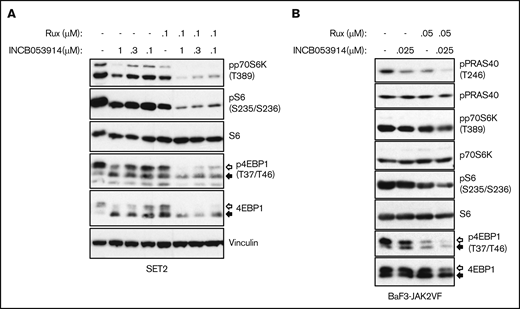
The combination of INCB053914 and ruxolitinib augments the suppression of biomarker phosphorylation indicative of mTORC signaling. Cell lysates of SET2 cells (A) and BaF3-JAK2VF cells (B) incubated with INCB053914 and ruxolitinib alone and in combination as indicated for 4 hours (A) or 8 hours (B) were immunoblotted to assess the effects on levels of the indicated proteins. The white and black arrows indicate the migration of hyperphosphorylated and hypophosphorylated 4EBP1 protein, respectively. Note that the vinculin blot in panel A and Figure 3B are the same because these were the same drug-treated cells and subsequent lysates analyzed in each figure.
The combination of INCB053914 and ruxolitinib augments the suppression of biomarker phosphorylation indicative of mTORC signaling. Cell lysates of SET2 cells (A) and BaF3-JAK2VF cells (B) incubated with INCB053914 and ruxolitinib alone and in combination as indicated for 4 hours (A) or 8 hours (B) were immunoblotted to assess the effects on levels of the indicated proteins. The white and black arrows indicate the migration of hyperphosphorylated and hypophosphorylated 4EBP1 protein, respectively. Note that the vinculin blot in panel A and Figure 3B are the same because these were the same drug-treated cells and subsequent lysates analyzed in each figure.
Neoplastic growth of hematopoietic progenitors from MPN patients is highly sensitive to INCB053914, which significantly augments inhibition induced by ruxolitinib
Hematopoietic progenitor cells of MPN patients can form erythroid colonies in methylcellulose in the absence of erythropoietin (Epo), and this hallmark neoplastic growth is used to assess anti-MPN therapeutics.36,43,45 We recently reported low nanomolar potency of INCB053914 against neoplastic erythroid colony growth of primary MPN patient cells in this assay, but the effect of INCB053914 with ruxolitinib was not established.40 To determine the effect of INCB053914 in combination with ruxolitinib on Epo-independent colony growth of primary MPN cells, we plated cells from JAK2V617F-positive patients in the absence of drug, in the presence of each drug individually, and in the presence of the 2 drugs combined using 25 to 50 nM of ruxolitinib (the approximate IC50 in this assay).36 Low nanomolar (5-20 nM) INCB053914 showed effective inhibition in all 8 primary MPN samples tested (Figure 5A). Importantly, although variability in responses was observed, all samples were sensitive to both drugs, and in a majority of the samples, synergy with ruxolitinib was observed (Figure 5A). In some samples, residual colony formation was nearly eliminated by the combination, including 1 sample in which only 5 nM INCB053914 with ruxolitinib all but abolished colony formation. Colony-forming unit-granulocyte-macrophage (CFU-GM) colony formation was also inhibited by INCB053914 and ruxolitinib, and the combination of drugs resulted in superior inhibition in several samples (Figure 5B). Additional information regarding the patients from which these samples were obtained is listed in supplemental Table 1.
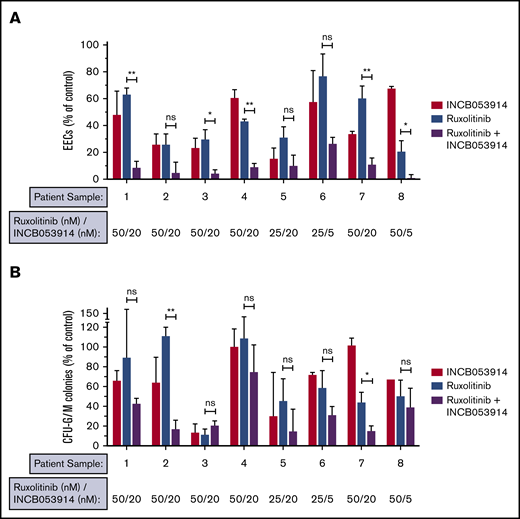
INCB053914 and ruxolitinib can synergize to inhibit the neoplastic growth of primary MPN cells. Primary peripheral blood mononuclear cells from JAK2V617F-positive MPN patients were plated in methylcellulose containing stem cell factor, interleukin-3, and GM-CSF, and colony formation was assessed in the presence of DMSO (0.1%), INCB053914, ruxolitinib, or the combination of the 2 drugs at the indicated concentrations. Epo-independent erythroid colony (EEC) formation (A) and CFU-G/M colony formation (B) were enumerated and are expressed as a percent of the number of colonies obtained with DMSO treatment. Sample 1 is from an essential thrombocythemia patient; samples 2, 3, 4, 6, 7, and 8 are from polycythemia vera patients; and sample 5 is from a patient with myelofibrosis. Additional information on the patients from which these samples were obtained is provided in supplemental Table 1. Samples 3 and 6 are from the same patient but samples were obtained about 6 months apart. Lower INCB053914 (5 nM) was assessed in the second sample (sample 6) because 20 nM was highly effective initially (sample 3). The sample remained sensitive to the combination even at this lower INCB053914 concentration. Error bars indicate SD. *P < .05; **P < .01. ns, not significant by unpaired Student t test.
INCB053914 and ruxolitinib can synergize to inhibit the neoplastic growth of primary MPN cells. Primary peripheral blood mononuclear cells from JAK2V617F-positive MPN patients were plated in methylcellulose containing stem cell factor, interleukin-3, and GM-CSF, and colony formation was assessed in the presence of DMSO (0.1%), INCB053914, ruxolitinib, or the combination of the 2 drugs at the indicated concentrations. Epo-independent erythroid colony (EEC) formation (A) and CFU-G/M colony formation (B) were enumerated and are expressed as a percent of the number of colonies obtained with DMSO treatment. Sample 1 is from an essential thrombocythemia patient; samples 2, 3, 4, 6, 7, and 8 are from polycythemia vera patients; and sample 5 is from a patient with myelofibrosis. Additional information on the patients from which these samples were obtained is provided in supplemental Table 1. Samples 3 and 6 are from the same patient but samples were obtained about 6 months apart. Lower INCB053914 (5 nM) was assessed in the second sample (sample 6) because 20 nM was highly effective initially (sample 3). The sample remained sensitive to the combination even at this lower INCB053914 concentration. Error bars indicate SD. *P < .05; **P < .01. ns, not significant by unpaired Student t test.
INCB053914 and ruxolitinib suppress JAK2V617F-driven tumor growth, and INCB053914 antagonizes ruxolitinib persistent myeloproliferation in vivo
To assess the efficacy of INCB053914 and ruxolitinib combination therapy in vivo, we first used a JAK2V617F-driven xenograft model. We have previously established the pharmacodynamics of INCB053914 in some in vivo models of hematologic malignancies.40 CB17 SCID mice were injected subcutaneously with SET2 cells, and tumors were allowed to form. Treatment with INCB053914 (100 mg/kg twice per day) or a low dose of ruxolitinib (30 mg/kg twice per day) alone had little effect on the growth of SET2 cell tumors (Figure 6A). Tumors of mice treated with vehicle or single agents continued to increase through the 14 days of treatment, with no significant effect of either drug at any time point. The observed lack of inhibition by ruxolitinib was likely a result of the low dose used (30 mg/kg twice per day), which is half the amount generally used for in vivo MPN models. However, tumors of mice treated with the combination of INCB053914 and ruxolitinib did not increase over the first 11 days of treatment (Figure 6A) and exhibited only a slight increase after 14 days of treatment, which was not statistically significant (P = .24 by unpaired Student t test).
![INCB053914 synergizes with ruxolitinib to inhibit MPN model cell tumor formation and antagonizes the development of MPN that persists during ruxolitinib treatment of a murine MPN model. (A) CB17 SCID mice were subcutaneously injected with SET2 cells and tumors were allowed to form for 13 days. At that point, mice were randomly assigned to generate 4 cohorts with equal average tumor sizes. Mice were treated with vehicle (n = 10, starting tumor size mean ± standard error of the mean [SEM] = 129 ± 25 mm3), ruxolitinib (30 mg/kg) (n = 7; mean, 140 ± 25 mm3), INCB053914 (100 mg/kg) (n = 9; mean, 130 ± 28 mm3 mg/kg), or a combination of the 2 drugs (n = 10; mean, 128 ± 18 mm3) twice per day by oral gavage. The average percent increase in tumor size for each treatment is shown over time (mean ± SEM). A statistically significant difference between each monotherapy and the combination was observed. (B) The MPL-W515L bone marrow transplant mouse model of MPN was used to assess the effects of INCB053914 alone and in combination with ruxolitinib on the development of MPN in vivo. Mouse bone marrow cells were retrovirally infected with virus containing the MPL-W515L MPN-driving oncogene and injected into mice whose bone marrow was ablated with 5-fluorouracil. Blood cell counts were performed 7 days after transplantation, and on that day, 4 cohorts (n = 9 each) with equal mean platelet numbers were generated for treatment with vehicle (mean platelet count ± SEM = 673 ± 34 × 109/L), INCB053914 (100 mg/kg; mean platelet count, 673 ± 32 × 109/L), ruxolitinib (60 mg/kg; mean platelet count, 667 ± 29 × 109/L), and a combination of those doses of INCB053914 and ruxolitinib (mean platelet count, 665 ± 26 × 109/L) twice per day orally. Blood counts were then performed after 7, 14, 21, and 35 days of treatment. White blood cell (WBC) counts (B) and platelet counts (C) are shown over days of treatment (boxes indicate 25th to 75th percentile, whiskers indicate the range, and the horizontal line indicates the median). WBC counts at day 35 of treatment were not obtained because of machine error. (D) Spleen weights of vehicle- (n = 4) and INCB053914 monotherapy-treated (n = 1) animals were obtained when they became moribund on day 8 of treatment; the remaining vehicle- and INCB053914 monotherapy–treated animals were moribund on day 14 of treatment. Three mice each for the ruxolitinib monotherapy and the combination therapy cohorts were euthanized to assess disease progression (spleen weight) on day 21 of treatment. Treatment continued for 35 days total (day 42 following transplantation) at which point the experiment was stopped, all animals were euthanized, and spleens were weighed. Mean spleen weights are shown (± SEM) over time. (E) Scoring of reticulin bone marrow in ruxolitinib-treated mice and mice treated with the combination therapy (data points represent the average of the average of 3 fields of n = 4 mice ± SD). *P < .05; **P < .01; ***P < .001; ****P < .0001 by unpaired Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/22/10.1182_bloodadvances.2019000260/1/m_advancesadv2019000260f6.png?Expires=1769081475&Signature=KEUyVr8ohHCKmHnIH593d4hHuRKxhGQbEHclGM1PnpLwi-I20Cj1I9xHSlxZvkozucxeZQlTAzLDZZ6-zsSCtL7oHzo30Yfqj39AinZCa38SNVigmlGr3toEVoo4wmry1yNIzZOZh8BdAPsm-6wYgJtUIOHcX0pQ7x4ULjrten0a1kLQoIt5pQR8JK3Wvis1OmdGHKdevfFcrFhAZZo7ajZBEBC7L-4l8amC6mb2db3cM4NuraloIVAemKOZytlJxp4m~bYBmnpmnjp8HgzOM4pq3x8Bq~bFgzEPhVfth3GgYwLWXhy461FdAM8E~IQsSthK1Pw66Yy2zviJkJAVTA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
INCB053914 synergizes with ruxolitinib to inhibit MPN model cell tumor formation and antagonizes the development of MPN that persists during ruxolitinib treatment of a murine MPN model. (A) CB17 SCID mice were subcutaneously injected with SET2 cells and tumors were allowed to form for 13 days. At that point, mice were randomly assigned to generate 4 cohorts with equal average tumor sizes. Mice were treated with vehicle (n = 10, starting tumor size mean ± standard error of the mean [SEM] = 129 ± 25 mm3), ruxolitinib (30 mg/kg) (n = 7; mean, 140 ± 25 mm3), INCB053914 (100 mg/kg) (n = 9; mean, 130 ± 28 mm3 mg/kg), or a combination of the 2 drugs (n = 10; mean, 128 ± 18 mm3) twice per day by oral gavage. The average percent increase in tumor size for each treatment is shown over time (mean ± SEM). A statistically significant difference between each monotherapy and the combination was observed. (B) The MPL-W515L bone marrow transplant mouse model of MPN was used to assess the effects of INCB053914 alone and in combination with ruxolitinib on the development of MPN in vivo. Mouse bone marrow cells were retrovirally infected with virus containing the MPL-W515L MPN-driving oncogene and injected into mice whose bone marrow was ablated with 5-fluorouracil. Blood cell counts were performed 7 days after transplantation, and on that day, 4 cohorts (n = 9 each) with equal mean platelet numbers were generated for treatment with vehicle (mean platelet count ± SEM = 673 ± 34 × 109/L), INCB053914 (100 mg/kg; mean platelet count, 673 ± 32 × 109/L), ruxolitinib (60 mg/kg; mean platelet count, 667 ± 29 × 109/L), and a combination of those doses of INCB053914 and ruxolitinib (mean platelet count, 665 ± 26 × 109/L) twice per day orally. Blood counts were then performed after 7, 14, 21, and 35 days of treatment. White blood cell (WBC) counts (B) and platelet counts (C) are shown over days of treatment (boxes indicate 25th to 75th percentile, whiskers indicate the range, and the horizontal line indicates the median). WBC counts at day 35 of treatment were not obtained because of machine error. (D) Spleen weights of vehicle- (n = 4) and INCB053914 monotherapy-treated (n = 1) animals were obtained when they became moribund on day 8 of treatment; the remaining vehicle- and INCB053914 monotherapy–treated animals were moribund on day 14 of treatment. Three mice each for the ruxolitinib monotherapy and the combination therapy cohorts were euthanized to assess disease progression (spleen weight) on day 21 of treatment. Treatment continued for 35 days total (day 42 following transplantation) at which point the experiment was stopped, all animals were euthanized, and spleens were weighed. Mean spleen weights are shown (± SEM) over time. (E) Scoring of reticulin bone marrow in ruxolitinib-treated mice and mice treated with the combination therapy (data points represent the average of the average of 3 fields of n = 4 mice ± SD). *P < .05; **P < .01; ***P < .001; ****P < .0001 by unpaired Student t test.
INCB053914 synergizes with ruxolitinib to inhibit MPN model cell tumor formation and antagonizes the development of MPN that persists during ruxolitinib treatment of a murine MPN model. (A) CB17 SCID mice were subcutaneously injected with SET2 cells and tumors were allowed to form for 13 days. At that point, mice were randomly assigned to generate 4 cohorts with equal average tumor sizes. Mice were treated with vehicle (n = 10, starting tumor size mean ± standard error of the mean [SEM] = 129 ± 25 mm3), ruxolitinib (30 mg/kg) (n = 7; mean, 140 ± 25 mm3), INCB053914 (100 mg/kg) (n = 9; mean, 130 ± 28 mm3 mg/kg), or a combination of the 2 drugs (n = 10; mean, 128 ± 18 mm3) twice per day by oral gavage. The average percent increase in tumor size for each treatment is shown over time (mean ± SEM). A statistically significant difference between each monotherapy and the combination was observed. (B) The MPL-W515L bone marrow transplant mouse model of MPN was used to assess the effects of INCB053914 alone and in combination with ruxolitinib on the development of MPN in vivo. Mouse bone marrow cells were retrovirally infected with virus containing the MPL-W515L MPN-driving oncogene and injected into mice whose bone marrow was ablated with 5-fluorouracil. Blood cell counts were performed 7 days after transplantation, and on that day, 4 cohorts (n = 9 each) with equal mean platelet numbers were generated for treatment with vehicle (mean platelet count ± SEM = 673 ± 34 × 109/L), INCB053914 (100 mg/kg; mean platelet count, 673 ± 32 × 109/L), ruxolitinib (60 mg/kg; mean platelet count, 667 ± 29 × 109/L), and a combination of those doses of INCB053914 and ruxolitinib (mean platelet count, 665 ± 26 × 109/L) twice per day orally. Blood counts were then performed after 7, 14, 21, and 35 days of treatment. White blood cell (WBC) counts (B) and platelet counts (C) are shown over days of treatment (boxes indicate 25th to 75th percentile, whiskers indicate the range, and the horizontal line indicates the median). WBC counts at day 35 of treatment were not obtained because of machine error. (D) Spleen weights of vehicle- (n = 4) and INCB053914 monotherapy-treated (n = 1) animals were obtained when they became moribund on day 8 of treatment; the remaining vehicle- and INCB053914 monotherapy–treated animals were moribund on day 14 of treatment. Three mice each for the ruxolitinib monotherapy and the combination therapy cohorts were euthanized to assess disease progression (spleen weight) on day 21 of treatment. Treatment continued for 35 days total (day 42 following transplantation) at which point the experiment was stopped, all animals were euthanized, and spleens were weighed. Mean spleen weights are shown (± SEM) over time. (E) Scoring of reticulin bone marrow in ruxolitinib-treated mice and mice treated with the combination therapy (data points represent the average of the average of 3 fields of n = 4 mice ± SD). *P < .05; **P < .01; ***P < .001; ****P < .0001 by unpaired Student t test.
To assess the efficacy of INCB053914 and ruxolitinib in syngeneic, immunocompetent mice, we used a bone marrow transplant model of MPN that is induced by the MPN-driving MPL-W515L oncoprotein.42 Balb/c mice were transplanted with bone marrow retrovirally transduced to express MPL-W515L. Beginning 7 days posttransplant, cohorts of recipient mice were left untreated or treated twice per day with INCB053914 (100 mg/kg), ruxolitinib (60 mg/kg), or the 2 drugs in combination at the same doses as in the monotherapies. As a monotherapy, INCB053914 did not affect leukocytosis compared with that in vehicle-treated animals (Figure 6B). Ruxolitinib, however, prevented the leukocytosis that was observed in vehicle- and INCB053914-treated animals after 7 days of treatment. At this time point, ruxolitinib-treated mice did not exhibit an increase in white blood cells (WBCs). However, ruxolitinib-treated animals displayed an increase in WBCs at 14 days of treatment and had increased cell counts after 21 days of treatment that were similar to those observed 2 weeks earlier in animals treated with vehicle and INCB053914. Thus, although ruxolitinib treatment delayed leukocytosis, leukocytosis persisted and increased over time during ruxolitinib therapy (Figure 6B). The addition of INCB053914 to ruxolitinib as a combination therapy significantly inhibited this ruxolitinib-persistent leukocytosis after both 14 and 21 days of treatment (Figure 6B). Furthermore, the combination of ruxolitinib and INCB053914 prevented the increasing thrombocytosis observed in mice treated for 14 and 21 days with ruxolitinib alone (Figure 6C). Although thrombocytosis was evident in mice treated with the combination therapy after 35 days of treatment, significant suppression of platelet levels was still observed compared with mice treated with ruxolitinib only. Thus, administration of INCB053914 along with ruxolitinib inhibited thrombocytosis through 3 weeks of treatment, and suppression was still evident for an additional 2 weeks compared with ruxolitinib-treated animals (Figure 6C). Similar suppression of monocytes and neutrophils was observed with the combination therapy compared with ruxolitinib monotherapy (supplemental Figure 5A). The addition of INCB053914 to ruxolitinib also suppressed the persistent splenomegaly observed in ruxolitinib-treated mice through termination of the experiment after 35 days of treatment (Figure 6D). Fibrosis indicated by reticulin staining of bone marrow cells that was observed in ruxolitinib-treated animals was significantly reduced in mice treated with the combination therapy46 (Figure 6E; supplemental Figure 6A). Examination of hematoxylin and eosin–stained bone marrow sections suggested a trend that was not statistically significant toward fewer megakaryocytes in mice treated with the combination therapy compared with ruxolitinib alone (supplemental Figure 6B-C). INCB053914 did not suppress red blood cell, hemoglobin, or hematocrit levels compared with those in ruxolitinib-treated mice, and animal body weights were not affected by drug treatment, which suggests that the combination therapy was well tolerated (supplemental Figure 5B-C). These data indicate that INCB053914 can effectively antagonize the development of MPN that persists during ruxolitinib treatment.
Discussion
Combination therapies that target downstream components of JAK2 signaling or signaling pathways parallel to JAK2 signaling could enhance the efficacy of JAK2 inhibitors and provide more effective therapeutic options for MPN patients. Targeting parallel pathways could enhance the efficacy of JAK2 inhibitors if those pathways mediate redundant or compensatory growth and survival signals to JAK2 signaling. For example, Stivala et al47 recently demonstrated that growth factors such as PDGF can maintain ERK activation in MPN cells after JAK2 inhibition, and this contributes to ruxolitinib persistence. Targeting key proteins that function downstream of JAK2 could enhance JAK2 inhibitor efficacy because JAK2 inhibitors alone seem limited in their ability to suppress JAK2 signaling to the extent required to reduce allele burden in patients. Co-targeting key signaling components downstream of JAK2 may facilitate more complete suppression of JAK2 signaling and improved responses to JAK2 inhibition.
PIM kinases elicit both promitogenic and antiapoptotic effects in cells and function downstream of JAK/STAT signaling.9,12,13,28,34,35,48-55 Such activities of PIM kinases, along with the fact that aberrant expression of PIMs can contribute to lymphomagenesis,53,54,56-58 suggest that PIMs can positively contribute to neoplastic cell growth in MPNs, because MPNs are driven by aberrant JAK2 signaling induced by JAK2, Mpl, or CalR mutation.59,60 Thus, PIM kinases may provide a potential target for anti-MPN therapy, both as a single agent and in combination with JAK2 inhibition. We and others have shown that PIM inhibition can suppress MPN cell growth and synergize with JAK2 inhibition in both MPN model cell lines and primary cells from MPN patients. This includes the pan-PIM inhibitor AZD1208 and other structurally diverse PIM inhibitors.36,37 Recently, we reported the development of a pan-PIM inhibitor INCB053914 and its efficacy in multiple preclinical models of hematopoietic cancers.40 INCB053914 is 1 of a few PIM inhibitors that effectively target all 3 PIM family members which, given the functional similarity of PIMs, could be critical in preventing compensatory signaling by other PIM family members when challenged with a non–pan-PIM inhibitor.61,62
The data reported here suggest that INCB053914 is effective at targeting MPN cell growth and augments the potency of the JAK1/2 inhibitor ruxolitinib. This is demonstrated in JAK2V617F-driven MPN model cells as well as in primary cells when measuring the hallmark Epo-independent neoplastic growth of primary MPN cells. This latter measurement is widely used to assess anti-MPN therapeutics and combination therapies with JAK2 inhibition.36,43,45 The efficacy of INCB053914 to antagonize this neoplastic growth of primary cells was particularly impressive, with efficacy in the low nanomolar range (Figure 5A). This potency of INCB053914 is superior to that reported for other PIM inhibitors in this assay.36 INCB053914 in combination with ruxolitinib significantly suppressed neoplastic colony formation of primary MPN cells, with numerous patient samples showing synergistic suppression at the concentrations of drugs used (Figure 5A).
The combination of INCB053914 and ruxolitinib antagonized cell growth and viability compared with single-agent effects of the drugs. This was partly the result of enhanced apoptosis induced by the combination of drugs compared with the effects of single agents (Figure 3A). Mechanistically, PIM can inactivate the proapoptotic activity of BAD via phosphorylation of serine 112.12 This leads to 14-3-3 protein binding to BAD and prevents BAD from antagonizing the antiapoptotic function of Bcl-2 and Bcl-xL.63 Interestingly, in SET2 cells, we observe little effect on pBAD(S112) with concentrations of INCB053914 up to 1 μM (Figure 3B). In the same experiment, 0.1 μM ruxolitinib treatment led to a slight inhibition of pBAD(S112). However, the combination of just 0.1 μM of INCB053914 with ruxolitinib resulted in significant loss of pBAD(S112) (Figure 3B), which correlates with the observed increase in apoptosis with the combination of drugs (Figure 3A). Because ruxolitinib is known to induce apoptosis of JAK2V617F-driven cells, one interpretation of our data is that PIM inhibition enhances the efficacy of ruxolitinib to induce apoptosis in part by disrupting the regulation of the proapoptotic activity of BAD. This is consistent with previous work that has placed BAD inactivation as a key effector of JAK2V617F signaling.35,64 Although our data indicate that ruxolitinib seems to sensitize the levels of pBAD(S112) to PIM inhibition via INCB053914 (Figure 3B-C), the mechanism for this remains to be explored. It could be the result of decreased PIM expression that occurs in response to JAK2 inhibition.35,36 The degree of suppression of pSTAT5(Y694) levels in SET2 and BaF3-JAK2V617F cells (Figure 3B-C) did not correlate with the extent of growth suppression (Figure 2B-C). In addition, the combination of ruxolitinib and INCB053914 differentially altered pSTAT5(Y694) in these cell lines, because the combination did not alter pSTAT5(Y694) levels below the level achieved after ruxolitinib treatment alone in SET2 cells (Figure 3B), but did alter them in BaF3-JAK2V617F cells (Figure 3C). However, in both cell lines, the significant increase in apoptosis (Figure 3A) and growth inhibition (Figure 2B-C) in response to the combination of drugs correlated with combined suppression of both pSTAT5(Y694) and pBAD(S112). This suggests that JAK2-dependent signaling pathways may be differentially sensitive to JAK2 inhibition and differentially contribute to cell growth and survival in different MPN model cells.
Similarly, the combination of INCB053914 and ruxolitinib synergistically suppressed phosphorylation of biomarkers of mTORC1 activation (Figure 4). This is consistent with the ability of PIM to promote mTORC1 signaling by phosphorylating PRAS40, which antagonizes the ability of PRAS40 to inhibit mTORC110,11 and is consistent with the effects of other PIM inhibitors.36,37,44,61 Because mTORC1 regulates a large number of cellular pathways, including metabolism and protein synthesis,15 the cellular effects of the combination of ruxolitinib and INCB053914 is likely the result of the combination of altered regulation of numerous critical cellular processes. Targeting mTOR has also demonstrated positive anti-MPN effects as a single agent and in combination with JAK2 inhibition.45,65-69
The potency of the combination of INCB053914 and ruxolitinib was also manifested in in vivo MPN models driven by aberrant JAK2 signaling. The combination of INCB053914 and ruxolitinib antagonized the growth of SET2 cell xenograft tumors at doses of each drug that had little to no effect as monotherapies (Figure 6A). Likewise, the combination of INCB053914 and ruxolitinib suppressed myeloproliferation induced by retroviral transduction of the MPL-W515L MPN-driving mutation in an in vivo MPN model widely used for anti-MPN therapeutic studies. In this model, unlike INCB053914 monotherapy, ruxolitinib treatment alone suppressed both leukocytosis and thrombocytosis compared with vehicle control (Figure 6B-C). However, ruxolitinib-treated mice, while responding at first, developed an increase in WBCs and platelets over time, suggesting that the initial efficacy of treatment eventually gave way to a progressive myeloproliferative phenotype that persisted during ruxolitinib therapy. Importantly, INCB053914 antagonized this ruxolitinib persistence and significantly delayed the onset of progressive disease, including splenomegaly, through the end of the therapeutic assessment (Figure 6B-D). This is consistent with our observation that this drug combination could enhance inhibition of CFU-G/M colony formation of primary human MPN cells (Figure 5B).
We did not detect reticulin staining indicative of fibrosis in the bone marrow of vehicle- or INCB053914-treated mice (not shown), likely because of the rapid demise of these animals (supplemental Figure 5D) before clear fibrosis could occur. Animals treated with ruxolitinib, however, survived 3 weeks longer and exhibited clear reticulin fibrosis in their bone marrow. This bone marrow fibrosis was abolished in mice treated with the combination of ruxolitinib and INCB053914 (Figure 6E; supplemental Figure 6A). A trend toward a lower level of megakaryocytes in the bone marrow of mice treated with the combination compared with ruxolitinib was also detected (supplemental Figure 6B-C), which could be significant, given the role of megakaryocytes in driving MPN.70-72 Consistent with our observations that INCB053914 antagonized disease that persisted during ruxolitinib treatment in our in vivo model studies (Figure 6; supplemental Figures 5A and 6A), INCB053914 also effectively antagonized the growth of ruxolitinib persistent SET2 and BaF3-JAK2VF cells (supplemental Figure 7), supporting the notion that INCB053914 may antagonize ruxolitinib resistance in patients. The ability of PIM inhibition to antagonize a ruxolitinib persistent state further supports a role for PIM in targeted therapy resistance as described in other cancer models.17,25,26 In addition, we have previously demonstrated that forced PIM1 expression can induce JAK2V617F-driven ruxolitinib persistent growth.36 Future experiments to assess the effects of ruxolitinib and INCB053914 in additional MPN therapeutic models, such as JAK2V617F transgenic mice, could provide further insight into this combination therapy for MPN.73,74
The 3 PIM family members are widely expressed, and PIM triple knockouts remain viable and fertile, suggesting that pan-PIM inhibition may not have overt toxicity. However, PIM family members play roles in HSCs and in cytokine signaling, suggesting that pan-PIM inhibition may affect myeloid and lymphoid cell functions. INCB053914 treatment is tolerated in mice,40 as is the combination of INCB053914 and ruxolitinib in this study. However, because both drugs target critical signaling pathways in blood cell biology, dosing modification of each drug may be needed to mitigate adverse effects in patients treated with this combination. INCB053914 displays impressive specificity to PIM kinases, with only 1 other kinase, PAS kinase, being similarly targeted.40 PAS kinase–deficient mice develop and grow normally75 but display metabolic phenotypes in the liver and other tissues,76,77 suggesting that metabolic effects resulting from targeting PIM kinases with INCB053914 are a possibility.
In summary, our findings support the notion that INCB053914 may be effective against JAK2-driven MPNs in humans, especially in the context of combination therapy with ruxolitinib. INCB053914 may enhance the efficacy of ruxolitinib upfront and may antagonize the development of ruxolitinib persistence in patients. Our data support the clinical assessment of combining INCB053914 and ruxolitinib to improve the efficacy of JAK2-targeted therapy for MPN patients. INCB053914 is currently being investigated in patients with advanced hematologic malignancies (ClinicalTrials.gov identifier: NCT02587598), including testing its combination with ruxolitinib in myelofibrosis patients who are currently receiving ruxolitinib but have suboptimal responses.
Acknowledgments
The authors thank John Cleveland, Bijal Shah, Jianguo Tao, Eric Padron, and Christopher Letson for helpful discussions, and John Cleveland for critical review of the manuscript; the staff of the Flow Cytometry Core of the H. Lee Moffitt Cancer Center and Research Institute for their expertise; Becky Stewart, Denise Hertel, and Julian Oliver for their assistance with histopathology; and Brittney Fiorentino for assistance with acquisition of patient samples.
This work was supported in part by a sponsored research agreement between G.W.R., P.K.E.-B., and Incyte Corporation, by the Flow Cytometry Core at the H. Lee Moffitt Cancer Center and Research Institute, and by the H. Lee Moffitt Cancer Center Support Grant (P30-CA076292), from the National Institutes of Health, National Cancer Institute.
Authorship
Contribution: L.M., R.J.C., G.P., Q.T.L.-S., N.E.A., and G.W.R. designed and performed the research studies; L.M., R.J.C., G.P., L.Z., M.C.S., H.K.K., P.K.E.-B., and G.W.R. analyzed and interpreted data; and G.W.R. wrote the manuscript.
Conflict-of-interest disclosure: G.W.R. and P.K.E.-B. received funding from Incyte Corporation to support this work. R.J.C., M.C.S., and H.K.K. are employees and stockholders of Incyte Corporation. The remaining authors declare no competing financial interests.
Correspondence: Gary W. Reuther, Department of Molecular Oncology, Moffitt Cancer Center and Research Institute, 12902 Magnolia Dr, Tampa, FL 33612; e-mail: gary.reuther@moffitt.org.

