

Key Points
Anti-FVIII inhibitory antibody development is TFH-cell dependent.
FVIII restimulation can specifically induce FVIII-primed TFH-cell proliferation.

Abstract
The development of neutralizing anti-FVIII antibodies (inhibitors) is a major complication of FVIII protein replacement therapy in patients with hemophilia A (HA). Although multiple lines of evidence indicate that the immune response against FVIII is CD4 T-cell–dependent and many FVIII-derived CD4 epitopes have already been discovered, the role of T follicular helper (TFH) cells in FVIII inhibitor development is unknown. TFH cells, a newly identified subset of CD4 T cells, are characterized by expression of the B-cell follicle-homing receptor CXCR5 and PD-1. In this study, we show for the first time that IV FVIII immunization induces activation and accumulation and/or expansion of PD-1+CXCR5+ TFH cells in the spleen of FVIII-deficient (FVIIInull) mice. FVIII inhibitor-producing mice showed increased germinal center (GC) formation and increased GC TFH cells in response to FVIII immunization. Emergence of TFH cells correlated with titers of anti-FVIII inhibitors. Rechallenge with FVIII antigen elicited recall responses of TFH cells. In vitro FVIII restimulation resulted in antigen-specific proliferation of splenic CD4+ T cells from FVIII-primed FVIIInull mice, and the proliferating cells expressed the TFH hallmark transcription factor BCL6. CXCR5+/+ TFH-cell–specific deletion impaired anti-FVIII inhibitor production, confirming the essential role of CXCR5+/+ TFH cells for the generation of FVIII-neutralizing antibodies. Together, our results demonstrate that the induction of activated TFH cells in FVIIInull mice is critical for FVIII inhibitor development, suggesting that inhibition of FVIII-specific TFH-cell activation may be a promising strategy for preventing anti-FVIII inhibitor formation in patients with HA.
Introduction
Hemophilia A (HA) is an X-linked, recessive, bleeding disease caused by a deficiency of factor VIII (FVIII). Current standard treatment is based on IV infusion of FVIII protein. One major complication of FVIII replacement therapy is the development of neutralizing anti-FVIII inhibitory antibodies (inhibitors) against FVIII.1 Up to 30% of patients with severe HA develop inhibitors, which seriously complicates treatment and increases morbidity and mortality from this disease.2,3
Although several genetic and nongenetic factors that contribute to the risk of developing inhibitors have been identified, it remains largely unknown why some patients never generate a clinically significant immune response, whereas others do.4-8 It has been reported that specific genetic mutations in HA patients correlate with a higher risk of inhibitor formation. Patients with large FVIII deletions have the highest rate of inhibitor formation, as the absence (or severe truncation) of the FVIII protein may prevent a patient’s immune system from initiating central tolerance to FVIII.9 Several polymorphic immune response genes (eg, interleukin-10 [IL-10], cytotoxic T-lymphocyte–associated protein-4 [CTLA4], and tumor necrosis factor-α [TNFα]) have been found to be associated with the risk of FVIII inhibitor development.6,10 This evidence suggests that both central and peripheral mechanisms of immunological tolerance are involved in preventing inhibitor occurrence in HA patients.
Multiple lines of evidence suggest that the FVIII immune response is CD4 T-cell dependent. In patients with an established humoral response to FVIII, HIV infection leads to the disappearance of FVIII inhibitors when CD4 T-cell counts decline, demonstrating the requirement for CD4 T cells in this process.11 Previous studies demonstrated that B cells producing anti-FVIII inhibitors undergo isotype switching and affinity maturation processes. A large proportion of FVIII inhibitors belong to the immunoglobulin G1 (IgG1) or IgG4 subclass, and the class switch to IgG4 is found only in patients with inhibitors, but not in healthy individuals or patients without inhibitors.12 Anti-FVIII IgG with inhibitory activity has an up to 100-fold higher affinity for FVIII than IgG without inhibitory activity.13 Isotype switching and affinity maturation are dependent on specific CD4 T-cell help, suggesting that the CD4 T-cell help is necessary for FVIII inhibitor development.
Activation of FVIII-specific CD4 T cells requires the interaction of the CD4 T-cell receptor with peptide-bound major histocompatibility complex II (MHCII) on the surface of antigen-presenting cells. CD4 T-cell epitopes derived from FVIII protein have been identified by measuring proliferation of CD4 T cells stimulated with synthetic overlapping peptides,14-17 generation of FVIII-specific CD4 T-cell hybridomas,18 and MHCII tetramer-guided epitope mapping.19-21 Determination of the repertoire of naturally presented peptides presented on MHCII of antigen-presenting cells by mass spectrometry has been successfully used to identify FVIIII CD4 T-cell epitopes.22,23 The increased repertoire of identified naturally presented FVIII CD4 epitopes indicates the important involvement of CD4 T cells in FVIII inhibitor development.
T follicular helper (TFH) cells are a newly identified subset of CD4 T cells that specialize in providing cognate help to B cells and are fundamentally essential for the generation of T-cell–dependent B-cell responses.24-26 Without cognate TFH-cell help, activated B cells are unable to generate and maintain the germinal center (GC) response that is required for efficient somatic hypermutation of immunoglobulin genes and the selective processes that facilitate affinity maturation of antibodies. TFH cells express the C-X-C chemokine receptor-5 (CXCR5), as well as the inducible costimulator (ICOS), IL-21, and the transcription factor B-cell lymphoma-6 (BCL6). Considering the importance of TFH cells for B-cell antibody responses, we studied the activation and induction of TFH cells and their role in inhibitor production in FVIII-immunized mice.
Methods
Mice
FVIII-deficient (FVIIInull) mice with a targeted disruption of exon 17 of the FVIII gene27 were originally obtained from Haig Kazazian (University of Pennsylvania) and crossed onto a C57BL/6J background. VWFnullFVIIInull mice, which are deficient in FVIII as well as VWF, the carrier protein for FVIII, were generated by the Shi laboratory by crossing FVIIInull onto VWFnull.28 Wild-type (WT) C57BL/6J mice, CD4-deficient (CD4−/−) mice,29 and CXCR5-deficient (CXCR5−/−) mice30 were purchased from the Jackson Laboratory (Bar Harbor, ME). All animals used in this study were on the C57BL/6J background, except VWFnullFVIIInull mice that were on a B6/129S mixed genetic background. All animals were kept in pathogen-free microisolator cages at the animal facilities operated by the Medical College of Wisconsin. Isoflurane or ketamine was used for anesthesia. Animal studies were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin.
Inhibitor immune response studies
FVIIInull mice were immunized IV with recombinant human B-domain–deleted FVIII (rhF8, Xyntha; Pfizer, New York, NY) at a dose of 50 to 100 U/kg weekly for 5 to 6 weeks. Five to 7 days after the last immunization, blood samples were collected. The titers of anti-FVIII inhibitors were determined by a modified Bethesda assay, as described in our previous reports.31,32 C57BL/6J WT and mixed bone marrow (BM) chimeric mice, which had normal levels of endogenous murine FVIII, were immunized with recombinant human full-length FVIII (rhfF8, Kogenate FS; Bayer Pharma, Whippany, NJ) at a dose of 200 U/kg weekly for 4 weeks. One week after the last immunization, blood samples were collected for assays. In some experiments, CD4 T cells were depleted by the administration of anti-CD4 antibody GK1.5 (BioXCell, West Lebanon, NH) on days −4 and −1 before the first immunization and 1 day before every other immunization.
Generation of BM chimeric mice
BM chimeric mice were generated by transplantation of mixed BM cells from CXCR5−/− or WT and CD4−/− mice into lethally irradiated (1100 rads) CD4−/− recipients. BM cells were collected from the femurs, tibia, and humeri of donor mice, and red cells were lysed. Each recipient mouse received a total of 107 cells in a volume of 300 μL phosphate-buffered saline via lateral tail vein injection. The ratio of donor BM cells in these chimeras was 80% of CD4−/− cells and 20% of WT or CXCR5−/− cells. Mice were monitored for CD4 T-cell reconstitution in the peripheral blood by flow cytometry analysis.
Flow cytometry analysis
The spleens or lymphoid nodes (LNs) were isolated from rhF8-immunized, FVIIInull mice. Single-cell suspensions of spleen or LNs were prepared by standard gentle mechanical disruption. Cells were stained for B220, CD3, CD4, CD44, CD62L, ICOS, CD25, PD-1, CXCR5, CD40L, Fas, GL7, Ki-67, CTLA4, BCL-6, Tbet, and GATA-3. Normal rat IgG2a κ, mouse IgG2b κ, and mouse IgG1 antibodies were used as isotype controls. For detection of Ki-67, Bcl6, Tbet, GATA-3, CTLA4, and Foxp3, surface-stained cells were fixed and permeabilized with the Foxp3/TF Staining Buffer Set, followed by incubation with corresponding fluorochrome-conjugated antibodies. Cells were analyzed by an LSRII or an LSRFortessa X-20 flow cytometer, and data were analyzed with FlowJo software. Doublets were gated out during FlowJo analysis to exclude possible B-/T-cell conjugates. The details of antibodies used in this study are provided in the supplemental Data.
T-cell proliferation assay
T-cell proliferation assays were performed according to the procedures described in our previous reports.32,33 In brief, isolated splenocytes were labeled with CellTrace Violet and cultured with various concentrations of rhF8 or the unrelated protein rhFIX (rhF9) for 96 hours. Cells were harvested and stained for viability and for the cell markers CD4, CD3, CD19, CXCR5, BCL6, Tbet, and GATA3. Isotype antibodies were used as controls in parallel. Samples were analyzed by flow cytometry. The details of T-cell proliferation assay are provided in the supplemental Data.
Statistical analysis
Data are presented as the mean ± standard deviation. Statistical comparisons of 2 experimental groups were evaluated by 2-tailed Student t test if data distribution passed the normality test. The Mann-Whitney U test was used for comparison if data distribution failed the normality test. Linear regression statistical analysis was performed with Prism 7 (GraphPad Software, La Jolla, CA). P < .05 was considered statistically significant.
Results
Depletion of CD4 T cells during FVIII immunization prevents anti-F8 inhibitor induction
We first sought to determine the requirement of CD4 T-cell help in the development of neutralizing anti-FVIII inhibitors in FVIIInull mice by IV FVIII immunization. To accomplish this, CD4 T cells were depleted in FVIIInull mice before each rhF8 infusion (supplemental Figure 1A). Anti-FVIII inhibitor titers were determined by Bethesda assay 1 week after the last immunization. All non-CD4–depleted FVIIInull control mice developed various titers of inhibitors after rhF8 immunization, but no inhibitors were detected in animals if CD4 T cells were depleted during FVIII immunization (supplemental Figure 1B). This confirms that CD4 help is required for FVIII inhibitor production.
Characterization of activated TFH cells in FVIII-immunized mice
Because TFH cells are important for efficient B-cell GC reactions,25,26 we next assessed TFH activation in the spleen of FVIII-immunized mice. Figure 1A depicts our gating strategy for determining the activated TFH-cell population by flow cytometry analysis. Beginning with CD4+ (CD3+CD4+CD19−) live cells, effector helper CD4+ cells were identified by excluding Foxp3+ CD4 cells and evaluating expression of CD44 and CD62L (CD44+CD62L−). Activated TFH cells were further defined by coexpression of CXCR5 and PD-1. To confirm the identity of the CXCR5hiPD-1hi from effector CD4+ T cells as activated TFH cells, we assessed their expression of the transcription factor BCL6. The BCL6 expression level on CXCR5hiPD-1hi cells was significantly higher than levels on naive CD4 or CXCR5loPD-1lo effector CD4 T cells (Figure 1B-C). Expression of other TFH-associated markers, including the costimulatory molecules ICOS and CD40L, were also compared. Similar to BCL6, increased expression of CD40L was found only on CXCR5hiPD-1hi cells. CXCR5loPD-1lo effector CD4 T cells expressed an increased level of ICOS compared with naive CD4 T cells, but CXCR5hiPD-1hi cells expressed the highest level of ICOS (Figure 1B-C). The percentage of Ki-67 expression on activated CXCR5hiPD-1hi TFH cells was significantly higher than CXCR5loPD-1lo effector cells, indicating that TFH cells are more proliferative than non-TFH effectors (Figure 1D).
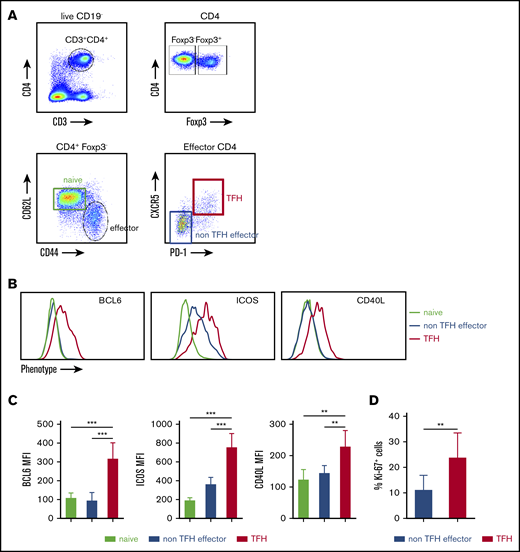
FVIII immunization induces activated TFH cells in spleen. FVIIInull mice were given 5 rounds of weekly IV FVIII immunization injection. Five days after the last immunization, the mice were euthanized, and splenocytes and plasma were collected for analysis. Age-matched naive FVIIInull mice were used as controls. Anti-FVIII inhibitor titers were determined by chromogenic-based Bethesda assay. Splenocytes were stained for CD3, CD4, CD19, CD44, CD62L, Foxp3, CXCR5, PD-1, and Ki-67 and analyzed by flow cytometry. (A) Gating strategy for the identification of activated TFH cells. CD4+ Th cells were identified by excluding Foxp3+ CD4+ cells inside the singlet live CD19−CD3+CD4+ cell population. Effector CD4+ Th cells were identified by CD44 and CD62L expression (CD44+CD62L−). Effector CD4+ cells were analyzed for CXCR5 and PD-1 coexpression to identify TFH cells. (B) shows representative histograms and (C) shows quantification of BCL6, ICOS, and CD40L expression on naive, CXCR−PD-1− effector (non-TFH effector cells), and TFH cells (n = 6 mice per group). (D) Ki-67 cell cycling staining on CXCR5−PD-1− effector and TFH cells (n = 6 mice per group). **P < .01; ***P < .001.
FVIII immunization induces activated TFH cells in spleen. FVIIInull mice were given 5 rounds of weekly IV FVIII immunization injection. Five days after the last immunization, the mice were euthanized, and splenocytes and plasma were collected for analysis. Age-matched naive FVIIInull mice were used as controls. Anti-FVIII inhibitor titers were determined by chromogenic-based Bethesda assay. Splenocytes were stained for CD3, CD4, CD19, CD44, CD62L, Foxp3, CXCR5, PD-1, and Ki-67 and analyzed by flow cytometry. (A) Gating strategy for the identification of activated TFH cells. CD4+ Th cells were identified by excluding Foxp3+ CD4+ cells inside the singlet live CD19−CD3+CD4+ cell population. Effector CD4+ Th cells were identified by CD44 and CD62L expression (CD44+CD62L−). Effector CD4+ cells were analyzed for CXCR5 and PD-1 coexpression to identify TFH cells. (B) shows representative histograms and (C) shows quantification of BCL6, ICOS, and CD40L expression on naive, CXCR−PD-1− effector (non-TFH effector cells), and TFH cells (n = 6 mice per group). (D) Ki-67 cell cycling staining on CXCR5−PD-1− effector and TFH cells (n = 6 mice per group). **P < .01; ***P < .001.
FVIII immunization increases the frequency of activated TFH cells in mice that produce anti-FVIII inhibitors
FVIIInull mice were given 5 weekly rounds of FVIII IV immunization. Five days after the last immunization, inhibitor titers were measured, and representative animals with inhibitors and no inhibitors were euthanized for splenocyte analysis. FVIII immunization induced a marked increase in the percentage of CD44+CD62L− effector CD4 cells in spleens of inhibitor-producing mice compared with saline controls or non–inhibitor-producing animals (14% ± 0.8%, 11% ± 0.25%, and 10% ± 1.3%, respectively; Figure 2A). The percentage of CXCR5hiPD-1hi TFH cells within the effector CD4 population also significantly increased in spleens of inhibitor-producing mice compared with saline-treated controls and non–inhibitor-producing mice (19.6% ± 1.6%, 8.1% ± 1.2%, and 7.6% ± 1.4%, respectively; Figure 2B). The percentage of activated TFH cells in total CD4 cells (Figure 2C) and the absolute number of activated TFH cells per spleen (Figure 2D) in inhibitor-producing mice were significantly higher than in the saline controls and non–inhibitor-producing mice (2.23% ± 0.26%, 0.65% ± 0.13%, and 0.61% ± 0.24%, respectively, and 14.4 ± 2.0 × 104, 3.55 ± 0.8 × 104, and 3.5 ± 0.9 × 104, respectively).
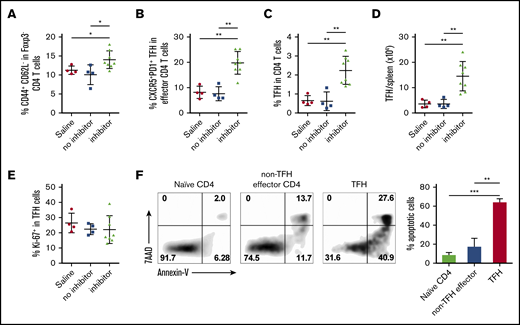
FVIII immunization increases the frequency of activated TFH cells in mice that produce inhibitors. FVIIInull mice were given 5 rounds of weekly IV FVIII immunizations. Five days after the last immunization, inhibitor titers were measured by Bethesda assay. Representative animals with inhibitors and no inhibitors were euthanized for TFH analysis. Age-matched naive FVIIInull mice were used as controls. Splenocytes were stained for CD3, CD4, CD19, CD44, CD62L, Foxp3, CXCR5, PD-1, and Ki-67, and analyzed by flow cytometry. (A) Percentage of effector helper cells (CD44+CD62L−) among Foxp3−CD4 T cells in saline-injected control mice, non-inhibitor–producing mice, and inhibitor-producing mice. (B) Percentage of CXCR5+PD-1+ TFH cells among effector CD4 helper cells. (C) Percentage of activated TFH cells among total CD4 T cells. (D) Total number of activated TFH cells per spleen. (E) Percentage of Ki-67+ in activated TFH cells. (F) Representative samples and summary data (n = 4 mice per group) of apoptotic cells in naive, non-TFH effector CD4 T cells and TFH cells from FVIII-immunized mice. *P < .05; **P < .01; ***P < .001.
FVIII immunization increases the frequency of activated TFH cells in mice that produce inhibitors. FVIIInull mice were given 5 rounds of weekly IV FVIII immunizations. Five days after the last immunization, inhibitor titers were measured by Bethesda assay. Representative animals with inhibitors and no inhibitors were euthanized for TFH analysis. Age-matched naive FVIIInull mice were used as controls. Splenocytes were stained for CD3, CD4, CD19, CD44, CD62L, Foxp3, CXCR5, PD-1, and Ki-67, and analyzed by flow cytometry. (A) Percentage of effector helper cells (CD44+CD62L−) among Foxp3−CD4 T cells in saline-injected control mice, non-inhibitor–producing mice, and inhibitor-producing mice. (B) Percentage of CXCR5+PD-1+ TFH cells among effector CD4 helper cells. (C) Percentage of activated TFH cells among total CD4 T cells. (D) Total number of activated TFH cells per spleen. (E) Percentage of Ki-67+ in activated TFH cells. (F) Representative samples and summary data (n = 4 mice per group) of apoptotic cells in naive, non-TFH effector CD4 T cells and TFH cells from FVIII-immunized mice. *P < .05; **P < .01; ***P < .001.
To interrogate the increased accumulation of TFH cells inside spleens of inhibitor-producing mice, we examined their proliferation and survival. First, we determined the proliferative status of the effector TFH cells by monitoring Ki-67 expression on TFH cells. Although activated TFH cells were dividing more actively than were non-TFH effector cells (Figure 1D), the percentage of activated TFH cells undergoing proliferation in inhibitor-producing mice was similar to that in control and non–inhibitor-producing animals (Figure 2E). Second, we measured the apoptosis status of activated TFH cells in FVIII-primed mice. We found that the frequencies of both early (annexin-V+7AAD−) and late (annexin-V+7AAD+) apoptotic cells increased significantly in activated TFH cells, compared with non-TFH effector and naive CD4 T cells (Figure 2F). These data demonstrate that activated TFH cells in FVIII-primed mice have a proapoptosis phenotype, suggesting that the long-term survival of TFH cells is not necessary for activated TFH-cell expansion and accumulation in FVIII inhibitor–producing mice.
FVIII inhibitor–producing mice have increased GC formation and increased GC TFH cells in response to FVIII immunization
CD19+Fas+GL-7+ cells were used to define antigen-activated GC B cells. We found that increased frequency of activated TFH cells in mice that produce FVIII inhibitors was aligned with a significant increase in the fraction of GC B cells (Figure 3A). We also examined the GC TFH-cell subpopulation by evaluating its GL-7 expression. The percentage and total number of GL-7+ TFH cells in spleen tissue were significantly increased in inhibitor-producing mice versus non–inhibitor-producing mice (14.6% ± 2.8% vs 10.5% ± 3%, and 2.2% ± 1.2 × 104 vs 0.44% ± 0.3% × 104, respectively; Figure 3B). It has been shown that expression of GL-7 on TFH cells is associated with a mature GC TFH-cell phenotype.34,35 We found that compared with GL-7low TFH cells, GL-7+ GC TFH cells expressed higher levels of the costimulatory molecules ICOS and CD40L (Figure 3C). These data demonstrate that the GC reaction occurs in HA mice after IV FVIII infusion.
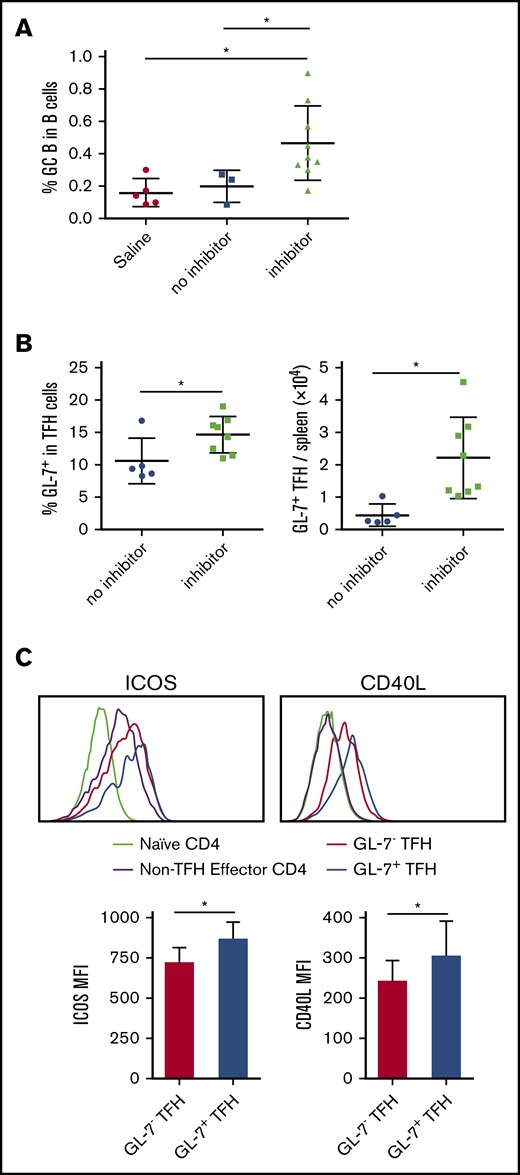
FVII inhibitor-producing mice have increased GC formation and increased GC TFH cells in response to F8 immunization. FVIIInull mice were given 5 weekly IV FVIII injections. Five days after the last immunization, inhibitor titers were measured by Bethesda assay. Representative animals with inhibitors and no inhibitors were euthanized for splenocyte analysis. Splenocytes were stained for CD3, CD4, CD19, CD44, CD62L, B220, Foxp3, CXCR5, PD-1, Fas, GL-7, ICOS, and CD40L and analyzed by flow cytometry. (A) Percentage of B220+ B cells bearing a GC phenotype (Fas+ GL-7+) in saline-injected control mice, non–inhibitor-producing mice and inhibitor-producing mice. (B) Percentage and total number of activated TFH cells expressing GL-7 in in spleen. (C) Representative histograms and quantification (n = 6 per group) of expression of ICOS and CD40L on naive, CXCR5−PD-1− effector, GL-7− TFH, and GL-7+ TFH cells. *P < .05. MFI, mean fluorescence intensity.
FVII inhibitor-producing mice have increased GC formation and increased GC TFH cells in response to F8 immunization. FVIIInull mice were given 5 weekly IV FVIII injections. Five days after the last immunization, inhibitor titers were measured by Bethesda assay. Representative animals with inhibitors and no inhibitors were euthanized for splenocyte analysis. Splenocytes were stained for CD3, CD4, CD19, CD44, CD62L, B220, Foxp3, CXCR5, PD-1, Fas, GL-7, ICOS, and CD40L and analyzed by flow cytometry. (A) Percentage of B220+ B cells bearing a GC phenotype (Fas+ GL-7+) in saline-injected control mice, non–inhibitor-producing mice and inhibitor-producing mice. (B) Percentage and total number of activated TFH cells expressing GL-7 in in spleen. (C) Representative histograms and quantification (n = 6 per group) of expression of ICOS and CD40L on naive, CXCR5−PD-1− effector, GL-7− TFH, and GL-7+ TFH cells. *P < .05. MFI, mean fluorescence intensity.
Emergence of CXCR5hiPD-1hi TFH cells correlates with the development of FVIII inhibitor responses
TFH activation induced by FVIII immunization was also confirmed in VWFnullFVIIInull mice, in which there was no VWF/FVIII interaction in vivo upon rhF8 infusion. In VWFnullFVIIInull mice, some animals generated high titers of FVIII inhibitors after IV FVIII immunization. Thus, we compared TFH-cell activation of mice that produce high titers of inhibitors (≥250 BU/mL) with that of mice that produce low-to-modest titers. Again, the percentage of CXCR5hiPD-1hi TFH cells within effector CD4 cells or total CD4 cells in the spleens of mice with inhibitors were significantly higher than in the saline controls (Figure 4A-B). Compared with the low-titer group, the high-titer group had a significant increase in the percentage of CXCR5hiPD-1hi TFH cells within the effector CD4-cell subset (24.7% ± 5.8% vs 16.8% ± 4.2%) and the percentage of activated TFH cells within the total CD4 T-cell population (3.72% ± 0.21% vs 1.83% ± 0.5%). We next evaluated the relationship between TFH activation and inhibitor responses induced by FVIII immunization. Magnitudes of the TFH-cell responses correlated significantly with the FVIII inhibitor titers (Figure 4C). These data indicate that there is a functional relationship between TFH-cell activation and FVIII inhibitor production after FVIII immunization.
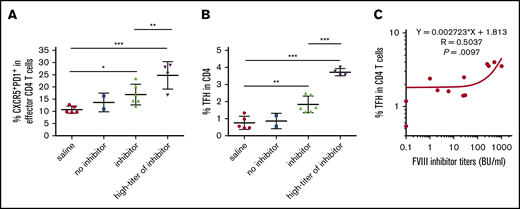
Emergence of PD-1+CXCR5+TFH cells correlates with the development of FVIII inhibitors. FVIII and VWF double-knockout mice were given weekly IV FVIII immunizations. One week after the last immunization, the mice were euthanized. Splenocytes and plasma were collected for analysis. Age-matched saline-treated mice were used as a control. Plasma inhibitor titers were determined by Bethesda assay. Splenocytes were analyzed by flow cytometry. (A) Percentage of CXCR5+PD-1+ TFH cells among effector CD4-helper cells. Anti-FVIII inhibitor titers greater than 250 BU/mL were defined as the high-titer inhibitor group. (B) Percentage of activated TFH cells among whole CD4 T cells. Anti-FVIII inhibitor titers greater than 250 BU/mL were defined as the high-titer inhibitor group. (C) Dot-plot shows the correlation between the titer of FVIII inhibitor and the percentage of activated TFH cells among CD4 T cells. Statistical significance was analyzed by linear regression. The r and P values are indicated in the figure. *P < .05; **P < .01; ***P < .001.
Emergence of PD-1+CXCR5+TFH cells correlates with the development of FVIII inhibitors. FVIII and VWF double-knockout mice were given weekly IV FVIII immunizations. One week after the last immunization, the mice were euthanized. Splenocytes and plasma were collected for analysis. Age-matched saline-treated mice were used as a control. Plasma inhibitor titers were determined by Bethesda assay. Splenocytes were analyzed by flow cytometry. (A) Percentage of CXCR5+PD-1+ TFH cells among effector CD4-helper cells. Anti-FVIII inhibitor titers greater than 250 BU/mL were defined as the high-titer inhibitor group. (B) Percentage of activated TFH cells among whole CD4 T cells. Anti-FVIII inhibitor titers greater than 250 BU/mL were defined as the high-titer inhibitor group. (C) Dot-plot shows the correlation between the titer of FVIII inhibitor and the percentage of activated TFH cells among CD4 T cells. Statistical significance was analyzed by linear regression. The r and P values are indicated in the figure. *P < .05; **P < .01; ***P < .001.
Ex vivo, FVIII-specific, proliferating CD4 T cells have the TFH phenotype
To examine specificity of the TFH cells for FVIII antigen, we used an ex vivo T-cell proliferation assay. Similar to what we previously reported,32,36 we confirmed that rhF8 restimulation induced the FVIII-specific proliferation of splenic CD4 T cells from rhF8-primed FVIIInull mice (Figure 5A). When CD4 T cells were further gated into CXCR5+ (TFH) and CXCR5− (non-TFH) populations, there were 31% daughter (differentiated) cells in the CXCR5+ population, but only 3% in the CXCR5− population after 10 U/mL rhF8 restimulation for 96 hours (Figure 5B). The CXCR5+ CD4 T cells had a higher proliferation index than the CXCR5− CD4 T cells: 2.24 vs 1.3, respectively. FVIII-specific proliferating CD4 cells were shown to express high levels of BCL6, as compared with nonproliferating cells. The CXCR5 mean fluorescence intensity of proliferating CD4 cells was also higher than that of nonproliferating cells (Figure 5C). In addition to BCL6, the expression of Tbet and GATA3, master transcription factors expressed in Th1 and Th2 cells, respectively, among proliferating CD4 T cells was also analyzed. Almost all FVIII-specific proliferating CD4 T cells were TFH transcription factor BCL6+, but proliferating CD4 T cells did not express the Th2 transcription factor GATA3, and less than 50% of proliferating CD4 T cells expressed the Th1 transcription factor Tbet (Figure 5D).
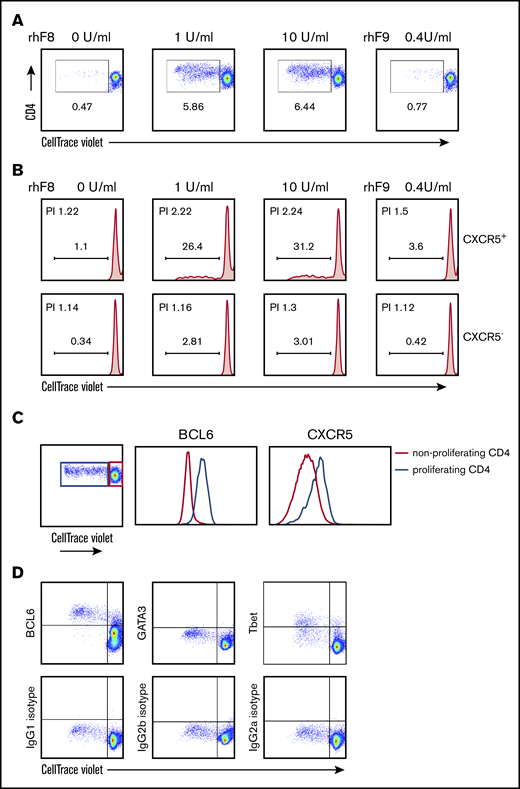
FVIII-specific proliferating CD4 T cells have a TFH phenotype. Splenocytes from rhF8-immunized FVIIInull mice were labeled with CellTrace Violet and stimulated with rhF8 for 96 hours. Nonstimulated or unrelated protein rhF9-stimulated splenocytes were used as controls in parallel. Cells were stained for mouse CD3, CD4, CXCR5, BCL6, GATA3, and Tbet. Proliferating daughter cells were analyzed by flow cytometry. (A) CD4+ T-cell proliferation is depicted. Representative plots from 4 experiments are shown. (B) CXCR5+ and CXCR5− T-cell proliferation is depicted. Representative plots from 4 experiments are shown. (C) BCL6 and CXCR5 expression levels on proliferating CD4 T cells (blue line) induced by ex vivo FVIII stimulation were compared with nonproliferating CD4 T cells (red line). (D) Transcription factor BCL6, GATA3, and Tbet expression on FVIII-induced proliferating CD4 T cells (top row). Corresponding isotype control antibody staining was used to define gating (bottom row). PI, proliferation index.
FVIII-specific proliferating CD4 T cells have a TFH phenotype. Splenocytes from rhF8-immunized FVIIInull mice were labeled with CellTrace Violet and stimulated with rhF8 for 96 hours. Nonstimulated or unrelated protein rhF9-stimulated splenocytes were used as controls in parallel. Cells were stained for mouse CD3, CD4, CXCR5, BCL6, GATA3, and Tbet. Proliferating daughter cells were analyzed by flow cytometry. (A) CD4+ T-cell proliferation is depicted. Representative plots from 4 experiments are shown. (B) CXCR5+ and CXCR5− T-cell proliferation is depicted. Representative plots from 4 experiments are shown. (C) BCL6 and CXCR5 expression levels on proliferating CD4 T cells (blue line) induced by ex vivo FVIII stimulation were compared with nonproliferating CD4 T cells (red line). (D) Transcription factor BCL6, GATA3, and Tbet expression on FVIII-induced proliferating CD4 T cells (top row). Corresponding isotype control antibody staining was used to define gating (bottom row). PI, proliferation index.
CXCR5 deficiency in CD4 T cells impairs anti-FVIII inhibitor induction
Expression of CXCR5 allows cells to enter the B-cell follicles directly by chemotaxis toward a gradient of C-X-C motif chemokine ligand 13 (CXCL13), the cognate ligand for CXCR5.36,37 Without CXCR5, TFH cells are unable to localize to the B-cell follicle and cannot interact with GC B cells. To determine the requirement for CXCR5+ TFH-cell migration during FVIII immunization, we generated mixed BM chimeras with a CD4 T-cell–specific CXCR5 deficiency (CD4-CXCR5−/−). Because those BM chimeric mice have normal levels of endogenous murine FVIII, we used a higher dose of rhfF8 to immunize the animals. FVIII inhibitor induction in mice that have normal levels of murine endogenous FVIII is also CD4 dependent. As shown in supplemental Figure 2, none of the WT mice developed detectable anti-FVIII inhibitors if CD4 T cells were depleted during FVIII immunization. CD4-CXCR5+/+ and CD4-CXCR5−/− chimeras were generated as depicted in Figure 6A. CD4 T-cell reconstitution between the 2 groups was similar as determined by flow cytometry (Figure 6B). Ten weeks after bone marrow transplantation, recipients were given 4 rounds of weekly rhfF8 immunization, and anti-FVIII inhibitor titers were determined by Bethesda assay (Figure 6C). Compared with CD4-CXCR5+/+ control mice, there was a significant decrease in the titer of FVIII inhibitors in CD4-CXCR5−/− mice after FVIII immunization (Figure 6D). This demonstrates that CXCR5 expression on FVIII-primed CD4 T cells is essential for efficient FVIII inhibitor induction.
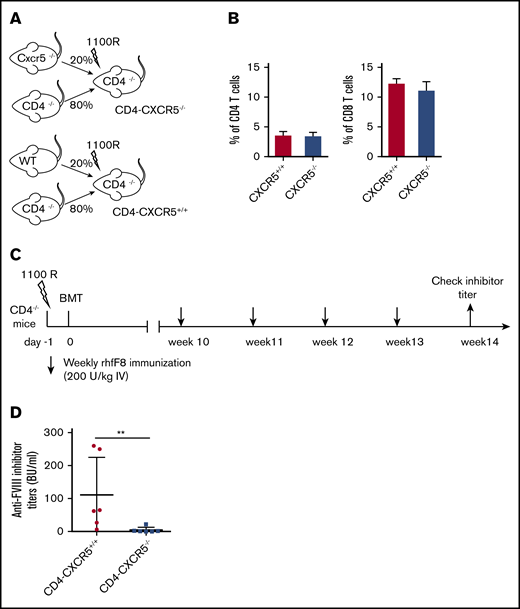
CD4 T-cell–specific CXCR5 deficiency impairs anti-FVIII inhibitor induction. (A) Schematic diagram for generating mixed BM chimeras. Mixed BM chimera in irradiated CD4−/− mice reconstituted with 80% BM from CD4−/− mice plus 20% from WT mice or 80% from CD4−/− mice plus 20% from CXCR5−/− mice. (B) Flow cytometry analysis of CD4 and CD8 T-cell reconstitution in blood at 10 weeks after BM transplantation. Mixed-BM chimeric mice were immunized with rhF8, as shown in the experimental schematic in panel C. (D) One week after the fourth injection with rhF8, plasma samples were collected for a Bethesda assay to determine the FVIII inhibitor titers. The data shown are for 6 mice per group. **P < .01.
CD4 T-cell–specific CXCR5 deficiency impairs anti-FVIII inhibitor induction. (A) Schematic diagram for generating mixed BM chimeras. Mixed BM chimera in irradiated CD4−/− mice reconstituted with 80% BM from CD4−/− mice plus 20% from WT mice or 80% from CD4−/− mice plus 20% from CXCR5−/− mice. (B) Flow cytometry analysis of CD4 and CD8 T-cell reconstitution in blood at 10 weeks after BM transplantation. Mixed-BM chimeric mice were immunized with rhF8, as shown in the experimental schematic in panel C. (D) One week after the fourth injection with rhF8, plasma samples were collected for a Bethesda assay to determine the FVIII inhibitor titers. The data shown are for 6 mice per group. **P < .01.
Rechallenge with FVIII antigen induces recall responses of TFH cells
We next evaluated whether FVIII-specific memory TFH cells would be detected when rechallenged with FVIII antigen in vivo. Five matched pairs of FVIII-primed mice with similar inhibitor titers were selected (Figure 7A). After resting for 8 to 10 weeks, the mice were given FVIII rechallenge or saline injection as the control. Five days after FVIII boost, the recall TFH cell reaction in spleens was analyzed. In saline-injected, FVIII-primed mice, the percentage of CXCR5+PD-1+ TFH cells among effector CD4 cells was similar to unprimed saline-injected control mice (Figure 2B). However, the percentage of CXCR5+PD-1+ TFH cells within the effector CD4 T-cell compartment, the percentage of activated TFH within total CD4 T cells, and the absolute number of TFH cells per spleen in FVIII-rechallenged animals were significantly higher than those obtained from saline-injected, primed mice (19.3% ± 4.1% vs 10.8% ± 2.5%; 4.6% ± 2% vs 2.4% ± 1.4%; and 36.7% ± 11 ×104 vs 14.8% ± 3.8% ×104, respectively; Figure 7B-D). These data suggest that memory FVIII-specific TFH cells persist and can induce efficient recall TFH responses after rfF8 rechallenge.
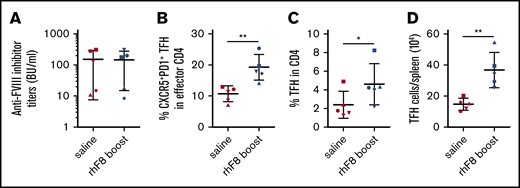
Rechallenge with FVIII antigen induces recall responses of TFH cells. Eight to 10 weeks after the last round of IV FVIII immunization, pairs of mice bearing similar inhibitor titers were rechallenged with 50 U/kg rhF8 or saline. Five days after boost, the mice were euthanized. Splenocytes were stained for CD3, CD4, CD19, CD44, CD62L, Foxp3, CXCR5, and PD-1 and analyzed by flow cytometry. (A) FVIII inhibitor titers. (B) Percentage of CXCR5+PD-1+ TFH cells among effector CD4 helper cells. (C) Percentage of activated TFH cells among total CD4 T cells. (D) Total number of activated TFH cells per spleen was compared between saline-treated and rhF8-boosted animals. Paired mice of similar inhibitor titers are represented by the same symbol. *P < .05; **P < .01.
Rechallenge with FVIII antigen induces recall responses of TFH cells. Eight to 10 weeks after the last round of IV FVIII immunization, pairs of mice bearing similar inhibitor titers were rechallenged with 50 U/kg rhF8 or saline. Five days after boost, the mice were euthanized. Splenocytes were stained for CD3, CD4, CD19, CD44, CD62L, Foxp3, CXCR5, and PD-1 and analyzed by flow cytometry. (A) FVIII inhibitor titers. (B) Percentage of CXCR5+PD-1+ TFH cells among effector CD4 helper cells. (C) Percentage of activated TFH cells among total CD4 T cells. (D) Total number of activated TFH cells per spleen was compared between saline-treated and rhF8-boosted animals. Paired mice of similar inhibitor titers are represented by the same symbol. *P < .05; **P < .01.
Discussion
CD4 T-cell help is critical for the generation and maintenance of GCs, and TFH cells are the CD4 T-cell subset needed for this process.26,38,39 Multiple lines of evidence indicate that the immune response to FVIII is CD4 T-cell dependent.40 However, according to current knowledge, CD4 T cells have been characterized by at least 7 distinct subsets, including Th1, Th2, Th9, Th17, Th22, TFH, and regulatory T (Treg) cells.41,42 It has long been thought that FVIII immune responses are directed by the Th1/Th2 pathway.43-46 The functional properties of TFH cells in FVIII inhibitor development have never been explored. In the current study, we confirmed that anti-FVIII inhibitor development in HA mice is CD4 T-cell dependent. We have demonstrated for the first time that IV FVIII immunization induces activation and accumulation of antigen-specific TFH cells in spleen of HA mice. FVIII inhibitor production upon FVIII infusion is impaired when CXCR5+/+ TFH cells are deleted. Our data strongly suggest that TFH cells play an important role in FVIII inhibitor development.
In our study, activated TFH cells were defined as CD44+CD62L−PD-1hiCXCR5hiCD4+ T cells. These cells have mature TFH-cell phenotypes with expression of BCL6, ICOS, and CD40L. The percentage and absolute number of activated TFH cells significantly increased in the spleens of mice that produced FVIII inhibitors. Increased splenic TFH generation correlated with GC formation and FVIII inhibitor development. When other secondary lymphoid organs (mesenteric and inguinal LNs) were analyzed, the percentage and absolute number of activated TFH cells in mice that produced inhibitors were similar to those in naive controls (data not shown). In the current study, FVIII was administered by IV injection to mimic clinical treatment. The spleen is a highly organized blood-filtration system that plays a primary role in the development of immunity to many blood-borne antigens. Previous studies has shown that FVIII accumulates in the marginal sinus of the spleen soon after its administration, and splenic marginal zone macrophages, CD11c+CD8α− dendritic cells, and B cells all play important roles in initiating inhibitor formation.47-50 In our current study, activated TFH cells accumulated only in the spleen and not in other secondary lymphoid organs, suggesting that the spleen is the primary site for TFH activation when FVIII is administered IV. GL-7 expression on TFH cells is associated with a mature GC TFH-cell phenotype, and maintenance of its expression in GC TFH cells depends on the establishment of cognate T-/B-cell interactions within the follicle.51 We found an increased frequency of GL-7-expressing TFH cells in the spleen of mice that produce anti-FVIII inhibitors. We believe that the high frequency of activated GL-7-expressing TFH cells is indicative of adequate T-/B-cell interactions in the spleens of mice that produce anti-FVIII inhibitors.
Our study shows a marked increase in the number of activated TFH cells in spleens after repeated IV FVIII immunization. With Ki-67 staining, we found that the proliferative status of activated TFH cells from inhibitor-producing mice was similar to that in control mice. This suggests that the increase in activated TFH cells in spleens of inhibitor-producing mice could be related to increased TFH-cell differentiation and/or survival, rather than cell proliferation. However, our further experiments showed that activated TFH cells in FVIII-primed mice had a proapoptotic phenotype, which suggests that increased survival of TFH cells is not the reason for TFH cell expansion and accumulation in inhibitor-producing mice after FVIII immunization. As both increased proliferation and better survival cannot explain the TFH-cell expansion, increased TFH-cell differentiation is a potential responsible mechanism. The percentage of effector CD4 T cells significantly increased in mice that received FVIII immunization, suggesting that more naive CD4 T cells are differentiated into effector cells and possibly more CD4 T cells are differentiated into TFH cells in inhibitor-producing mice compared with naive or non–inhibitor-producing animals. It has been shown that Th1, Th2, and Th17 cells can be reprogrammed to obtain TFH cell characteristics and differentiate into TFH cells, both in vitro and in vivo.52-54 TFH-cell differentiation from the Th1 and/or Th2 subsets when mice received repeated IV infusions of FVIII is another potential mechanism responsible for TFH-cell expansion. Although most activated TFH cells have a proapoptotic phenotype similar to that of terminally differentiated effector cells, in vivo FVIII rechallenge efficiently induced a TFH-cell recall response. Furthermore, our in vitro T-cell proliferation assay confirmed that rhF8-primed TFH cells can specifically respond to rhF8 restimulation and proliferate effectively. These data suggest that not all of the activated TFH cells in FVIII-primed mice are terminally differentiated, short-lived cells. Indeed, the evidence suggests that some TFH cells can stay in the GC, survive, and develop into memory TFH cells.38 55 How TFH cells are regulated for their fates during FVIII immune responses is still unclear and should be further investigated.
Because CXCR5 mediates the follicular localization of TFH cells, we determined the requirement for CXCR5+ TFH-cell migration during FVIII immunization to verify the role of CXCR5+ TFH in anti-FVIII inhibitor generation. Using an experimental mouse model with specific depletion of CXCR5+/+ TFH cells, we demonstrated that CXCR5+/+ TFH cells are critical for the development of FVIII-neutralizing antibodies. Thus, identification of TFH cells involved in the FVIII immune response may help to develop novel strategies to prevent inhibitor formation in patients with HA. Considering their essential role in the anti-FVIII immune response, TFH cells should be a better target for immune modulation to help induce tolerance in HA. Interrupting TFH-cell differentiation by blocking CD40–CD40L interactions or IL-21 signaling, or by delivering miR-146a11, already shown to improve disease outcomes in lupus-prone mice,56 are strategies that could be used. Modulating the TFH pathway could be a new intervention to prevent anti-FVIII inhibitor development.
In conclusion, the data reported here demonstrate that the induction of TFH cells in FVIIInull mice is critical for FVIII-induced inhibitor development and that the FVIII immune response is GC dependent. Our findings in the current study may help in developing novel therapeutics to prevent or reverse FVIII immune responses in the treatment of patients with HA.
Acknowledgments
The authors thank Haig Kazazian (the University of Pennsylvania) for the FVIIInull mice.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant HL-102035 (Q.S.); National Hemophilia Foundation Bridge Award (Q.S.); and generous gifts from the Children’s Hospital of Wisconsin Foundation (Q.S.); and the Midwest Athletes Against Childhood Cancer Fund (Q.S.).
Authorship
Contribution: W.J. designed the study, performed experiments, analyzed the data, and wrote the manuscript; J.C. designed the study, performed the experiments, and analyzed the data; Y. Cai and Y. Chen performed the experiments; J.A.S. performed the experiments and made comments on the manuscript; B.D.J. provided reagents and made comments on the manuscript; W.C. contributed to the concept of the study and the study design and made comments on the manuscript; and Q.S. directed the study, interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Qizhen Shi, Department of Pediatrics, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: qshi@versiti.org.

