TO THE EDITOR:
Acute myeloid leukemia (AML) is characterized by a diverse cytogenetic and mutational landscape.1 AML subgroups defined by the World Health Organization (WHO) display characteristic gene expression signatures enabling transcriptome-based classification.2 As part of the Leucegene project, we previously reported the gene expression profiles of several AML subgroups and described a unified gene expression signature of the RUNX1-RUNX1T1 and the rare RUNX1-CBFA2T3 AML subgroups, revealing that alterations sharing a unified gene expression profile can share similar oncogenic deregulations.3
Using a similar strategy, here we define the transcriptomic profile of AML with mutations in nucleophosmin (NPM1), which is the most frequent WHO-defined subgroup.4 NPM1 is a nucleolar chaperone protein involved in several biological pathways, including ribosome biogenesis.5 NPM1 shuttling between nucleus and cytoplasm is determined by motifs that include the C-terminal nucleolar localization signal (NoLS), a bipartite nuclear localization signal, and 2 N-terminal nuclear export signals (NES).5 NPM1 mutations occur almost exclusively in exon 12, disrupting the NoLS and creating a new C-terminal NES, resulting in NPM1 aberrant cytoplasmic localization.5 The same phenotype is observed in the presence of rarely occurring mutations in exons 9 to 11.6-10 NPM1 mutations can be identified using immunohistochemistry4 and flow cytometry,6 but sequencing-based approaches limited to 3′ located exons are most commonly performed. Adequate identification of NPM1 mutations is critical for stratification of patients into favorable or intermediate risk groups, following on the presence or absence of concomitant FLT3 internal tandem duplications (ITDs), impacting therapeutic strategies and minimal residual disease monitoring.11,12 Using the NPM1 transcriptomic profile, we identified transcriptionally similar samples carrying exon 5 (NPM1e5) mutations that also result in aberrant cytoplasmic localization of NPM1.
This study is part of the Leucegene project, approved by the Research Ethics Boards of Université de Montréal, Maisonneuve-Rosemont Hospital, and the Centre Hospitalier Universitaire Ste-Justine. All 430 AML samples were collected with informed consent between 2001 and 2015 according to Quebec Leukemia Cell Bank procedures.13 Workflow for sequencing, mutation analysis, and transcripts quantification has been described previously and is detailed in the supplementary Methods.13 Additionally Freebayes v1.3.1 was used for identification of mutations from RNA-sequencing,14 and FLT3-ITDs were identified by k-mer counting.15 EPCY was used to identify differentially expressed transcripts that are predictive of NPM1 mutation status (https://github.com/iric-soft/epcy).
Using this workflow, we identified 125 samples with NPM1 mutations, all located in exon 12 (NPM1e12), representing 29% (125/430) of the Leucegene cohort. Of these samples, 94 (75%), 10 (8%), 7 (6%), 2 (1%), and 2 (1%) consisted of types A (TCTG), D (CCTG), B (CATG), K (CCAG), and ZM (CAGA) mutations, respectively. The remaining 10 samples (8%) had unique mutations (supplemental Table 1).
We identified the most discriminative transcripts between NPM1-mutated AML (n = 125) and all other samples (n = 305), corresponding to 285 overexpressed and 198 underexpressed genes (Figure 1A; supplemental Table 2). This gene expression signature included many of the previously reported deregulated genes in NPM1-mutated AML including overexpression of HOXA and HOXB genes, MEIS1 and NKX2-3, as well as low expression of CD34 (Figure 1A).16 Using this signature, we identified, as expected, strong transcriptomic similarities in samples from WHO-defined cytogenetic or mutational subgroups, including NPM1-mutated AML (Figure 1B). NPM1e12 samples were grouped in 3 main clusters (clusters 3, 7, and 10 [orange bars in Figure 1B]). Interestingly, clusters 7 and 10, which together were composed of 93% of NPM1e12 samples, also contained 7 samples in which no NPM1 mutations were detected, possibly suggesting that cryptic mutations in NPM1 could be missed by our approach (Figure 1B). Based on this hypothesis, we queried the NPM1 complete coding sequence in all samples directly in unmapped reads using the km algorithm15 developed by our group. This approach identified all previously annotated NPM1e12 together with 2 additional samples with 18 and 21 bp in frame insertions affecting exon 5. The 2 additional samples, located in NPM1-enriched clusters 7 and 10, represented 0.5% (2/430) of the cohort and 1.6% (2/127) of the NPM1-mutated subset. These mutations were previously missed because large indels are challenging to identify in mapped RNA-sequencing data.17 Both mutations were confirmed independently using exome and Sanger sequencing (supplemental Figure 1), and somatic origin was confirmed in 1 sample with available germline exome data. No NPM1 alteration or recurrent mutation were identified in the remaining 5 NPM1wt samples from clusters 7 and 10 (detailed in supplemental Table 4). This indicated a partial overlap in the gene expression signature of NPM1-mutated samples and a subset of other AML samples (eg, high HOXA/B and low CD34 gene expression).
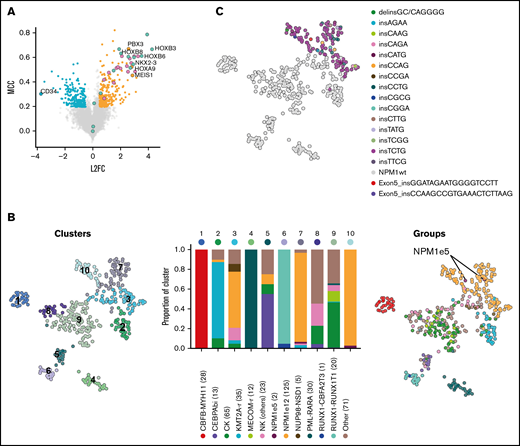
Gene expression profile and clustering of NPM1-mutated AML. (A) Differentially expressed and predictive genes in NPM1e12 AML (n = 125) compared with control AML (n = 305) using EPCY. Overexpressed (orange) and underexpressed (blue) genes are defined by Log 2 Fold Change (L2F) >|0.5| and NPM1e12 predictive power is defined by Matthews Correlation Coefficient (MCC) >0.2. Selected genes are labeled, and HOXA/B genes are colored separately. (B) Uniform manifold approximation and projection (UMAP) performed on the complete log transformed Leucegene cohort (n = 430) using the most predictive genes from supplemental Table 2 followed by principal component analysis and colored by Louvain-based clusters (left)19 or mutational/cytogenetic group (right). Barplot shows the proportion of genetic groups per cluster; the number of samples per group is indicated in parentheses. (C) UMAP colored by NPM1 mutation type.
Gene expression profile and clustering of NPM1-mutated AML. (A) Differentially expressed and predictive genes in NPM1e12 AML (n = 125) compared with control AML (n = 305) using EPCY. Overexpressed (orange) and underexpressed (blue) genes are defined by Log 2 Fold Change (L2F) >|0.5| and NPM1e12 predictive power is defined by Matthews Correlation Coefficient (MCC) >0.2. Selected genes are labeled, and HOXA/B genes are colored separately. (B) Uniform manifold approximation and projection (UMAP) performed on the complete log transformed Leucegene cohort (n = 430) using the most predictive genes from supplemental Table 2 followed by principal component analysis and colored by Louvain-based clusters (left)19 or mutational/cytogenetic group (right). Barplot shows the proportion of genetic groups per cluster; the number of samples per group is indicated in parentheses. (C) UMAP colored by NPM1 mutation type.
Transcriptomic differences within NPM1-mutated AML were predominantly influenced by cellular identity, inferred using the French American British classification, rather than by the NPM1 mutation subtype (Figure 1C; supplemental Figure 2). NPM1e5 AML carried FLT3-ITDs (n = 2), DNMT3A R882H (n = 2), IDH1 (n = 1) mutations, which are all also frequently associated with NPM1e12 mutations (supplemental Table 3). NPM1e5 AMLs thus share the mutational and transcriptomic landscape of exon 12 mutations.
NPM1 exon 12 mutations disrupt the balance between nucleolar/nuclear localization and nuclear export signals. We next investigated whether a similar imbalance was at play in novel exon 5 mutations. Although exon 5 mutations do not impact the C-terminal nucleolar localization signal, mutated sequences were predicted to introduce a novel NES compared with the endogenous protein (Figure 2A). In line with this hypothesis, we demonstrated aberrant cytoplasmic localization of NPM1 in primary NPM1e5 AML samples similar to NPM1e12 AML samples (Figure 2B). We further showed that the mutation is sufficient to cause aberrant cytoplasmic localization of NPM1 by overexpressing patient-specific mutation sequences in HEK-293t cell lines, in which NPM1 localization was comparable to that observed in the primary AML cells (Figure 2C). Altogether, these observations confirmed that the introduction of a novel NES by NPM1e5 mutations results in aberrant cytoplasmic localization of the mutated protein.
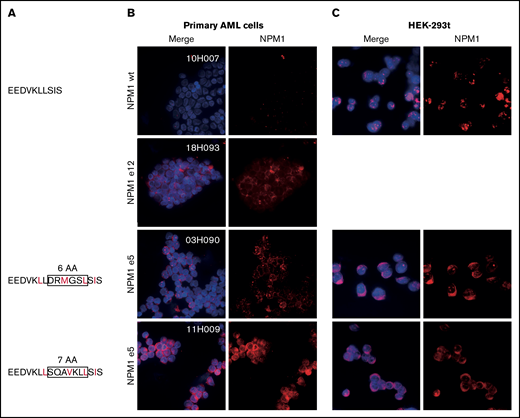
Aberrant cytoplasmic localization of NPM1e5 mutations. (A) NPM1 exon 5 protein sequences. Rectangles indicate amino acids insertions, and predicted NES are indicated in red. (B) NPM1 localization in primary NPM1 wild-type (WT) and NPM1 mutated samples. (C) Overexpression of WT and mutated patient-specific sequences in HEK-293t cell lines. Images were acquired on a Leica DMi8 microscope using 100× magnification with oil immersion. Brightness and contrast were adjusted with ImageJ.
Aberrant cytoplasmic localization of NPM1e5 mutations. (A) NPM1 exon 5 protein sequences. Rectangles indicate amino acids insertions, and predicted NES are indicated in red. (B) NPM1 localization in primary NPM1 wild-type (WT) and NPM1 mutated samples. (C) Overexpression of WT and mutated patient-specific sequences in HEK-293t cell lines. Images were acquired on a Leica DMi8 microscope using 100× magnification with oil immersion. Brightness and contrast were adjusted with ImageJ.
Martelli et al also recently reported four NPM1e5 mutations, initially suspected in abnormal cytoplasmic localization of NPM1 in samples without NPM1 exon 12 mutations.18 In this independent cohort, NPM1e5 mutations were found in 0.4% of AML, a frequency similar to that found in our study (0.5%). Interestingly, all 6 NPM1e5 mutations reported to date in both cohorts are different, standing in sharp contrast to NPM1e12 mutations in which types A, B, and D represent ∼90% of known mutations. Our findings reveal that NPM1e5 mutations share a unified transcriptomic signature with NPM1e12 mutations and a similar cytoplasmic localization.
In summary, our results provide additional evidence that novel NPM1 exon 5 mutations, similar to exon 12 mutations, lead to aberrant cytoplasmic protein localization. We provide the first evidence of transcriptomic similarities between these mutations and others affecting exon 12. NPM1e5 may be missed by standard clinical testing targeting exon 12 only, as well as with approaches that fail to detect large indels. NPM1 mutation detection must include exon 5 screening and use adapted bioinformatic approaches. Identification of rare NPM1 mutations, including NPM1e5, is important for genetic risk assessment and measurable residual disease monitoring. The detection of these rare mutations could also be useful for determining the optimal treatment for these patients, such as venetoclax-based therapies, which provide favorable responses in NPM1-mutated AML.
Acknowledgments: The authors wish to thank Muriel Draoui for project coordination, Sophie Corneau for sample coordination, Jennifer Huber and Raphaëlle Lambert at the Institute for Research in Immunology and Cancer (IRIC) genomics platform for sequencing. The authors acknowledge the invaluable contribution of Quebec Leukemia Cell Bank (BCLQ) members Giovanni D’angelo, Claude Rondeau, and Sylvie Lavallée and that of IRIC bioinformatic platform members Geneviève Boucher, Patrick Gendron, and Xiao Ju.
This work was supported by the Government of Canada through Genome Canada and the Ministère de l’Economie, de l’Innovation et des Exportations du Québec through Génome Québec.
V.-P.L. is supported by the Fondation Charles Bruneau and holds a Fonds de Recherche en Santé du Québec clinician scientist award. G.S. holds the Bégin-Plouffe Chair in blood stem cell chemogenomics of the Faculty of Medicine, Université de Montréal. J.H. holds the Industrielle-Alliance research chair in leukemia at Université de Montréal. M.V. was supported by a scolarship from the Institut de valorisation de donnéees (IVADO) The BCLQ is supported by grants from the Cancer Research Network of the Fonds de recherche du Québec–Santé. RNA-Seq read mapping and transcript quantification were performed on the supercomputer Briaree from Université de Montréal, managed by Calcul Québec and Compute Canada. This research was enabled in part by support provided by Calcul Québec (www.calculquebec.ca) and the Digital Research Alliance of Canada (alliancecan.ca).
Contribution: V.L. contributed to project conception, analyses, experiments and cowrote the manuscript; V.-P.L. contributed to project conception, analyses, and coordination and wrote the manuscript; M.V. analyzed NPM1 and cohort mutations; E.B. performed experiments; G.S. contributed to project conception; J.H. contributed to project conception and provided all the AML samples; S.L. was responsible for supervision of Leucegene bioinformatics team and codeveloped the km approach and EPCY; E.A. codeveloped EPCY; and A.F. codeveloped and performed km analyses.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Vincent-Philippe Lavallée, Centre de Recherche du Centre Hospitalier Universitaire Sainte-Justine, 3175 Chemin de la Côte-Sainte-Catherine, H3T 1C5, Montréal, QC, Canada; e-mail: vincent-philippe.lavallee@umontreal.ca.