

Key Points
Expression of Hippo component TAZ is downregulated in MM through hypermethylation.
TAZ reexpression, exogenously or pharmacologically, causes apoptosis and enhances sensitivity to anti-MM therapies by downregulating MYC.

Abstract
Multiple myeloma (MM) is an incurable blood cancer that is often characterized by amplification and overexpression of the MYC oncogene. Despite efforts, direct targeting of MYC is not yet possible; therefore, alternative strategies to inhibit MYC activity are necessary. TAZ is a transcriptional coactivator downstream of the Hippo-signaling pathway that functions as an oncogene in many solid tumors. However, its role in hematological malignancies is largely unexplored. Here, we show that, in contrast to solid tumors, expression of TAZ is lower in hematological malignancies, and that high expression of TAZ correlates with better patient outcomes. We further show that TAZ is hypermethylated in MM patient samples and in a panel of MM cell lines. Genetic overexpression of TAZ or pharmacological upregulation of TAZ by treatment with the demethylating agent decitabine induces apoptosis. Importantly, TAZ-induced apoptosis is independent of canonical Hippo components LATS1 or the TEA-domain family of transcription factors. Instead, RNA-sequencing analysis revealed that overexpression of TAZ represses a MYC transcriptional program and we show that increased TAZ expression correlates with decreased MYC expression in both cell-line models and patient samples. Furthermore, promoter derepression of TAZ expression sensitizes MM cell lines through a reciprocal reduction in MYC expression using additional therapeutics such as bortezomib, trichostatin A, and panobinostat. Our findings uncover an unexpected role for TAZ in MM tumorigenesis and provide a compelling rationale for exploring the therapeutic potential of upregulating TAZ expression to restore sensitivity to specific therapeutics in MM.
Introduction
Multiple myeloma (MM) is characterized by the malignant accumulation of monoclonal plasma cells. Early transformative events in the germinal B center result in structural genetic rearrangements and copy-number alterations that render cells more sensitive to secondary events that drive disease progression.1,2 Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM) represent asymptomatic precursors of fully active MM.3 Evolution through these phases results in increasing clonal heterogeneity due to epigenetic changes, secondary genetic events, or interactions with the bone marrow microenvironment.4
A common secondary event during MM progression involves amplification of the MYC oncogene by secondary translocations.5 These complex translocations place the MYC gene next to active superenhancers in its partner loci that drive MYC expression.6 MYC exerts its neoplastic effects by acting as either a transcriptional activator or repressor depending on its binding partners.7,8 The MYC protein binds to 10% to 15% of the genome and is pivotal in regulating cell-cycle progression, cell growth, and metabolism, and in mediating an immune response.7 In MM, a MYC activation signature was identified in 67% of active MM patients and was less frequently observed in patients with MGUS or SMM.9 Given the addiction to MYC in MM, several different approaches have been developed to inhibit this oncogene, including both direct and indirect targeting of MYC.7 Some of these approaches such as lenalidomide10,11 and bromodomain and extraterminal motif (BET) inhibitors12 indirectly target MYC transcription; improved knowledge of mechanisms responsible for MYC deregulation in MM will enable the identification of additional vulnerabilities and better therapeutic options in MYC-driven tumors.
TAZ, encoded by the WWTR1 gene, is a transcriptional coactivator that is best described for its role within the Hippo signaling pathway. The core signaling cassette includes the 20-like MST1/2 that phosphorylate and activate LATS1/2, which leads to subsequent phosphorylation and cytoplasmic retention of 2 transcriptional coactivator paralogs, YAP and TAZ. Unphosphorylated TAZ or YAP translocate to the nucleus to bind the TEA-domain (TEAD) family of transcription factors. In solid tumors, YAP and TAZ primarily induce a prosurvival, antiapoptotic transcriptional program.13
Although YAP and TAZ have been shown to be dispensable for hematopoiesis,14 the role of the Hippo-signaling pathway in hematological malignancies is less well defined. Whereas several studies show that loss of LATS2 expression in patient samples15-17 or upregulation of YAP or TAZ in cell-line models18-20 promote blood cancer progression, much like in solid tumors, several studies suggest an alternative role for these Hippo-pathway components. For example, higher levels of LATS2 were observed in newly diagnosed acute myeloid leukemia patients21 or chronic myeloid leukemia patients22 compared with healthy controls. Similarly, chemical activation of YAP inhibited cell growth and tumor growth in myeloma mouse models.23 Cottini et al showed that high expression of YAP correlated with improved survival outcomes in MM patients and provided a mechanism by which c-Abl phosphorylates YAP to induce a proapoptotic phenotype in myeloma, leukemia, and lymphoma cell-line models.24 The currently understood role of TAZ in MM has been limited to the study of its effects on the osteogenic potential of the mesenchymal stem cell. Compared with healthy controls, mesenchymal stem cells from MM patients have reduced osteogenic potential and lower levels of TAZ expression.25 This study suggests that TAZ plays a protective role by lessening the development of osteolytic lesions commonly seen in MM patients.
To gain better insight into the role of TAZ in MM, we used cell-line models and patient data sets to show that TAZ elicits an antitumorigenic and proapoptotic function by selectively repressing MYC expression and its transcriptional program. We further show that demethylating the TAZ promoter upregulates TAZ expression and subsequently sensitizes cells to commonly used antimyeloma therapeutics.
Materials and methods
Cell lines and reagents
For detailed culture conditions for KMM1, KMS11, JJN3, U266, and A549 cells, see supplemental Materials and methods. To generate cell lines expressing wild-type or mutant TAZ, cells were transduced using a spinoculation protocol with lentivirus-expressing wild-type TAZ (TAZ), TAZ with point mutations (W152A, P155A) in the WW domain (TAZ-WWm), a TEAD-binding mutant (TAZ-F52A/F53A) or empty vector control (WPI). Lentiviral plasmids were generously provided by Xiaolong Yang (Queen’s University, Kingston, ON, Canada). Decitabine (DAC), bortezomib (BTZ), trichostatin A (TSA), cycloheximide, and MG132 were purchased from Sigma; panobinostat (Pano) was obtained from Caymann Chemicals.
Cell-proliferation and cell-viability assays
For cell-proliferation assays and viability assays, immediately after spinoculation, 1 × 104 cells were plated in triplicate in 24-well plates. Using the trypan blue exclusion assay, total cell viable number and percentage of cell death were calculated. For combination treatments, 5 × 103 cells were plated in 96-well plates. After 24 hours, cells were treated with DAC for 48 hours, followed by treatment with BTZ, TSA, or Pano for 72 hours. Viability was measured using the PrestoBlue assay (Thermo Fisher).
Immunoblotting and antibodies
Cells were lysed in radioimmunoprecipitation assay lysis buffer (50 mM Tris, 150 mM NaCl, 1% NP40, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate supplemented with protease inhibitors) and quantified using the Pierce BCA Protein Assay kit (ThermoScientific). Commercial antibodies were as follows: TAZ and β-actin (Abcam); YAP, STAT1, baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5), and signaling lymphocytic activation molecule F7 (SLAMF7) (Santa Cruz Biotechnology); LATS1, MST1, BIM, and cysteine-rich angiogenic inducer 61 (CYR61) (Cell Signaling Technology); and MYC (ThermoFisher).
Gene expression profiling
RNA was extracted 4 days after viral transduction using the RNeasy kit following the manufacturer’s instructions (Qiagen). The poly A library was prepared using the RNA Seq V2 kit (ThermoFisher). Barcoded libraries were equally pooled and amplified onto ion sphere particles supplied by the Ion Pi HiQ OT2 kit (Life Technologies). Ion sphere particles loaded with libraries were sequenced on the Ion Torrent Proton sequencer using Ion PI chip V3. Detailed methods for library preparation can be found in supplemental Materials and methods.
The raw read files were mapped to the human reference genome GRCh37/hg19 using the RNA-sequencing (RNA-seq) analysis plugin (v5.4.0.1) from Ion Torrent Suite data (v5.4.0). Differentially expressed genes included those with P ≤ .05 and log2 fold change over 2 or log2 fold change under −2. A heatmap of differentially expressed genes was created using log2 values. Gene-set enrichment analysis (GSEA) was performed using the publicly available software from the Broad Institute26 using the Hallmark data set (h.all.v6.2.symbols.gmt).27 A false discovery rate ≤0.05 was considered significant.
Quantitative PCR
RNA was isolated as described. Complementary DNA was synthesized from 1 μg of RNA using qScript cDNA supermix (Quanta Biosciences). Quantitative polymerase chain reaction (PCR; qPCR) analysis with PerfeCTa SYBR green supermix, low ROX (Quanta Biosciences) was run on a ViiA7 real-time PCR machine (ThermoFisher Scientific) using 18S ribosomal RNA as internal control. Relative messenger RNA (mRNA) was calculated using the ΔΔCT method. qPCR primers are listed in supplemental Table 1.
Genomic DNA extraction and methylation-specific PCR
Genomic DNA was extracted using the PureLINK Genomic DNA MiniKit (Thermo Fisher Scientific). Bisulfite modification of 500 ng of genomic DNA was performed using the EZ DNA Methylation kit (Zymo Research). Methylation-specific PCR (MSP) was performed using platinum Taq polymerase (Invitrogen) following conditions outlined in supplemental Table 2. Universal methylated human DNA standard (Zymo Research) and human HCT116 DKO nonmethylated DNA standard (Zymo Research) served as controls.
Statistical analysis
Results
Expression of TAZ predicts survival outcomes in patients with MM
To explore the role of TAZ in MM, we probed several gene expression data sets. In patient samples, there was a consistent and significant decrease in TAZ expression as disease progresses from healthy plasma cells through precursor MGUS and SMM phases to fully active MM (P < .001) (Figure 1A; supplemental Figure 1). Dividing MM patients into groups based on TAZ expression, individuals with low expression had significantly shorter overall survival compared with those with high expression (P = .0304) (Figure 1B).

Decreased TAZ expression in MM cell lines and patient samples predicts worse outcomes. (A) Expression data comparing normal plasma cells (NPCs) from healthy subjects (n = 22), MGUS (n = 44), SMM (n = 12), and MM (n = 351), combining data from GSE5900 and GSE2658 using probe set 202134_s_at. Data are presented using box-and-whisker plots according to the Tukey method. The Student t test was used to evaluate significance: **P ≤ .01 and ***P ≤ .001. (B) Overall survival curve relative to TAZ expression in individuals affected by MM, obtained from http://www.canevolve.org/AnalysisResults/AnalysisResults.html and based on GSE2658 (log-rank test). Optimal cutoff determined using Cutoff Finder (http://molpath.charite.de/cutoff).28 (C) Immunoblot analysis of HMCLs for protein expression of TAZ, YAP, LATS1, and MST1 relative to A549 cells. (D) mRNA expression of TAZ, YAP, LATS1, and MST1 relative to A549 cells in HMCLs. A.U., arbitrary unit; HR, hazard ratio; ns, not significant.
Decreased TAZ expression in MM cell lines and patient samples predicts worse outcomes. (A) Expression data comparing normal plasma cells (NPCs) from healthy subjects (n = 22), MGUS (n = 44), SMM (n = 12), and MM (n = 351), combining data from GSE5900 and GSE2658 using probe set 202134_s_at. Data are presented using box-and-whisker plots according to the Tukey method. The Student t test was used to evaluate significance: **P ≤ .01 and ***P ≤ .001. (B) Overall survival curve relative to TAZ expression in individuals affected by MM, obtained from http://www.canevolve.org/AnalysisResults/AnalysisResults.html and based on GSE2658 (log-rank test). Optimal cutoff determined using Cutoff Finder (http://molpath.charite.de/cutoff).28 (C) Immunoblot analysis of HMCLs for protein expression of TAZ, YAP, LATS1, and MST1 relative to A549 cells. (D) mRNA expression of TAZ, YAP, LATS1, and MST1 relative to A549 cells in HMCLs. A.U., arbitrary unit; HR, hazard ratio; ns, not significant.
Similarly, in cell-culture systems, among 4 human myeloma cell lines (HMCLs), TAZ was undetectable (KMM1, KMS11, U266) or only weakly expressed (JJN3) at both protein (Figure 1C) and mRNA levels (Figure 1D) compared with the non–small cell lung cancer cell line A549. By probing the cancer cell line encyclopedia for mRNA expression of TAZ, a noteworthy pattern emerged: TAZ was consistently upregulated in tumor cell lines of epithelial origin but was profoundly downregulated in hematological malignancies (supplemental Figure 2A). Moreover, at the protein level, TAZ is not expressed in several leukemia or lymphoma cell lines (supplemental Figure 2B). Other Hippo-pathway components are also differentially expressed compared with solid tumors. As previously described,24 YAP is also only weakly expressed or undetectable in HMCLs. In contrast, upstream regulators of the Hippo pathway, LATS1 and MST1, which are normally suppressed in solid tumors,30-32 were expressed at higher levels in HMCLs compared with the lung cancer cell line A549 (Figure 1C-D). These results suggest that the role of the Hippo pathway, particularly TAZ, in tumorigenesis is fundamentally different in MM and other hematological malignancies compared with their previously established oncogenic roles in solid tumors.
TAZ is hypermethylated in HMCLs and patient samples
Because TAZ was expressed at neither the mRNA nor the protein level and is rarely mutated in hematopoietic and lymphoid tissues,33 we hypothesized that TAZ was hypermethylated. Hypermethylation is a common mechanism by which cancer cells turn off tumor-suppressor genes, and in MM, the transition from MGUS to active MM is marked by gene-specific hypermethylation at CpG islands.34 An analysis of published CpG methylation microarray data sets35 revealed that methylation of TAZ was greater in a subset of MM patients compared with healthy controls or patients with MGUS (Figure 2A). Probing cell-line data from the cancer cell line encyclopedia also revealed that TAZ is more highly methylated in cell lines derived from patients with hematological malignancies compared with those from solid tumors (supplemental Figure 3).
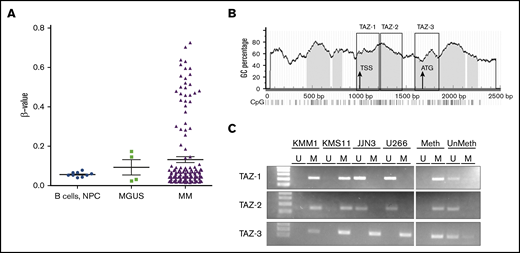
TAZ is hypermethylated in MM. (A) Methylation β values of normal plasma and B cells (n = 9) and plasma cells from MGUS, SMM (n = 4), or MM (n = 161) are shown as mean ± standard error of the mean (SEM) using data from GSE21304.35 β values range from 0 to 1.0, equivalent to 0% to 100% methylation. (B) Prediction of CpG islands in TAZ promoter using MethPrimer (www.urogene.org/methprimer/) with regions highlighted for MSP. (C) MSP of 3 regions within the TAZ promoter reveals hypermethylation in KMM1, KMS11, JJN3, and U266 cell lines. Universally methylated (Meth) and unmethylated (UnMeth) DNA served as MSP controls. GC, guanine and cytosine nucleotides; M, methylated; TSS, transcription start site; U, unmethylated.
TAZ is hypermethylated in MM. (A) Methylation β values of normal plasma and B cells (n = 9) and plasma cells from MGUS, SMM (n = 4), or MM (n = 161) are shown as mean ± standard error of the mean (SEM) using data from GSE21304.35 β values range from 0 to 1.0, equivalent to 0% to 100% methylation. (B) Prediction of CpG islands in TAZ promoter using MethPrimer (www.urogene.org/methprimer/) with regions highlighted for MSP. (C) MSP of 3 regions within the TAZ promoter reveals hypermethylation in KMM1, KMS11, JJN3, and U266 cell lines. Universally methylated (Meth) and unmethylated (UnMeth) DNA served as MSP controls. GC, guanine and cytosine nucleotides; M, methylated; TSS, transcription start site; U, unmethylated.
To validate that TAZ is hypermethylated in our cell-line models, we used MSP to evaluate the presence or absence of methylation in cytosine guanine dinucleotide (CpG) islands within the promoter of the WWTR1 gene. These regions cover the transcriptional start site (TAZ-1, TAZ-2) and a CpG island at the first exon (TAZ-3) (Figure 2B). TAZ is methylated across all 3 regions in KMM1 and KMS11 cells. In contrast, JJN3 and U266 are only methylated in the region around the ATG start site.
Reexpression of TAZ induces cell death in HMCLs
Because TAZ was not expressed in most HMCLs, we used a lentiviral-mediated expression system to restore TAZ expression and investigate its function. In comparison with wild-type KMM1 cells or KMM1 cells expressing empty vector (WPI), cells expressing TAZ failed to proliferate (Figure 3A). Whereas KMM1 or WPI cells had a doubling time of 2.05 ± 0.30 days and 2.46 ± 0.17 days, respectively, the doubling time of TAZ-expressing cells was almost 4 times longer at 8.28 ± 1.63 days (P < .001). Neither a TEAD-binding mutant (F52A/F53A) nor a WW-domain mutant unable to bind LATS1/2 or other PPxY-containing transcription factors36 were able to rescue this effect, suggesting that the regulation of proliferation by TAZ is independent of its roles within the canonical Hippo-signaling pathway.
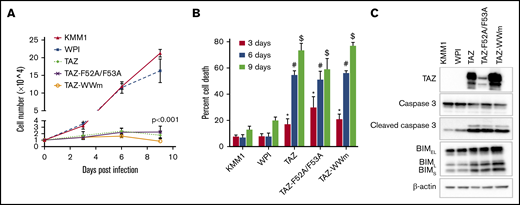
Overexpression of TAZ induces cell death. (A) Proliferation assay of KMM1 cells transduced with empty vector (WPI), wild-type TAZ (TAZ), or TAZ mutants unable to bind TEAD (TAZ-F52A/F53A) or LATS1/2 (TAZ-WWm). Data are mean ± standard deviation (SD) of triplicates. Single-factor analysis of variance (ANOVA) of doubling times indicated significance between all TAZ-expressing lines and either KMM1 or WPI cell lines. (B) Cell-viability assay of cell lines described in panel A after 3, 6, or 9 days posttransduction. Data are mean ± SD of triplicates. *P ≤ .05 of TAZ, #P ≤ .05 of TAZ-F52A/F53A, or $P ≤ .05 of TAZ-WWm compared with KMM1 or WPI cell lines, respectively, using a Student t test. (C) Immunoblot of cell lines described in panel A for markers of apoptosis including cleaved caspase 3 and the 3 isoforms of BIM (BIMEL, BIML, BIMS). β-actin was used as a loading control.
Overexpression of TAZ induces cell death. (A) Proliferation assay of KMM1 cells transduced with empty vector (WPI), wild-type TAZ (TAZ), or TAZ mutants unable to bind TEAD (TAZ-F52A/F53A) or LATS1/2 (TAZ-WWm). Data are mean ± standard deviation (SD) of triplicates. Single-factor analysis of variance (ANOVA) of doubling times indicated significance between all TAZ-expressing lines and either KMM1 or WPI cell lines. (B) Cell-viability assay of cell lines described in panel A after 3, 6, or 9 days posttransduction. Data are mean ± SD of triplicates. *P ≤ .05 of TAZ, #P ≤ .05 of TAZ-F52A/F53A, or $P ≤ .05 of TAZ-WWm compared with KMM1 or WPI cell lines, respectively, using a Student t test. (C) Immunoblot of cell lines described in panel A for markers of apoptosis including cleaved caspase 3 and the 3 isoforms of BIM (BIMEL, BIML, BIMS). β-actin was used as a loading control.
Given that TAZ-expressing cells failed to proliferate, we next asked whether this was due to an increase in cell death. Strikingly, TAZ-expressing cells displayed significantly increased cell death in comparison with wild-type KMM1 or cells expressing the empty vector (WPI). Cell death started as early as 4 days posttransduction and increased through 9 days when 73.40% ± 5.30% of TAZ-expressing cells were no longer viable compared with only 12.91% ± 2.67% and 19.92% ± 2.70% cell death in KMM1 and WPI cells, respectively. As expected, neither the TEAD-binding mutant nor the WW-domain mutants were able to rescue the cell-death phenotype observed in TAZ-expressing cells (Figure 3B). From these results, the lack of proliferation in TAZ-expressing cells is due to increased cell death. Importantly, this effect of TAZ on cell proliferation and cell death was also observed in another HMCL, KMS11 (supplemental Figure 4), highlighting the importance of TAZ in regulating MM cell survival across cellular contexts.
We further confirmed that TAZ induces an antisurvival, proapoptotic response at the molecular level (Figure 3C). Expression of TAZ or its mutants (TAZ-F52A/F53A, TAZ-WWm) resulted in a decrease in total caspase 3 levels with a concomitant increase in cleaved caspase 3. Similarly, all 3 isoforms of the proapoptotic BIM protein were upregulated upon TAZ or mutant TAZ expression. These results, combined with our data showing that the expression of TAZ is lost during MM progression, point to a tumor-suppressor role for TAZ in MM.
TAZ downregulates MYC targets
To better understand the mechanism mediating TAZ-induced cell death, we performed whole-transcriptome analysis and unbiased GSEA. We characterized the transcriptional consequences of reexpressing TAZ in KMM1 cells by comparing cells expressing TAZ (TAZ) to control (WPI). A total of 146 genes were upregulated and 114 genes downregulated by at least log2-fold (supplemental Figure 5). Using the Hallmarks collection from the Molecular Signatures Database (MSigDB),27 GSEA not only revealed an upregulation of apoptotic targets, (Figure 4A), which is consistent with our phenotypic observations (Figure 3B), but also revealed a significant downregulation of MYC targets when TAZ is overexpressed (Figure 4B,D). Importantly, 2 different MYC target sets from the MSigDB Hallmark collection were anticorrelated with TAZ expression. Additional MYC gene sets were also negatively correlated with TAZ expression including Schuhmacher_MYC_Targets_UP and Schlosser_MYC_Targets_and Serum_Response_Up (Figure 4D). These results highlight the reproducibility and specificity of the effect of TAZ on MYC targets. Moreover, targeted GSEA of other key pathways involved in MM pathogenesis such as NF-κB, STAT3, or SMAD37 signaling failed to show enrichment, providing additional evidence that TAZ specifically regulates MYC in MM. In plasma cells from MGUS or MM patients, TAZ expression negatively correlated with MYC expression, where patients with higher TAZ expression were more likely to have reduced MYC expression (P < .001) (Figure 4E; supplemental Figure 6). Together, these results suggest that TAZ confers a selective repression of transcriptional networks regulated by MYC.
![Transcriptomic analysis reveals TAZ as negative regulator of MYC. GSEA reveals enrichment of apoptotic genes (A) and downregulation of MYC targets (B-C) using the MSigDB Hallmark collection in TAZ-overexpressing KMM1 cells relative to WPI-expressing cells. (D) Table of gene sets enriched in TAZ-expressing KMM1 cells, highlighting the number of genes in each set (n), the normalized enrichment score (NES), and test of statistical significance (false discovery rate [FDR] q value). (E) Expression of TAZ and MYC are anticorrelated in MM patient samples combining data from GSE5900 and GSE2658 using probe sets 202134_s_at (TAZ) and 202431_s_at (MYC). (F) Validation of TAZ targets at the mRNA levels using qPCR. Data are mean ± SD of triplicate samples. **P ≤ .01 using the Student t test. (G) Validation of TAZ targets at the protein level. β-actin was used as a loading control.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/22/10.1182_bloodadvances.2019000374/2/m_advancesadv2019000374f4.png?Expires=1769093739&Signature=3YTAc3c0HJkLDqP1WSjAdreiGNAZbpwGPTs3uFPjIp-WyBAn4xyrYEgZs~TPHPEgfY5CGMoSfOT~ypdHTPDKERvj5TFS00rK7PmNf97JtTNJyzINF0J5~CBqj30F-82g9iCnZw060rO4uqeF~8e8Y-KARNzRq~-ySNKw3cBaMP3v6NNikaS9ZE7e9RxXujL4bQVODXdhpE~l3DrJtV-9KoAwgtkU0-t-8mMpMtpsqVzt0~lvFh0ECjpDB~WgOfUrQT-CEJLyhoR6EY6ZwvRX9twBG6iYm4rOoMOQnR~8~AiUYhPVLkVP5XfS-GVId0b2aeSvxeCDlA4AGjVWu8Rg1Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transcriptomic analysis reveals TAZ as negative regulator of MYC. GSEA reveals enrichment of apoptotic genes (A) and downregulation of MYC targets (B-C) using the MSigDB Hallmark collection in TAZ-overexpressing KMM1 cells relative to WPI-expressing cells. (D) Table of gene sets enriched in TAZ-expressing KMM1 cells, highlighting the number of genes in each set (n), the normalized enrichment score (NES), and test of statistical significance (false discovery rate [FDR] q value). (E) Expression of TAZ and MYC are anticorrelated in MM patient samples combining data from GSE5900 and GSE2658 using probe sets 202134_s_at (TAZ) and 202431_s_at (MYC). (F) Validation of TAZ targets at the mRNA levels using qPCR. Data are mean ± SD of triplicate samples. **P ≤ .01 using the Student t test. (G) Validation of TAZ targets at the protein level. β-actin was used as a loading control.
Transcriptomic analysis reveals TAZ as negative regulator of MYC. GSEA reveals enrichment of apoptotic genes (A) and downregulation of MYC targets (B-C) using the MSigDB Hallmark collection in TAZ-overexpressing KMM1 cells relative to WPI-expressing cells. (D) Table of gene sets enriched in TAZ-expressing KMM1 cells, highlighting the number of genes in each set (n), the normalized enrichment score (NES), and test of statistical significance (false discovery rate [FDR] q value). (E) Expression of TAZ and MYC are anticorrelated in MM patient samples combining data from GSE5900 and GSE2658 using probe sets 202134_s_at (TAZ) and 202431_s_at (MYC). (F) Validation of TAZ targets at the mRNA levels using qPCR. Data are mean ± SD of triplicate samples. **P ≤ .01 using the Student t test. (G) Validation of TAZ targets at the protein level. β-actin was used as a loading control.
Importantly, many of the transcriptional targets identified in the RNA-seq analysis were also validated by qPCR (Figure 4F) or western blot (Figure 4G). Previous studies identified many of the TAZ-regulated genes as targets of MYC. For example, in large genome-wide screens for MYC targets, MYC was shown to bind to the promoters of CXCR4, CXCL10, CYR61, STAT1, ID1, TGFB2, and IRF1, among others38,39 . In other studies, MYC upregulated BIRC5 in chronic myeloid leukemia cells40 or downregulated the WNT pathway inhibitor DKK1.41 Our results show that TAZ specifically represses this MYC transcriptional network in KMM1 cells.
TAZ reduced MYC mRNA expression by 1.502 ± 0.058-fold (P = .0071) compared with control WPI-expressing KMM1 cells (Figure 4F). More striking, however, is the significant downregulation of MYC at the protein level (Figure 4G). Further analysis showed that TAZ regulates MYC at both the transcriptional level (supplemental Figure 7A) as well as at the posttranscriptional level (supplemental Figure 7B) by increasing phosphorylation at T58 (supplemental Figure 7C), priming MYC for protein degradation (supplemental Figure 7D).
DAC upregulates TAZ to antagonize a MYC transcriptional program
DAC is a hypomethylating agent that inhibits DNA methyltransferases (DNMTs). It is approved for the treatment of myelodysplastic syndrome and acute myeloid leukemia42 and has shown promise in preclinical MM studies.43 Because TAZ is hypermethylated in HMCLs (Figure 2C), we assessed whether DAC could upregulate TAZ expression. At both the mRNA and protein levels, DAC induced TAZ expression in KMM1 (Figure 5A), KMS11 (Figure 5B), JJN3 (Figure 5C), and U266 (Figure 5D) cells in a dose-dependent manner. At the highest dose of DAC, TAZ mRNA levels were upregulated 21.30 ± 0.36-fold in KMM1 cells, 32.33 ± 4.98-fold in KMS11 cells, 29.82 ± 5.60-fold in JJN3 cells, and 29.14 ± 0.83-fold in U266 cells. In all cell lines, increased mRNA levels correlated with elevated protein levels. Moreover, in response to DAC, increased TAZ protein levels correlated with decreased MYC protein levels in KMM1, KMS11, and JJN3 cells. Unlike many other HMCLs, U266 cells do not express MYC.44 Finally, in cells that already express TAZ, DAC treatment was less effective (supplemental Figure 8A), suggesting that the upregulation of TAZ is responsible, in part, for the downregulation of MYC and sensitivity to DAC.
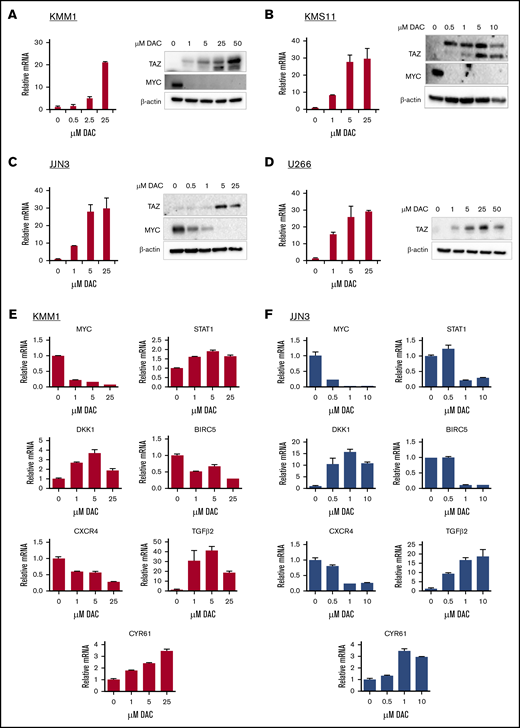
Demethylating agent DAC upregulates TAZ and represses MYC transcriptional program. KMM1 (A), KMS11 (B), JJN3 (C), and U266 (D) were treated with increasing concentrations of DAC and evaluated for TAZ and MYC expression at the mRNA and/or protein levels. KMM1 (E) or JJN3 (F) cells were treated with increasing concentrations of DAC and evaluated for deregulation of MYC targets at the mRNA level. Data are mean ± SD of triplicate samples.
Demethylating agent DAC upregulates TAZ and represses MYC transcriptional program. KMM1 (A), KMS11 (B), JJN3 (C), and U266 (D) were treated with increasing concentrations of DAC and evaluated for TAZ and MYC expression at the mRNA and/or protein levels. KMM1 (E) or JJN3 (F) cells were treated with increasing concentrations of DAC and evaluated for deregulation of MYC targets at the mRNA level. Data are mean ± SD of triplicate samples.
Pharmacological upregulation of TAZ by DAC also antagonized the MYC transcriptional program identified in our RNA-seq analysis. In KMM1 cells, treatment with DAC upregulated DKK1, CYR61, STAT1, and TGFβ2 while downregulating BIRC5 and CXCR4 mRNA levels in a dose-dependent manner (Figure 5E). Similarly, these targets were differentially expressed after DAC treatment in JJN3 cells (Figure 5F). The differential expression of MYC targets upon DAC treatment correlated with the increase in TAZ and decrease in MYC expression.
Upregulation of TAZ sensitizes HMCLs to targeted therapeutics
Given that TAZ is hypermethylated and that its reexpression induced cell death, we next sought to establish whether upregulation of TAZ by DAC could sensitize cells to other antimyeloma therapeutics. BTZ is a proteasome inhibitor that improves survival outcomes when used in combination with other agents in previously untreated MM patients45 or when used alone or in combination in relapsed or refractory MM patients.46 In both KMM1 and JJN3 cells, treatment with increasing concentrations of DAC for 48 hours prior to exposure to BTZ enhanced sensitivity to BTZ. These observations are supported by the calculated combination index (CI) where CI values <1 indicate synergy (Figure 6A-B). The increased sensitivity to BTZ correlated with increased expression of TAZ and decreased expression of MYC (Figure 6C-D). Histone deacetylases (HDACs) have also emerged as relevant therapeutic targets in MM when used in combination with other therapies such as BTZ.47 TSA inhibits multiple HDAC classes and serves as proof in concept that HDAC inhibitors can inhibit MM growth in vitro. As before, in both KMM1 and JJN3 cells, treatment with increasing concentrations of DAC prior to exposure to TSA enhanced sensitivity to TSA and was synergistic (Figure 6E-F). The increased sensitivity to TSA and synergistic relationship between DAC and TSA correlated with enhanced expression of TAZ and the potent downregulation MYC expression (Figure 6G-H). These results were repeated with the more clinically relevant HDAC inhibitor, Pano, which has been approved for third-line treatment of MM48 (supplemental Figure 7). As further evidence that TAZ regulates sensitivity to specific therapeutics, we showed that TAZ-overexpressing cells treated with BTZ (supplemental Figure 8B), TSA (supplemental Figure 8C), or Pano (supplemental Figure 8D) were more sensitive than control (WPI) cells. These results suggest that upregulating TAZ may be a novel approach to enhance sensitivity to existing or novel therapeutics.
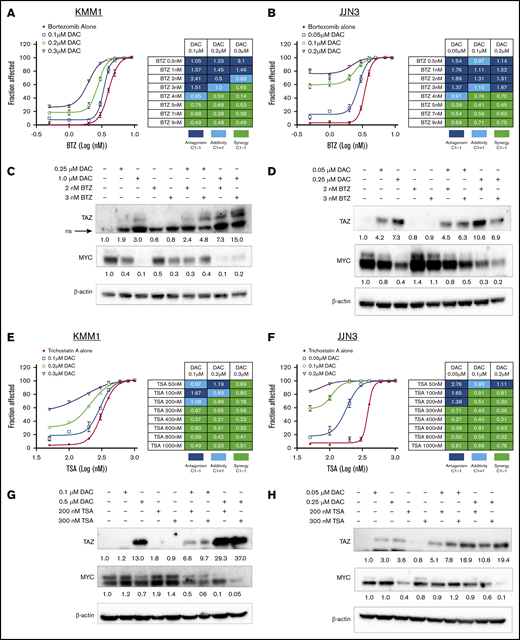
Upregulation of TAZ sensitizes MM cells to chemotherapeutics. KMM1 (A) and JJN3 (B) cells were treated with increasing concentrations of DAC for 48 hours followed by exposure to BTZ for 72 hours before cell viability was assessed. CI values were calculated using CompuSyn software. Immunoblot analysis of TAZ and MYC expression in KMM1 (C) and JJN3 (D) cells treated with DAC for 48 hours followed by BTZ exposure for 24 hours. KMM1 (E) and JJN3 (F) cells were treated with increasing concentration of DAC for 48 hours followed by exposure to TSA for 72 hours before cell viability was assessed and CI values calculated. Immunoblot analysis of TAZ and MYC expression in KMM1 (G) and JJN3 (H) cells treated with DAC for 48 hours followed by TSA exposure for 24 hours. Densitometry was performed and relative expression, normalized to β-actin, is shown below the lanes indicating expression compared with untreated cells. ns, nonspecific.
Upregulation of TAZ sensitizes MM cells to chemotherapeutics. KMM1 (A) and JJN3 (B) cells were treated with increasing concentrations of DAC for 48 hours followed by exposure to BTZ for 72 hours before cell viability was assessed. CI values were calculated using CompuSyn software. Immunoblot analysis of TAZ and MYC expression in KMM1 (C) and JJN3 (D) cells treated with DAC for 48 hours followed by BTZ exposure for 24 hours. KMM1 (E) and JJN3 (F) cells were treated with increasing concentration of DAC for 48 hours followed by exposure to TSA for 72 hours before cell viability was assessed and CI values calculated. Immunoblot analysis of TAZ and MYC expression in KMM1 (G) and JJN3 (H) cells treated with DAC for 48 hours followed by TSA exposure for 24 hours. Densitometry was performed and relative expression, normalized to β-actin, is shown below the lanes indicating expression compared with untreated cells. ns, nonspecific.
Discussion
Using MM cell-line models and clinical samples, we identify an unexpected role for TAZ in MM. We show that TAZ expression decreases during MM progression and that its reexpression induces a proapoptotic response that is in marked contrast to its previously established oncogenic role in solid tumors.13 Our observations suggest that TAZ elicits this tumor-suppressor function by decreasing MYC expression and its transcriptional program. We provide evidence that pharmacological upregulation of TAZ sensitizes HCMLs to antimyeloma therapeutics.
Our results showed that expression of TAZ predicts patient outcomes, where patients with higher expression of TAZ have better survival outcomes (Figure 1B). This correlates with our finding that reexpression of TAZ induced cell death (Figure 3; supplemental Figure 4). Recent studies have shown that, in specific contexts, TAZ or its paralog YAP act noncanonically by inducing apoptosis. Like TAZ, reexpression of YAP induces cell death in MM and other hematological cell lines through a TEAD-independent mechanism.24 Other studies show that uncontrolled YAP and TAZ activity after genetic deletion of LATS1/2 inhibits colon cancer cell growth particularly in cell-suspension conditions49 or leads to hypertranscription and massive cell death in cells that are particularly sensitive to DNA damage.50 This cell death occurs through the selective regulation of unique target genes. A better understanding of the context-specific regulation and downstream effects of TAZ is necessary.
Our transcriptome analysis revealed enriched repression of MYC targets (Figure 4B-D), suggesting that TAZ-induced cell death in MM is partly due to downregulated MYC. Indeed, we show that reexpression of TAZ leads to a reduction in both MYC mRNA (Figure 4F; supplemental Figure 7A) and protein (Figure 4G; supplemental Figure 7B). Although previous studies have shown that YAP and/or TAZ upregulate MYC expression through both TEAD-dependent51-53 and TEAD-independent mechanisms,54,55 in our study, TAZ downregulates MYC. Other genes identified in our transcriptome analysis also suggest context-dependent regulation by YAP/TAZ. Whereas YAP upregulates BIRC5 to elicit its prosurvival function,52,53 in MM TAZ downregulates BIRC5 (Figure 4F) to promote apoptosis. Not only do these studies suggest that YAP and TAZ function independently, but they also suggest that the regulation of their target genes may also be context dependent.
Because of the important roles MYC plays in regulating cell growth, MYC is tightly controlled at both the transcriptional and posttranscriptional levels. Although transcription of MYC is driven by growth factors, the protein levels of MYC are regulated in response to nutrient supply and metabolic stress.56 TAZ-mediated downregulation of MYC occurs at the mRNA (Figure 4F), and robustly at the protein (Figure 4G), level. We further show that control of MYC expression by TAZ involves posttranslational mechanisms (supplemental Figure 7C-D). FBW7 is one of the best-characterized E3 ligases that targets MYC for degradation after phosphorylation of T58 by glycogen synthase kinase β.57,58 The precise mechanisms mediating TAZ-dependent loss of MYC expression in MM remain important areas of study and may lead to enhanced efficacy of therapies that target MYC. TAZ regulation of MYC expression, especially posttranscriptionally, provides the rationale for therapeutic combinations involving upregulating TAZ and anti-MYC therapies that primarily target transcriptional vulnerabilities such as lenalidomide and BET inhibitors.59
Deregulated epigenetics is a major contributor to MM pathogenesis. In 1273 newly diagnosed MM patients, a recent study identified epigenetic mutations in 24.4% of cases.60 Our results show that not only does TAZ expression decrease as myeloma progresses from healthy plasma cells to fully active MM (Figure 1A), but the methylation status of TAZ also increases (Figure 2A). The high variability of TAZ methylation in MM suggests that alternative mechanisms may also be responsible for regulating TAZ expression in MM. These mechanisms may involve microRNA silencing of TAZ, as has been shown in other cancers,61 or other epigenetic modifiers. In several studies, including some genome-wide screens, TAZ was found to be upregulated in response to silencing the histone demethylase JMJD1a62 or treatment with epigenetic inhibitors such as BTZ,63 enhancer of zeste homolog 2 (EZH2) inhibitors,64 the panHDAC inhibitor Pano,65 and the BET inhibitor JQ1.66 We show that the TAZ promoter is specifically methylated (Figure 2C) and that globally inhibiting DNMTs increased TAZ expression in HMCLs (Figure 5A-D). However, given the interplay between methylation, acetylation, and other epigenetic modifications, the precise regulation of TAZ expression may involve several different epigenetic classes.
Because epigenetic processes are reversible, pharmacological inhibitors of DNMTs and HDACs have become attractive therapeutic strategies. Not only did DAC increase TAZ expression, but treatment also decreased MYC expression (Figure 5A-D) and its transcriptional program (Figure 5E-F) in HMCLs. Previous studies have shown that DAC downregulates MYC expression in chronic myeloid leukemia67 or Burkitt lymphoma68 cell lines, and in myelodysplastic syndrome patient samples69 ; however, the mechanisms mediating this repression are not well described. Our results suggest that the effects of DAC are at least in part mediated through TAZ because cells expressing TAZ, where MYC expression is already repressed, are more resistant to DAC treatment (supplemental Figure 8A).
The development of rational combination therapies may improve the efficacy of DAC in the treatment of MM. Although BTZ alone, or related proteasome inhibitor carfilzomib, has been shown to increase TAZ expression,20,63,70 our data showed that sequential dosing of DAC followed by BTZ not only enhanced the efficacy of these therapeutics but also correlated with significant upregulation of TAZ and downregulation of MYC expression in HMCL models (Figure 6). Similarly, in both our study (Figure 6E-H) and others, the combination of DAC with HDAC inhibitors is more efficacious due to the synergistic deregulation of specific cell-survival genes.65,71-73 Precise mechanisms responsible for this synergy remain to be elucidated and are likely to involve numerous signaling molecules63,65,71,73,74 and to be cell type dependent. Our model suggests that these antimyeloma therapies upregulate TAZ, leading to decreased MYC expression and its transcriptional program and subsequent cell death. This mechanism may be most relevant in hematological malignancies where TAZ expression is lower (supplemental Figure 2) and its promoter more heavily methylated (supplemental Figure 3).
In conclusion, we have shown that TAZ expression correlates with clinical outcomes and that TAZ expression is regulated at the epigenetic level in MM. Using cell-line models, genetic or pharmacological upregulation of TAZ induced cell death and inhibited MYC expression and some of its transcriptional targets. We also provided evidence that upregulating TAZ sensitizes cells to additional antimyeloma therapeutics. This provides the rationale for the development of novel TAZ-based therapeutics to improve the clinical efficacy of existing MM therapeutics.
Acknowledgments
This work was supported by a Canadian Cancer Society Research Chair and a Canadian Institutes of Health Research–Strategy for Patient-Oriented Research Mentorship Chair (T.R.), as well as a fellowship award from the Beatrice Hunter Cancer Research Institute, the New Brunswick Health Research Foundation, and Lung Cancer Canada (S.G.).
Authorship
Contribution: S.G. conceived, designed, performed, and analyzed the experiments; S.G. and T.R. wrote the manuscript; G.W., S.C., and N.C. performed the RNA-seq experiments; M.L. performed the MSP and drug combination experiments; and J.W. analyzed publicly available data sets.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tony Reiman, Department of Oncology, Saint John Regional Hospital, University of New Brunswick, 400 University Ave, Saint John, NB E2L 4L2, Canada; e-mail: anthony.reiman@horizonnb.ca.

