

Key Points
The toxicity of allo-HCT in patients with prior CAR-T therapy was not higher than what is expected in these high-risk patients.
In ALL patients, there seems to be a benefit from earlier utilization of allo-HCT after CAR-T therapy.

Abstract
Allogeneic hematopoietic cell transplantation (allo-HCT) is offered to selected patients after chimeric antigen receptor–modified T-cell (CAR-T) therapy. Lymphodepleting chemotherapy and CAR-T therapy have immunosuppressive and immunomodulatory effects that could alter the safety profile of subsequent allo-HCT. We reviewed our experience with 32 adults (acute lymphoblastic leukemia [ALL], n = 19; B-cell non-Hodgkin lymphoma [NHL]/chronic lymphocytic leukemia [CLL], n = 13) who received an allo-HCT after CAR-T therapy, with a focus on posttransplant toxicities. Myeloablative conditioning (MAC) was used in 74% of ALL patients and 39% of NHL/CLL patients. The median time from CAR-T therapy to allo-HCT was 72 days in ALL patients and 122 days in NHL/CLL patients. Cumulative incidences of grade 3-4 acute graft-versus-host disease (GVHD) and chronic GVHD were 25% and 10%, respectively. All patients had neutrophil recovery (median, 18.5 days) and all but 3 had platelet recovery (median, 12 days). Twenty-two percent had viral or systemic fungal infection within 100 days after allo-HCT. The 100-day and 1-year cumulative incidences of NRM were 16% and 21%, respectively, for ALL patients and 15% and 33%, respectively, for NHL/CLL patients. In ALL patients, later utilization of allo-HCT after CAR-T therapy was associated with higher mortality. In NHL/CLL patients, MAC was associated with higher mortality. Toxicities did not exceed the expected incidences in this high-risk population.
Introduction
Treatment with autologous CD19-specific chimeric antigen receptor T-cell (CAR-T) therapy has shown promising efficacy in patients with relapsed or refractory acute lymphoblastic leukemia (ALL), B-cell non-Hodgkin lymphoma (NHL), and chronic lymphocytic leukemia (CLL).1-6 Allogeneic hematopoietic cell transplantation (allo-HCT) is often offered to ALL patients to consolidate remission achieved after CAR-T therapy. In NHL/CLL, allo-HCT is primarily used in patients who have refractory disease after CAR-T therapy or relapse after an initial response if a subsequent remission is achieved with additional treatment.7,8 Lymphodepletion (LD) chemotherapy and CAR-T therapy have immunosuppressive and immunomodulatory effects, and their impact on the immune system and endothelium could affect the safety profile of allo-HCT.9,10 We report safety and toxicity data from patients undergoing allo-HCT after receiving CAR-T therapy.
Methods
Patients with ALL, NHL, or CLL who received CAR-T therapy on an investigator-initiated phase 1/2 clinical trial (NCT01865617)4-6 and subsequently underwent allo-HCT were included in this analysis. Graft-versus-host disease (GVHD) estimates were calculated using cumulative incidence, with relapse or death as competing risks. A Cox proportional-hazards model was used to study associations between clinical factors and overall mortality and nonrelapse mortality (NRM). We considered the following variables for univariate analysis: prior cytokine-release syndrome (CRS) or neurotoxicity (NT), time between CAR-T infusion and allo-HCT, hematopoietic cell transplantation comorbidity index (HCT‐CI), conditioning regimen intensity, and disease-directed therapy between CAR-T therapy and allo-HCT. The study was approved by the institutional review board of Fred Hutch and was conducted in accordance with the Declaration of Helsinki.
Results
Between 2014 and 2017, 32 patients (ALL, n = 19; NHL, n = 8; CLL, n = 5) underwent allo-HCT after ≥1 CAR-T infusion. The median age at transplant was 46 years (range, 23-74). Three ALL patients (16%) had a previous allo-HCT, and 5 NHL patients (38%) had undergone autologous transplant before CAR-T therapy. Twenty-six patients (81%) had cyclophosphamide and fludarabine and 6 patients (19%) had cyclophosphamide-based LD without fludarabine before their first CAR-T therapy. Nine patients received a second CAR-T infusion a median of 50 days after the first infusion (range, 14-307; 8 with LD, 1 without LD). Efficacy and toxicity of CAR-T treatment are summarized in Table 1.
Transplant characteristics in patients with prior CD19 CAR-T treatment
| ALL (n = 19) | NHL/CLL (n = 13) | Entire cohort (N = 32) | |
|---|---|---|---|
| Age, median (range), y | 39 (23-74) | 52 (37-65) | 46 (23-74) |
| Dose of first CAR-T therapy, cells/kg | |||
| 2 × 105 | 8 (42) | 2 (15) | 10 (31) |
| 2 × 106 | 11 (58) | 9 (70) | 20 (63) |
| 2 × 107 | 0 (0) | 2 (15) | 2 (6) |
| LD regimen for first CAR-T therapy | |||
| Cy-Flu | 16 (84) | 10 (77) | 26 (81) |
| Cy-based without Flu | 3 (16) | 3 (23) | 6 (19) |
| Best response to first CAR-T therapy* | |||
| CR | 18 (95) | 2 (15) | 20 (62) |
| PR | 0 (0) | 5 (39) | 5 (16) |
| SD | 0 (0) | 2 (15) | 2 (6) |
| PD | 1 (5) | 4 (31) | 5 (16) |
| CRS (grade) after first CAR-T therapy† | |||
| 0 | 7 (37) | 3 (23) | 10 (31) |
| 1 | 4 (21) | 5 (39) | 9 (28) |
| 2 | 7 (37) | 4 (31) | 11 (35) |
| 3 | 1 (5) | 1 (8) | 2 (6) |
| NT (grade) after first CAR-T therapy‡ | |||
| 0 | 12 (63) | 5 (39) | 17 (53) |
| 1 | 1 (5) | 2 (15) | 3 (9) |
| 2 | 2 (11) | 4 (31) | 6 (19) |
| 3 | 4 (21) | 2 (15) | 6 (19) |
| 4 | 0 (0) | 0 (0) | 0 (0) |
| Second CAR-T infusion, n | 2 | 7 | 9 |
| Best response to second CAR-T therapy | |||
| CR | 2 (100) | 1 (14) | 3 (34) |
| PR | 0 (0) | 2 (28.5) | 2 (22) |
| SD | 0 (0) | 2 (28.5) | 2 (22) |
| PD | 0 (0) | 2 (28.5) | 2 (22) |
| CRS (grade) after second CAR-T therapy | |||
| 0 | 2 (100) | 4 (57) | 6 (67) |
| 1 | 0 (0) | 2 (29) | 2 (22) |
| 2 | 0 (0) | 1 (14) | 1 (11) |
| 3 | 0 (0) | 0 (0) | 0 (0) |
| NT (grade) after second CAR-T therapy | |||
| 0 | 2 (100) | 5 (72) | 7 (77) |
| 1 | 0 (0) | 0 (0) | 0 (11) |
| 2 | 0 (0) | 1 (14) | 1 (11) |
| 3 | 0 (0) | 0 (0) | 0 (0) |
| 4 | 0 (0) | 1 (14) | 1 (11) |
| HCT-CI | |||
| 0 | 4 (21) | 2 (15) | 6 (19) |
| 1 | 2 (11) | 5 (39) | 7 (22) |
| 2 | 5 (26) | 3 (23) | 8 (25) |
| 3 | 3 (15) | 0 (0) | 3 (9.5) |
| 4 | 2 (11) | 1 (8) | 3 (9.5) |
| 5 | 2 (11) | 2 (15) | 4 (12) |
| 6 | 1 (5) | 0 (0) | 1 (3) |
| Donor type | |||
| MRD | 3 (16) | 2 (15) | 5 (16) |
| MUD | 9 (50) | 8 (62) | 17 (53) |
| mMURD | 1 (4) | 1 (8) | 2 (6) |
| Haploidentical | 1 (4) | 2 (15) | 3 (9) |
| UCT | 5 (26) | 0 (0) | 5 (16) |
| Cell type | |||
| PBSC | 13 (69) | 13 (100) | 26 (81) |
| BM | 1 (5) | 0 (0) | 1 (3) |
| Cord | 5 (26) | 0 (0) | 5 (16) |
| Conditioning regimen | |||
| MAC | 14 (74) | 5 (39) | 19 (59) |
| RIC | 2 (10) | 3 (23) | 5 (16) |
| NMA | 3 (16) | 5 (38) | 8 (25) |
| GVHD prophylaxis | |||
| CNI + MMF | 5 (26) | 6 (46) | 11 (35) |
| CNI + MMF + sirolimus | 1 (5) | 2 (15) | 3 (9) |
| CNI + MTX | 9 (48) | 3 (23) | 12 (38) |
| CNI + MTX + abatacept | 3 (16) | 0 (0) | 3 (9) |
| CNI + MMF + PtCy | 1 (5) | 1 (8) | 2 (6) |
| NCI + ATG | 0 (0) | 1 (8) | 1 (3) |
| ALL (n = 19) | NHL/CLL (n = 13) | Entire cohort (N = 32) | |
|---|---|---|---|
| Age, median (range), y | 39 (23-74) | 52 (37-65) | 46 (23-74) |
| Dose of first CAR-T therapy, cells/kg | |||
| 2 × 105 | 8 (42) | 2 (15) | 10 (31) |
| 2 × 106 | 11 (58) | 9 (70) | 20 (63) |
| 2 × 107 | 0 (0) | 2 (15) | 2 (6) |
| LD regimen for first CAR-T therapy | |||
| Cy-Flu | 16 (84) | 10 (77) | 26 (81) |
| Cy-based without Flu | 3 (16) | 3 (23) | 6 (19) |
| Best response to first CAR-T therapy* | |||
| CR | 18 (95) | 2 (15) | 20 (62) |
| PR | 0 (0) | 5 (39) | 5 (16) |
| SD | 0 (0) | 2 (15) | 2 (6) |
| PD | 1 (5) | 4 (31) | 5 (16) |
| CRS (grade) after first CAR-T therapy† | |||
| 0 | 7 (37) | 3 (23) | 10 (31) |
| 1 | 4 (21) | 5 (39) | 9 (28) |
| 2 | 7 (37) | 4 (31) | 11 (35) |
| 3 | 1 (5) | 1 (8) | 2 (6) |
| NT (grade) after first CAR-T therapy‡ | |||
| 0 | 12 (63) | 5 (39) | 17 (53) |
| 1 | 1 (5) | 2 (15) | 3 (9) |
| 2 | 2 (11) | 4 (31) | 6 (19) |
| 3 | 4 (21) | 2 (15) | 6 (19) |
| 4 | 0 (0) | 0 (0) | 0 (0) |
| Second CAR-T infusion, n | 2 | 7 | 9 |
| Best response to second CAR-T therapy | |||
| CR | 2 (100) | 1 (14) | 3 (34) |
| PR | 0 (0) | 2 (28.5) | 2 (22) |
| SD | 0 (0) | 2 (28.5) | 2 (22) |
| PD | 0 (0) | 2 (28.5) | 2 (22) |
| CRS (grade) after second CAR-T therapy | |||
| 0 | 2 (100) | 4 (57) | 6 (67) |
| 1 | 0 (0) | 2 (29) | 2 (22) |
| 2 | 0 (0) | 1 (14) | 1 (11) |
| 3 | 0 (0) | 0 (0) | 0 (0) |
| NT (grade) after second CAR-T therapy | |||
| 0 | 2 (100) | 5 (72) | 7 (77) |
| 1 | 0 (0) | 0 (0) | 0 (11) |
| 2 | 0 (0) | 1 (14) | 1 (11) |
| 3 | 0 (0) | 0 (0) | 0 (0) |
| 4 | 0 (0) | 1 (14) | 1 (11) |
| HCT-CI | |||
| 0 | 4 (21) | 2 (15) | 6 (19) |
| 1 | 2 (11) | 5 (39) | 7 (22) |
| 2 | 5 (26) | 3 (23) | 8 (25) |
| 3 | 3 (15) | 0 (0) | 3 (9.5) |
| 4 | 2 (11) | 1 (8) | 3 (9.5) |
| 5 | 2 (11) | 2 (15) | 4 (12) |
| 6 | 1 (5) | 0 (0) | 1 (3) |
| Donor type | |||
| MRD | 3 (16) | 2 (15) | 5 (16) |
| MUD | 9 (50) | 8 (62) | 17 (53) |
| mMURD | 1 (4) | 1 (8) | 2 (6) |
| Haploidentical | 1 (4) | 2 (15) | 3 (9) |
| UCT | 5 (26) | 0 (0) | 5 (16) |
| Cell type | |||
| PBSC | 13 (69) | 13 (100) | 26 (81) |
| BM | 1 (5) | 0 (0) | 1 (3) |
| Cord | 5 (26) | 0 (0) | 5 (16) |
| Conditioning regimen | |||
| MAC | 14 (74) | 5 (39) | 19 (59) |
| RIC | 2 (10) | 3 (23) | 5 (16) |
| NMA | 3 (16) | 5 (38) | 8 (25) |
| GVHD prophylaxis | |||
| CNI + MMF | 5 (26) | 6 (46) | 11 (35) |
| CNI + MMF + sirolimus | 1 (5) | 2 (15) | 3 (9) |
| CNI + MTX | 9 (48) | 3 (23) | 12 (38) |
| CNI + MTX + abatacept | 3 (16) | 0 (0) | 3 (9) |
| CNI + MMF + PtCy | 1 (5) | 1 (8) | 2 (6) |
| NCI + ATG | 0 (0) | 1 (8) | 1 (3) |
Unless otherwise indicated, data are n (%).
ATG, anti-thymocyte globulin; BM, bone marrow; CNI, calcineurin inhibitor; CR, complete response; Cy, cyclophosphamide; Flu, fludarabine; MAC, myeloablative conditioning; MMF, mycophenolate mofetil; mMRD, mismatch related donor; MRD, matched related donor; MTX, methotrexate; MUD, matched unrelated donor; NMA, nonmyeloablative conditioning; PBSC, peripheral blood stem cell; PD, progressive disease; PR, partial response; PtCy, posttransplant cyclophosphamide; RIC, reduced-intensity conditioning; SD, stable disease; UCT, umbilical cord transplant.
Response assessment using the Lugano classification17 and the International Workshop on Chronic Lymphocytic Leukemia18 criteria.
CRS grading per modified Lee et al.19
NT grading per National Cancer Institute Common Toxicity Criteria for Adverse Events (CTCAE version 4.03).20
In ALL patients, the median time from CAR-T therapy to allo-HCT was 72 days (range, 28-138). Two patients (10%) received therapy between CAR-T therapy and allo-HCT. Before allo-HCT, all ALL patients had a morphologic complete response (CR), and 18 (95%) were minimal residual disease negative by flow cytometry (sensitivity, 1:10 000). Eight of 10 ALL patients (80%) with an identified index clone by immunoglobulin heavy (IGH) chain sequencing had no malignant clone in marrow before allo-HCT. In NHL/CLL patients, the median time to allo-HCT was 112 days (range, 55-456). Nine patients (69%) received interim therapy. At the time of allo-HCT, all patients (n = 13) had evidence of disease, and 6 patients (46%) had bulky lymphadenopathy (≥5 cm).
Nineteen patients (59%; 74% of ALL and 38% of NHL/CLL) received myeloablative conditioning (MAC). Stem cell graft sources included HLA-matched unrelated peripheral blood stem cells (PBSCs; 53%), HLA-matched related PBSCs (16%), umbilical cord blood (UCB; 16%, all ALL), haploidentical PBSCs (9%), and HLA-mismatched unrelated PBSCs (6%). The median HCT-CI was 2; 19 patients (59%) had HCT-CI ≥2. Relevant transplant characteristics are summarized in Table 1.
The median follow-up after allo-HCT was 35 months. All patients achieved neutrophil engraftment (>1000 per cubic millimeter). The median times to an absolute neutrophil count ≥500 and ≥1000 per cubic millimeter were 16 and 18.5 days, respectively. With the exception of 3 patients with a higher platelet goal (50 000-100 000) because of increased hemorrhagic risk, all patients achieved platelet engraftment (>70 000 per cubic millimeter without transfusion) by day 100 after hematopoietic cell transplantation. Median time to achieve a platelet count ≥ 20 000 and ≥70 000 per cubic millimeter were 12 and 14 days, respectively. All patients received platelet transfusions (median, 6.5 episodes) and red cell transfusions (median, 5.5 episodes) in the first 100 days. All patients achieved 100% donor CD3 and CD33 chimerism in blood at day 28. CAR-Ts were detected at the limit of detection by quantitative polymerase chain reaction in only 3 patients (1 after MAC) after allo-HCT.
One-year estimates of grade 2-4 and grade 3-4 acute GVHD were 69% (95% confidence [CI], 52-85) and 25% (95% CI, 10-40), respectively. The median time to acute GVHD was 27 days. Five (16%) patients developed chronic GVHD a median of 305 days after hematopoietic cell transplantation (mild, n = 2; moderate, n = 1; severe, n = 2), with a 1-year estimate of 10% (range, 0-21). B-cell aplasia was observed in 24 patients (75%) at allo-HCT but was not associated with the incidence of grade 3-4 acute GVHD (P = .38). Three patients (9%) developed thrombotic microangiopathy (TMA) with renal disease. One patients had experienced transient grade 3 NT after CAR-T infusion and then developed hemolytic uremic syndrome 79 days later (on day 17 posttransplant). The other 2 patients had no neurologic adverse events after CAR-T therapy. We did not identify associations between prior CRS and/or NT after CAR-T therapy and TMA after allo-HCT or between CRS/NT and overall mortality or NRM.
Viral and fungal infections are summarized in Table 2. Eleven patients (34%) had bacterial infections: coagulase-negative Staphylococcus (n = 3), Enterococcus spp. (n = 3), Escherichia coli (n = 2), Streptococcus mitis (n = 2), Clostridium difficile (n = 2), and Legionella pneumophila (n = 1). Six patients (18%) had invasive fungal infections, including 3 with documented aspergillosis. One patient had vitreous toxoplasmosis. Six ALL patients died from disease progression (n = 2), Aspergillus pneumonia (n = 1), bacterial sepsis (n = 1), GVHD (n = 1), and idiopathic pulmonary syndrome (n = 1). Six NHL/CLL patients died because of disease progression (n = 2), GVHD (n = 2), pulmonary emboli (n = 1), and fungal infection (n = 1) (Table 2). At a median follow-up of 36 months for ALL patients, 1-year estimate of overall survival (OS) was 58% (95% CI, 40-85), and the 100-day and 1-year NRM estimate rates were 16% and 21%, respectively. For NHL/CLL patients, at a median follow-up of 35 months, 1-year estimate of OS was 59% (95% CI, 37-95), and the 100-day and 1-year NRM rates were 15% and 33%, respectively (Figure 1).
Posttransplant complications in patients with prior CD19-targeted CAR-T treatment
| ALL (n = 19) | NHL/CLL (n = 13) | Entire cohort (N = 32) | |
|---|---|---|---|
| Acute GVHD | |||
| Grade 1 | 2 (10.5) | 1 (7.7) | 3 (9.4) |
| Grade 2 | 8 (42.1) | 6 (46.2) | 14 (43.8) |
| Grade 3 | 3 (15.8) | 2 (15.4) | 5 (15.6) |
| Grade 4 | 1 (5.3) | 2 (15.4) | 3 (9.4) |
| 1-y cumulative incidence of grade 2-4 (95% CI), % | 63 (40-86) | 77 (51-100) | 69 (52-85) |
| 1-y cumulative incidence of grade 3-4 (95% CI), % | 21 (2-40) | 31 (4-57) | 25 (10-40) |
| Chronic GVHD | |||
| No | 15 (78.9) | 12 (92.3) | 27 (84.4) |
| Yes | 4 (21.1) | 1 (7.7) | 5 (15.6) |
| 1-y cumulative incidence (95% CI), % | 16 (0-33) | 0 (0-0) | 10 (0-21) |
| Viral and fungal infections | |||
| Parainfluenza | 2 (10.5) | 0 (0.0) | 2 (6.2) |
| HSV-1 stomatitis | 1 (5.3) | 0 (0.0) | 1 (3.1) |
| Adenovirus hepatitis | 1(5.3) | 0 (0.0) | 1 (3.1) |
| CMV gastroenteritis | 1 (5.3) | 2 (15.4) | 3 (9.4) |
| RSV | 1 (5.3) | 0 (0.0) | 1 (3.1) |
| Aspergillosis | 3 (15.8) | 0 (0.0) | 3 (9.4) |
| Other fungal | 1 (5.3) | 2 (15.4) | 3 (9.4) |
| Toxoplasmosis | 1 (5.3) | 0 (0.0) | 1 (3.1) |
| Nonhematologic toxicities | |||
| Neurologic: PRESS | 1 (8) | 0 (0) | 1 (3) |
| Cardiac: atrial fibrillation | 1 (5) | 1 (8) | 2 (6) |
| Hepatic: cholecystitis | 1 (5) | 0 (0) | 1 (3) |
| Renal: TMS | 0 (0) | 3 (23) | 3 (9) |
| Renal: other renal | 2 (10.5) | 0 (0) | 2 (6) |
| GI: bleeding | 0 (0) | 1 (8) | 1 (3) |
| Pulmonary: DAH | 2 (10.5) | 1 (8) | 3 (9) |
| Pulmonary: PE | 0 (0) | 1 (8) | 1 (3) |
| Cause of death | |||
| Disease progression | 2 (10.5) | 2 (15.4) | 4 (12.5) |
| GVHD | 1 (5.3) | 2 (15.4) | 3 (9.4) |
| Fungal infection | 1 (5.3) | 1 (7.7) | 2 (6.2) |
| Sepsis | 1 (5.3) | 0 | 1 (3.1) |
| Pulmonary | |||
| PE | 0 | 1 (7.7) | 1 (3.1) |
| IPS | 1 (5.3) | 0 | 1 (3.1) |
| Total | 6 (31) | 6 (46) | 12 (37.5) |
| ALL (n = 19) | NHL/CLL (n = 13) | Entire cohort (N = 32) | |
|---|---|---|---|
| Acute GVHD | |||
| Grade 1 | 2 (10.5) | 1 (7.7) | 3 (9.4) |
| Grade 2 | 8 (42.1) | 6 (46.2) | 14 (43.8) |
| Grade 3 | 3 (15.8) | 2 (15.4) | 5 (15.6) |
| Grade 4 | 1 (5.3) | 2 (15.4) | 3 (9.4) |
| 1-y cumulative incidence of grade 2-4 (95% CI), % | 63 (40-86) | 77 (51-100) | 69 (52-85) |
| 1-y cumulative incidence of grade 3-4 (95% CI), % | 21 (2-40) | 31 (4-57) | 25 (10-40) |
| Chronic GVHD | |||
| No | 15 (78.9) | 12 (92.3) | 27 (84.4) |
| Yes | 4 (21.1) | 1 (7.7) | 5 (15.6) |
| 1-y cumulative incidence (95% CI), % | 16 (0-33) | 0 (0-0) | 10 (0-21) |
| Viral and fungal infections | |||
| Parainfluenza | 2 (10.5) | 0 (0.0) | 2 (6.2) |
| HSV-1 stomatitis | 1 (5.3) | 0 (0.0) | 1 (3.1) |
| Adenovirus hepatitis | 1(5.3) | 0 (0.0) | 1 (3.1) |
| CMV gastroenteritis | 1 (5.3) | 2 (15.4) | 3 (9.4) |
| RSV | 1 (5.3) | 0 (0.0) | 1 (3.1) |
| Aspergillosis | 3 (15.8) | 0 (0.0) | 3 (9.4) |
| Other fungal | 1 (5.3) | 2 (15.4) | 3 (9.4) |
| Toxoplasmosis | 1 (5.3) | 0 (0.0) | 1 (3.1) |
| Nonhematologic toxicities | |||
| Neurologic: PRESS | 1 (8) | 0 (0) | 1 (3) |
| Cardiac: atrial fibrillation | 1 (5) | 1 (8) | 2 (6) |
| Hepatic: cholecystitis | 1 (5) | 0 (0) | 1 (3) |
| Renal: TMS | 0 (0) | 3 (23) | 3 (9) |
| Renal: other renal | 2 (10.5) | 0 (0) | 2 (6) |
| GI: bleeding | 0 (0) | 1 (8) | 1 (3) |
| Pulmonary: DAH | 2 (10.5) | 1 (8) | 3 (9) |
| Pulmonary: PE | 0 (0) | 1 (8) | 1 (3) |
| Cause of death | |||
| Disease progression | 2 (10.5) | 2 (15.4) | 4 (12.5) |
| GVHD | 1 (5.3) | 2 (15.4) | 3 (9.4) |
| Fungal infection | 1 (5.3) | 1 (7.7) | 2 (6.2) |
| Sepsis | 1 (5.3) | 0 | 1 (3.1) |
| Pulmonary | |||
| PE | 0 | 1 (7.7) | 1 (3.1) |
| IPS | 1 (5.3) | 0 | 1 (3.1) |
| Total | 6 (31) | 6 (46) | 12 (37.5) |
Unless otherwise indicated, data are n (%).
CMV, cytomegalovirus; DAH, diffuse alveolar hemorrhage; GI, gastrointestinal; HSV, herpes simplex virus; IPS, idiopathic pulmonary syndrome; PE, pulmonary embolus; PRESS, posterior reversible encephalopathy syndrome; RSV, respiratory syncytial virus.
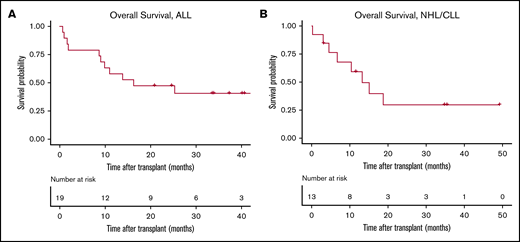
OS curves. Patients who received an allo-HCT after CAR-T therapy for a diagnosis of ALL (A) or NHL/CLL (B).
OS curves. Patients who received an allo-HCT after CAR-T therapy for a diagnosis of ALL (A) or NHL/CLL (B).
In ALL patients, longer time from CAR-T therapy to allo-HCT (≥80 vs <80 days) was associated with higher risk for death (hazard ratio [HR], 4.01; 95% CI, 1.14-14.0; P = .03) and higher NRM (HR, 4.4; 95% CI, 0.54-21.1; P = .19). On the other hand, in NHL/CLL patients, there was a trend toward lower NRM (HR, 0.14; 95% CI, 0.01-1.72; P = .12) and a lower risk for fungal or systemic viral infection (odds ratio, 0.04; 95% CI, 0.001-0.64; P = .04) when allo-HCT was done later (≥80 vs <80 days) after CAR-T therapy. Using a MAC regimen (vs reduced-intensity conditioning or nonmyeloablative conditioning) was associated with a higher risk for death (HR, 3.83; 95% CI, 0.91-16.6; P = .06) and NRM (HR, 8.3; 95% CI, 0.90-100; P = .06) in NHL/CLL patients. Higher HCT-CI was associated with a trend toward higher overall mortality (HR, 1.37; 95% CI, 0.9-2.01; P = .11) and NRM (HR, 1.43; 95% CI, 0.9-2.28; P = .12).
Discussion
Treatments, such as allo-HCT, may improve the durability of remissions achieved after CAR-T therapy, or they can be used after progression post–CAR-T treatment. Prior CAR-T therapy can potentially increase the posttransplant toxicity by inducing immunosuppression and endothelial damage. In this study, we focused on post–allo-HCT complications in patients with prior CAR-T treatment. The study demonstrated the feasibility of this approach and did not reveal a signal indicating an increased risk for specific adverse events after allo-HCT.
At our institution (Fred Hutch/University of Washington), allo-HCT is commonly offered as consolidative therapy to ALL patients who achieve a remission after CAR-T therapy. On the other hand, NHL/CLL patients are usually referred for allo-HCT only with refractory or relapsed disease after CAR-T therapy (Figure 2).
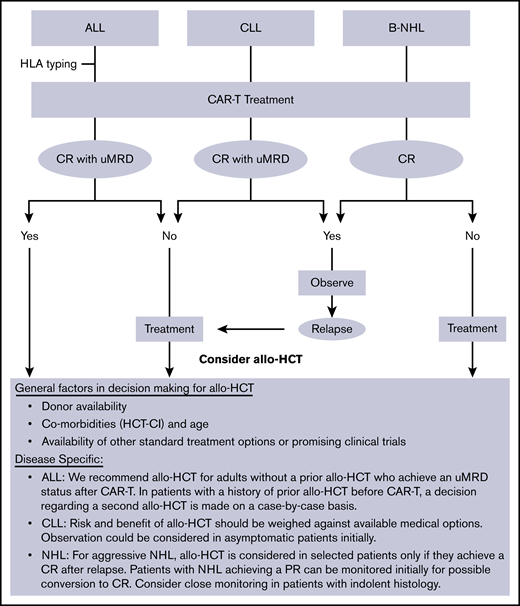
General institutional approach to utilization of allo-HCT in patients treated with CD19-targeted CAR-T therapy. PR, partial remission; uMRD, undetectable minimal residual disease.
General institutional approach to utilization of allo-HCT in patients treated with CD19-targeted CAR-T therapy. PR, partial remission; uMRD, undetectable minimal residual disease.
Given this differential approach in utilizing allo-HCT, which could potentially induce a bias in the interpretation of data when analyzed in combination, we reported the data separately for the 2 cohorts (ALL vs NHL/CLL). The main purpose of the study was to detect any possible adverse event that was disproportionately more common in this setting compared with the known post–allo-HCT benchmark. Although such risk was not observed in this study, these patients should be monitored closely for potential toxicities that may become evident as more patients are treated with allo-HCT after CAR-T therapy.
One notable finding in the ALL cohort was the improved outcomes in patients who received allo-HCT earlier rather than later after CAR-T therapy. These data support our current practice of early transplant discussions and HLA typing for suitable ALL patients undergoing CAR-T therapy. The benefit of performing an earlier allo-HCT was not seen in the NHL/CLL cohort. To explain this difference, it should be noted that fewer patients in the ALL group received interim therapy compared with NHL/CLL patients, who more commonly required additional treatment of disease control before allo-HCT (10.5% vs 69%). This may explain, at least in part, the higher toxicity of allo-HCT when it was done earlier in NHL/CLL patients.
In summary, the incidences of adverse events after allo-HCT were not above those expected in these high-risk patients and the incidence and kinetics of hematopoietic recovery, and the incidences of infections and GVHD were comparable to previous studies conducted in the absence of prior CAR-T therapy.11-16 This study represents a heterogenous group of patients with a variety of treatments and transplant details. This was inevitable given the relatively limited experience with CAR-T therapy and the even smaller number of patients with subsequent allo-HCT. Nevertheless, and although the results should be interpreted with caution, the data provide a platform for the design of prospective studies to address the efficacy, cost effectiveness, and timing of allo-HCT after CAR-T immunotherapy.
Acknowledgments
The authors thank the clinical providers, nurses, and research staff in the transplant and immunotherapy programs at Fred Hutch, Seattle Cancer Care Alliance, and the University of Washington.
The Fred Hutchinson Cancer Research Center receives research funding from Juno Therapeutics, a Celgene company.
Authorship
Contribution: M.S., J.G., K.A.H., J.M.V., D.G.M., and C.J.T. designed the research; M.S., J.G., K.A.H., J.M.V., F.M., K.A.H., A.L., A.V.H., M.L.S., S.C., X.C., R.D.C., B.G.T., A.K.G., B.M.S., D.G.M., and C.J.T. provided and analyzed data; M.S. and C.J.T. wrote the manuscript; and all authors reviewed, edited, and approved the final manuscript.
Conflict-of-interest disclosure: M.S. has acted as a consultant or played an advisory role for AbbVie, Genentech, AstraZeneca, Sound Biologics, Verastem Oncology, ADC Therapeutics, Pharmacyclics, and Atara Biotherapeutics and has received research funding from Mustang Biopharma, Celgene, Pharmacyclics, Gilead Sciences, Genentech, AbbVie, TG Therapeutics, BeiGene, Acerta Pharma, Merck, and Sunesis. R.D.C. has received research funding from Amgen, Incyte, Kite/Gilead, Merck, Pfizer, and Vanda Pharmaceuticals and has served as a consultant/advisor to Amgen and Pfizer. B.G.T. has received research funding and patent/royalties from Mustang Bio. A.K.G. has received grants and nonfinancial support from Teva Pharmaceutical Industries, Bristol-Myers Squibb, Merck, Takeda, TG Therapeutics, and Effector; has received grants, personal fees, and nonfinancial support from Seattle Genetics, Pfizer, Janssen, Gilead Sciences, Spectrum, Amgen, and Incyte; and has received personal fees from Aptevo Therapeutics, BRIM Biotechnology, Seattle Genetics, Amgen, Acerta, I-Mab Biopharma, and Sanofi. B.M.S. has acted as a consultant for Kiadis Pharma, Actinium Pharmaceuticals, Bristol-Meyers Squibb, and Frazier Healthcare Ventures; has received research funding from Bellicum Pharmaceuticals; has equity ownership in AnaptysBio, OncoResponse, Inipharm, Mavupharma Inc., EpiThany, and Blaze Bioscience; and has been employed by Mavupharma Inc. D.G.M. has received honoraria from Kite Pharma, Gilead Sciences, Celgene, Genentech, and Novartis; has received institutional research funding from Kite Pharma, Juno Therapeutics, and Celgene; and has received remuneration for travel and accommodations from A2 Biotherapeutics. C.J.T. has received research funding from Juno Therapeutics, a Celgene company, and Nektar Therapeutics; holds a patent licensed to Juno Therapeutics, a Celgene company; has served on advisory boards and has options in Caribou Biosciences, Eureka Therapeutics, and Precision Biosciences; and has served on advisory boards for Aptevo, Humanigen, Juno Therapeutics, a Celgene company, Kite Pharma, a Gilead Company, Nektar Therapeutics, Novartis, T-CURX, and Allogene Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Mazyar Shadman, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, D5-396, Seattle, WA 98109; e-mail: mshadman@fredhutch.org.

