Key Points
PDXs from risk-stratified AML samples are crucial for studying AML biology and testing novel therapeutics.
We characterize human AML engraftment in NSGS mice, offering a valuable platform for in vivo testing of targeted therapies.

Abstract
Acute myeloid leukemia (AML) is the most common acute leukemia in adults. Disease heterogeneity is well documented, and patient stratification determines treatment decisions. Patient-derived xenografts (PDXs) from risk-stratified AML are crucial for studying AML biology and testing novel therapeutics. Despite recent advances in PDX modeling of AML, reproducible engraftment of human AML is primarily limited to high-risk (HR) cases, with inconsistent or very protracted engraftment observed for favorable-risk (FR) and intermediate-risk (IR) patients. We used NSGS mice to characterize the engraftment robustness/kinetics of 28 AML patient samples grouped according to molecular/cytogenetic classification and assessed whether the orthotopic coadministration of patient-matched bone marrow mesenchymal stromal cells (BM MSCs) improves AML engraftment. PDX event-free survival correlated well with the predictable prognosis of risk-stratified AML patients. The majority (85-94%) of the mice were engrafted in bone marrow (BM) independently of the risk group, although HR AML patients showed engraftment levels that were significantly superior to those of FR or IR AML patients. Importantly, the engraftment levels observed in NSGS mice by week 6 remained stable over time. Serial transplantation and long-term culture-initiating cell (LTC-IC) assays revealed long-term engraftment limited to HR AML patients, fitter leukemia-initiating cells (LICs) in HR AML samples, and the presence of AML LICs in the CD34− leukemic fraction, regardless of the risk group. Finally, orthotopic coadministration of patient-matched BM MSCs and AML cells was dispensable for BM engraftment levels but favored peripheralization of engrafted AML cells. This comprehensive characterization of human AML engraftment in NSGS mice offers a valuable platform for in vivo testing of targeted therapies in risk-stratified AML patient samples.
Introduction
Acute myeloid leukemia (AML), the most common acute leukemia in adults, is a biologically and genetically heterogeneous group of disorders that is characterized by the rapid expansion and accumulation of poorly differentiated myeloid cells in the bone marrow (BM) and infiltrating tissues.1-3 Disease heterogeneity is well documented, and patients are stratified at disease presentation based on cytogenetic, molecular, and immunophenotypic data. Patient stratification is crucial for prognostication AQ1and informs treatment decisions.1 AML treatment has not improved substantially in the last 2 decades, and many patients fail to respond to standard-of-care chemotherapy.2,3 Robust AML patient-derived xenograft (PDX) models represent a unique in vivo tool for studying the pathogenesis of AML and for testing novel therapeutics. AML PDXs provide information about the mechanisms underlying disease initiation and progression, (epi)genetic clonal evolution, the biology of AML leukemia-initiating cells (LICs), BM niche-leukemia interactions, and chemoresistance.4-9 However, in immunodeficient mice, it is challenging to anticipate the engraftment of primary samples derived from patients with favorable-risk (FR) and intermediate-risk (IR) AML, which hinders the establishment of accurate PDX models encompassing AML heterogeneity.
Advanced strains of immunodeficient mice have improved PDX modeling of AML.10,11 In NSG mice, the most widely used immunodeficient mouse strain, reproducible xenoengraftment of human AML is primarily limited to high-risk (HR) cases; inconsistent or very protracted engraftment is observed for FR and IR patients.12 It was not until recently that a more consistent engraftment of FR/IR AML samples was achieved using NSGS mice, a more advanced immunodeficient mouse strain that results from crossing NSG and NSS mice; it provides an increased myeloid potential as a result of the transgenic expression of human stem cell factor, granulocyte-macrophage colony-stimulating factor, and interleukin-3.6,13,14
A reciprocal remodeling between the BM niche and leukemic cells is well established in several hematological malignancies.15-17 In fact, AML progression and chemoresistance are highly dependent on leukemia-BM stroma interactions.15,18 Several nonorthotopic humanized BM niche models have been explored for hematopoietic xenotransplantation.19 Furthermore, the impact of orthotopic cotransplantation (intra-BM transplantation) of BM mesenchymal stromal cells (MSCs) on the engraftment of primary human samples has been evaluated in part in NSG mice in the context of healthy hematopoietic stem and progenitor cells (HSPCs), multiple myeloma, and myelodysplastic syndromes (MDSs), with contradictory results.14,17,20-22 However, it remains unknown whether intra-BM coinjection of patient-matched bone marrow mesenchymal stromal cells (BM MSCs) impacts the engraftment of cells from humans with primary AML.
In this study, we characterized the engraftment robustness/kinetics of cells from 28 AML patients, grouped according to molecular/cytogenetic classification, in NSGS mice and assessed the contribution of patient-matched BM MSCs in AML engraftment. We report that PDX event-free survival (EFS) correlated well with the predictable prognosis of risk-stratified AML patients and that NSGS mice represent a robust model for a rapid read-out of the engraftment potential of cells from risk-stratified AML patients. Serial transplantation and long-term culture-initiating cell (LTC-IC) assays revealed that long-term engraftment was limited to HR patients and fitter LICs in HR AML samples vs FR or IR AML samples. Orthotopic coadministration of patient-matched BM MSCs and AML cells was dispensable for BM engraftment levels but favored peripheralization of engrafted AML cells. This comprehensive characterization of human AML cell engraftment in NSGS mice represents a robust platform for in vivo testing of targeted therapies for risk-stratified AML patient samples.
Methods
Patients
Fresh BM aspirates were obtained at the time of disease presentation from 28 patients (17 males and 11 females) with a mean age of 43 years (standard error of the mean [SEM], 5 years) and cytogenetically different AMLs. Patients were diagnosed in the following hospitals: Hospital Clinico San Carlos, Hospital Clínico Universitario Virgen de Arrixaca, Hospital Universitario de Gran Canaria, ICO-Hospital Germans Trias i Pujol, Hospital Infantil Universitario Niño Jesús, Hôpital Armand Trousseau, and Hospital de la Santa Creu i Sant Pau. Molecular-cytogenetic stratification was based on European LeukemiaNet3 and World Health Organization23 classifications. Accordingly, the disease was classified as FR AML (displaying favorable cytogenetics/molecular features), IR AML (normal karyotype and absence of mutations in TP53, NPM1, FLT3, and WT), or HR AML (displaying unfavorable cytogenetics/molecular features).3,23 The main cytogenetic/molecular diagnostics and other clinical-biological features of each patient are listed in Table 1. Blasts were immunophenotyped by flow cytometry using the monoclonal antibodies (mAbs) CD45-APC-H7 (clone 2D1), CD19-BV421 (clone HIB19), and CD34-PE-Cy7 (clone 8G12) (all from BD Biosciences), and CD33-PE (clone AC104.3E3; Miltenyi Biotec), following the manufacturer’s recommendations. The study was approved by the Institutional Review Board of the Clinic Hospital of Barcelona (HCB/2014/0687), and samples were collected upon signed informed consent.
Isolation and expansion of BM MSCs
AML MSCs were isolated from the BM of the 28 patients, as described previously in detail.24,25 AML MSC cultures were maintained in advanced Dulbecco’s modified Eagle medium (Gibco) supplemented with 10% fetal calf serum (Invitrogen), l-glutamine, and penicillin-streptomycin-amphotericin B (Gibco). Established AML MSC cultures were characterized extensively and used at passages 1 through 3.24-29
PDX models and follow-up
Seven- to 12-week-old NSGS mice (n = 275 with an equal distribution of males and females), housed under pathogen-free conditions, were used in this study. All experimental procedures were approved by the Animal Care Committee of The Barcelona Biomedical Research Park (HRH-16-0037-P2). BM mononuclear cells (MNCs) from AML patients were isolated by density gradient centrifugation using Ficoll-Hypaque (Amersham Biosciences). Subsequently, samples were T cell-depleted with an anti–CD3-OKT3 antibody (LABCLINICS). To do this, cells were incubated with the antibody (5.83 µg per 106 MNCs) on ice for 30 minutes just prior to intra-BM (IBM) transplantation. A total of 106 T-cell–depleted MNCs was IBM transplanted into sublethally irradiated (2 Gy) mice, as previously described,30-32 with or without 105 patient-matched established BM MSCs.24,25 At weeks 6 and 12, BM and peripheral blood (PB) were collected by aspiration from noninjected tibiae and facial vein bleeding, respectively, and leukemic engraftment was analyzed by flow cytometry. Mice were euthanized when disease symptoms were evident, leukemia engraftment was incompatible with animal welfare (>70%), or at week 18 (end point of the experiment).33 For EFS curves, an event was called when disease symptoms with detectable human graft in BM were observed or when the human graft was >70% in BM in the absence of disease symptoms. Spleens and livers were collected and analyzed when euthanized. For secondary transplantation, 106 leukemic cells from primary mice with leukemic graft > 60% in BM were IBM transplanted into irradiated secondary recipients. For limiting dilution experiments, irradiated mice were IBM transplanted with as few as 105 cells from HR AML patients.
Analysis of leukemic grafts
Cells retrieved from BM, PB, spleen, and liver were stained with HLA-ABC-FITC and CD45-APC-H7 mAbs and analyzed by flow cytometry to detect the human graft (HLA-ABC+CD45+ population). Within the human graft, the leukemia was immunophenotyped using CD33-PE, CD19-BV421, and CD34-PE-Cy7 mAbs, as described above, on a BD FACSCanto II cytometer running FACSDiva software (BD Biosciences). The cutoff to define engraftment was established at >0.1% in the BM. AML samples were categorized as high engrafters (>20%), medium engrafters (5-20%), low engrafters (0.1-5%), or nonengrafters (<0.1%) based on the percentage of leukemic cells in the BM. If a mouse was euthanized before the end point (week 18), the engraftment levels observed at the sacrifice point were included in the following time points of the curve, not altering the mean values for each risk group. Common leukemic genomic abnormalities, such as the inv(16), t(8;21), +8, and mixed lineage leukemia (MLL) rearrangements as MLL-AF9, were confirmed by fluorescence in situ hybridization (FISH) in engrafted PDX samples, as described previously.34-37 In mice engrafted with IR AML samples, the identity of the AML graft was confirmed using targeted next-generation sequencing (NGS), as previously described.38,39
Three-dimensional confocal microscopy of BM
Three-dimensional imaging of mouse femoral BM cavities was performed as described.40 Briefly, bones were collected, fixed, dehydrated, embedded in OCT, and snap frozen in liquid nitrogen. Thick slices (400-700 μm) of BM were obtained by iteratively sectioning both sides of the femur using a cryostat until the BM cavity was bilaterally exposed. Slices were blocked and incubated with primary antibodies against endomucin (clone V.7C7; sc-65495; Santa Cruz Biotechnology; 1:100) and human CD45 (clone MEM-28; Abcam; 1:200). Slices were then incubated with a secondary antibody cocktail solution (Jackson ImmunoResearch) including 2 μM 4',6-diamidino-2-phenylindole (DAPI). Finally, the slices were mounted on glass slides for imaging on a SP8 Leica confocal microscope equipped with hybrid detectors using 10× (HCX-PL FLUOTAR) and 20× (HC PL APO CS2) objectives. Imaging data were analyzed and videos were assembled using Imaris v8.2 software (Bitplane AG). A supplemental Video is available online.
FISH studies
PDX samples were processed as described.34-37 In brief, cells were resuspended in warm hypotonic solution (0.075 mM KCl) for 20 minutes at 37°C and fixed in cold methanol/acetic acid (3:1). Samples were spread onto methanol-cleaned slides and kept at −20°C until processing. Two-color FISH experiments were performed using XL CBFB or XL t(8;21) (both from Metasystems Inc.) or LSI MLL Break-Apart (Abbott Molecular) probes to detect inv(16), t(8;21), or MLL rearrangements, respectively. FISH was performed following standard procedures.34-37 Briefly, cells were denatured at 73°C in 70% formamide in 2× saline sodium citrate (SSC) for 2 minutes. Hybridization was carried out by adding 5 μL of the corresponding probe mixture to preparations and incubating the slides in a humid chamber at 37°C for 16 hours. Posthybridization washes were performed using 0.4× SSC with 0.3% NP-40 at 73°C, followed by 2× SSC with 0.1% NP-40 at room temperature, for 1 minute each. Slides were mounted with DAPI II solution (Abbott Molecular). Analyses were performed using a Nikon Ci-S/Ci-L epifluorescence microscope equipped with specific filters for DAPI, FITC, and Cy3 and a dual-band pass filter for FITC and Cy3. The primary chromosomal abnormality was confirmed in ≥20 informative nuclei per PDX sample.
Targeted NGS
A targeted-NGS study was performed on 2 independent IR AML xenografts. The NGS panel included 32 genes related to myeloid disorders, including those genes known to be mutated at disease presentation.38,39 Briefly, DNA was extracted from PDX IR AML cells, and a library was prepared using a QIAseq FX DNA Library Kit (QIAGEN). Target regions were then captured and amplified by hybridizing the DNA library with a Myeloid Solution Capture Kit (SOPHiA GENETICS). The captured and amplified DNA was sequenced using a MiSeq platform (Illumina). The results were analyzed using a Sophia DDM platform, as described elsewhere.38,39
LTC-IC assays
CD45+CD33+CD19− AML blasts from 6 patients (2 patients from each risk group; supplemental Table 1) were stained with CD33-PE (clone AC104.3E3; Miltenyi Biotec), CD45-FITC (clone HI30; BD Biosciences), and CD19-APC (clone HIB19; BD Biosciences) antibodies and sorted using a FACSAria Fusion Cell Sorter (BD Biosciences). Sorted blasts were plated using a limiting dilution analysis of 4 doses (250, 500, 1000 and 2000 cells) in 15 replicates in 96-well microplates containing a confluent irradiated (50 Gy) MS-5 monolayer. The AML blast-MS5 coculture was maintained in MyeloCult H5100 culture medium (STEMCELL Technologies) supplemented with recombinant human interleukin-3, granulocyte colony-stimulating factor, and thrombopoietin (MS-51 3GT) (20 ng/mL each; PeproTech), as previously described41 at 37°C in 5% CO2. After 5 weeks, the number of wells plated with 250 cells displaying cobblestone areas (CAs) were scored, and the long-term culture (LTC) medium was replaced by methylcellulose H4435 (STEMCELL Technologies). After an additional 2 weeks, each well was scored as positive/negative if colony-forming units were present/absent. The frequency of LICs was determined using ELDA software, as previously described (http://bioinf.wehi.edu.au/software/elda/).33,42,43
Statistical analysis
Data are expressed as mean ± SEM of different independent experiments unless otherwise specified. Statistical comparisons were performed using unpaired a Student t test. EFS curves were estimated according to the Kaplan-Meier method and were compared using a log-rank test. Statistical analyses, normality test (Shapiro-Wilk), and linear regression were performed using GraphPad Prism v6.0 (GraphPad Software). Statistical significance was defined as a P value < .05.
Results
Characterization of the engraftment of risk-stratified AML cells in NSGS mice
AML patient samples were classified as FR, IR, or HR, according to European LeukemiaNet/World Health Organization cytogenetics/molecular features (Table 1). To establish a suitable PDX model that would allow us to characterize the AML risk groups, 106 T-cell–depleted mice BM MNCs were IBM transplanted into irradiated NSGS mice (n = 112 mice), and the leukemic graft in BM was monitored by fluorescence-activated cell sorting (FACS) for 18 weeks (Figure 1A). NSGS mice transplanted with HR AML cells began to show symptoms of disease or to exhibit engraftment levels in BM that were incompatible with animal welfare (>70%) after week 5. Follow-up of the animals confirmed a significantly lower 18-week EFS in mice transplanted with HR AML samples than in mice transplanted with FR or IR AML samples (29% vs 75% and 83%, respectively; P < .0001; Figure 1B). This correlation between PDX EFS and a predictable prognosis in risk-stratified AML patients faithfully mimics the current risk-stratification of AML patients. We then quantified the proportion of engrafted mice and engrafted patient samples, categorizing AML samples as high (>20%), medium (5-20%), or low (0.1-5%) engrafters or nonengrafters (<0.1%). Of note, the majority of the mice showed engraftment in BM independently of the risk group (87%, 85%, and 94% for FR, IR, and HR AML, respectively; Figure 1C). However, although 80% of the HR AML patients showed high engraftment levels (>20%) only 40% and 25% of the FR AML and IR AML patients, respectively, showed such a robust engraftment (Figure 1C). No correlation was observed between the number of AML primary cells transplanted and the engraftment level (supplemental Figure 1). For mice that exhibited levels of engraftment < 1%, FACS analysis was confirmed by in situ 3-dimensional microscopy imaging (Figure 1D; supplemental Video).
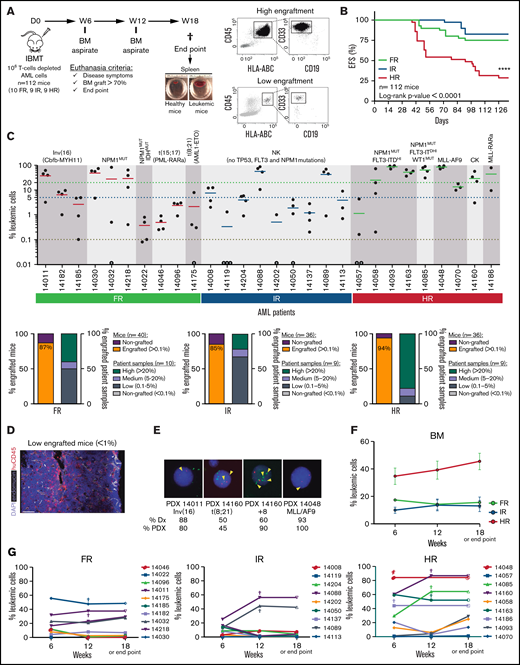
Characterization of risk-stratified AML engraftment in NSGS mice. (A) Experimental design (left panel) and representative FACS analysis of the engraftment (right panels). (B) Kaplan-Meier curves representing the 18-week EFS from each risk group (n = 28 AML primary samples and 112 mice). Mice were considered “dead” when they showed disease symptoms with detectable human graft in BM or when human graft reached 70% in BM. (C) Percentage of AML engraftment in BM at euthanization for all patient samples grouped by risk (upper panel). The horizontal dotted lines indicate the cutoff for engraftment levels >0.1% (red line), between 0.1% to 5% (blue line), and >20% (green line). Each circle represents a single mouse. Gray circles depict mice with <0.01% engraftment. The mean engraftment for the 3 or 4 mice used per patient sample is represented as a horizontal bar. Percentage of engrafted mice (leukemic cells > 0.1%) and percentage of engrafted patient samples (lower panels). Based on engraftment levels, patient samples were categorized as high (leukemic graft > 20%), medium (5-20%), or low (0.1-5%). (D) In situ 3-dimensional microscopy imaging of murine BM from a representative very low engrafted sample (0.8% by FACS; supplemental Video). (E) FISH for the indicated chromosomal abnormalities in PDX cells retrieved from mice at week 18. Magnification 100×. Yellow arrowheads depict the indicated chromosome abnormalities. (F) Engraftment kinetics in BM for each AML risk group at 6, 12, and 18 weeks (or at the end point). Data are mean (± SEM) engraftment levels for all patients within each risk group. (G) As in (F) but showing each patient and risk group independently. Each point indicates the mean engraftment for each patient sample (3-4 mice). Individual mice euthanized before week 18 are indicated by “†”; the engraftment value observed at euthanization was maintained at the end point. ****P < .0001, log-rank test. NK, normal karyotype; CK, complex karyotype; D, day; IBMT, intra-BM transplantation; W, week.
Characterization of risk-stratified AML engraftment in NSGS mice. (A) Experimental design (left panel) and representative FACS analysis of the engraftment (right panels). (B) Kaplan-Meier curves representing the 18-week EFS from each risk group (n = 28 AML primary samples and 112 mice). Mice were considered “dead” when they showed disease symptoms with detectable human graft in BM or when human graft reached 70% in BM. (C) Percentage of AML engraftment in BM at euthanization for all patient samples grouped by risk (upper panel). The horizontal dotted lines indicate the cutoff for engraftment levels >0.1% (red line), between 0.1% to 5% (blue line), and >20% (green line). Each circle represents a single mouse. Gray circles depict mice with <0.01% engraftment. The mean engraftment for the 3 or 4 mice used per patient sample is represented as a horizontal bar. Percentage of engrafted mice (leukemic cells > 0.1%) and percentage of engrafted patient samples (lower panels). Based on engraftment levels, patient samples were categorized as high (leukemic graft > 20%), medium (5-20%), or low (0.1-5%). (D) In situ 3-dimensional microscopy imaging of murine BM from a representative very low engrafted sample (0.8% by FACS; supplemental Video). (E) FISH for the indicated chromosomal abnormalities in PDX cells retrieved from mice at week 18. Magnification 100×. Yellow arrowheads depict the indicated chromosome abnormalities. (F) Engraftment kinetics in BM for each AML risk group at 6, 12, and 18 weeks (or at the end point). Data are mean (± SEM) engraftment levels for all patients within each risk group. (G) As in (F) but showing each patient and risk group independently. Each point indicates the mean engraftment for each patient sample (3-4 mice). Individual mice euthanized before week 18 are indicated by “†”; the engraftment value observed at euthanization was maintained at the end point. ****P < .0001, log-rank test. NK, normal karyotype; CK, complex karyotype; D, day; IBMT, intra-BM transplantation; W, week.
The presence of chromosomal abnormalities associated with FR and HR AML, such as inv(16), t(8;21), +8, and MLL-AF9, was detected by FISH in BM blasts from engrafted mice at euthanization (Figure 1E). Similarly, when the leukemic identity of the human graft from mice reconstituted with IR AML cells was analyzed by targeted NGS, the pathogenic mutations found in myeloid genes at diagnosis were recapitulated in the matched PDX samples (Table 2). This, together with the myeloid-skewed graft in the absence of a lymphoid graft, confirms that it stemmed from the original leukemic clone and not from residual healthy myeloid progenitor cells. Of interest, beyond the pathogenic mutations found in the diagnostic sample, we also observed that IR AML PDXs displayed multiple pathogenic mutations in myeloid genes that were absent in the diagnostic sample (Table 2), likely reflecting minor leukemic clones undetectable at diagnosis or the acquisition of de novo environmental/niche pressure–mediated myeloid mutations.
Previous studies have indicated that, by extending the time-to-engraftment read-out of AML primary cells in NSG mice, all types of AML samples, including FR AML, could be studied.5,44 Interestingly, when the engraftment kinetics were analyzed sequentially in BM at 6, 12, and 18 weeks, we observed that the engraftment levels increased only slightly from week 6 to week 18 for HR AML samples (from 34.6% ± 6% to 45.5% ± 6%; P > .05), whereas the engraftment levels observed at week 6 remained stable until week 18 for FR and IR AML samples, suggesting that the stable engraftment levels observed in NSGS mice by week 6 may shorten/facilitate5 downstream studies requiring in vivo primary AML-PDXs (Figure 1F-G). Overall, these results confirm that NSGS mice allow for a rapid and stable read-out of the engraftment of cells from risk-stratified AML patients.
Long-term engraftment of AML cells in NSGS mice is limited to those from HR AML patients who have more or fitter LICs
NSGS mice have been shown not to support a robust engraftment of healthy CD34+ HSPCs in long-term serial transplantation assays as a result of the potential exhaustion of the engraftment-initiating cells attributed to the long-term exposure to human myeloid cytokines.45-47 In contrast, the engraftment of MDS primary cells is sustained upon secondary transplantation in NSGS mice.14 To evaluate whether long-term engraftment of AML cells can be achieved in NSGS mice, 106 cells were isolated from the BM of primary recipients robustly engrafted (>60% in BM) with FR, IR, or HR AML samples and IBM transplanted into secondary NSGS recipients (n = 24 mice; Figure 2A). We observed, 18 weeks later, that all secondary mice transplanted with HR AML samples showed a robust BM engraftment similar to that observed in primary recipients (Figure 2B-C). However, for FR and IR AML samples, only 22% and 50%, respectively, of the secondary recipients had engraftment levels that were profoundly lower than those observed in the primary recipients for those 2 groups (Figure 2B). Secondary engraftment phenocopied primary myeloid engraftment (HLA-ABC+CD45+CD33+) in the absence of lymphoid engraftment (Figure 2C).
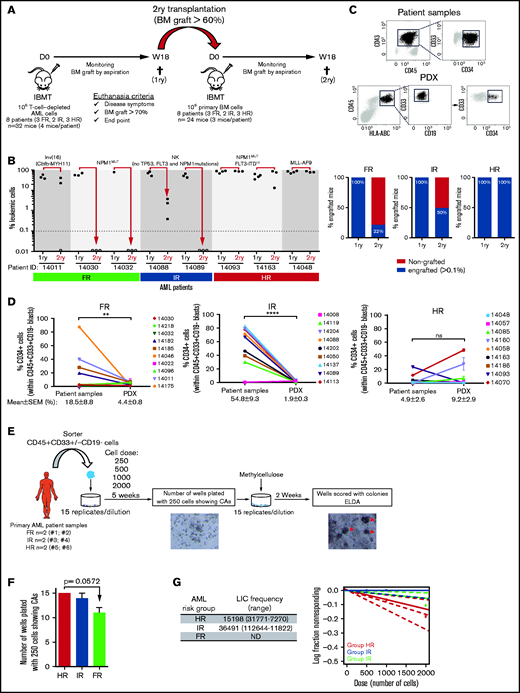
Secondary transplantation of risk-stratified AML cells in NSGS mice. (A) Experimental design. 1ry, primary; 2ry, secondary. (B) Percentage of BM engraftment in secondary (2ry) recipients (n = 24) at euthanization (18 weeks or sacrifice point) for each patient sample grouped by risk (left panel). Only the highly engrafted primary samples/mice were selected for serial transplantation (1-2 mice). The dotted line indicates the cutoff for engraftment levels > 0.1%. Each circle represents a single mouse. Gray circles depict mice with <0.01% engraftment. Percentage of engrafted 2ry recipients (leukemic cells > 0.1%) transplanted with BM cells from primary recipients of FR, IR and HR samples (right panel). (C) Representative FACS analysis of CD45, CD33, CD19, and CD34 expression in AML patient samples and PDXs. CD34 expression within the leukemic blasts is heterogeneous. (D) Proportion of CD34+ AML blasts before and after xenotransplantation in NSGS mice for all risk-stratified AML primary samples (n = 28). For primary AML samples, each point represents a single patient. (E) Schematic diagram of the LTC-IC assays. Magnification 40×. Red arrowheads depict the colonies. (F) Number of wells plated with 250 cells showing CAs after 5 weeks of blast:MS5 LTC. (G) Estimated LICs in FR, IR, and HR AML primary samples for cell doses of 250 to 2000 AML blasts. For PDX samples, each dot represents the mean ± SEM from all mice transplanted with that sample (n = 112 mice). Data are mean ± SEM. **P < .01, ****P < .0001, unpaired Student t test. D, day; IBMT, intra-BM transplantation; ND, not detected; NK, normal karyotype; ns, not significant; W, week.
Secondary transplantation of risk-stratified AML cells in NSGS mice. (A) Experimental design. 1ry, primary; 2ry, secondary. (B) Percentage of BM engraftment in secondary (2ry) recipients (n = 24) at euthanization (18 weeks or sacrifice point) for each patient sample grouped by risk (left panel). Only the highly engrafted primary samples/mice were selected for serial transplantation (1-2 mice). The dotted line indicates the cutoff for engraftment levels > 0.1%. Each circle represents a single mouse. Gray circles depict mice with <0.01% engraftment. Percentage of engrafted 2ry recipients (leukemic cells > 0.1%) transplanted with BM cells from primary recipients of FR, IR and HR samples (right panel). (C) Representative FACS analysis of CD45, CD33, CD19, and CD34 expression in AML patient samples and PDXs. CD34 expression within the leukemic blasts is heterogeneous. (D) Proportion of CD34+ AML blasts before and after xenotransplantation in NSGS mice for all risk-stratified AML primary samples (n = 28). For primary AML samples, each point represents a single patient. (E) Schematic diagram of the LTC-IC assays. Magnification 40×. Red arrowheads depict the colonies. (F) Number of wells plated with 250 cells showing CAs after 5 weeks of blast:MS5 LTC. (G) Estimated LICs in FR, IR, and HR AML primary samples for cell doses of 250 to 2000 AML blasts. For PDX samples, each dot represents the mean ± SEM from all mice transplanted with that sample (n = 112 mice). Data are mean ± SEM. **P < .01, ****P < .0001, unpaired Student t test. D, day; IBMT, intra-BM transplantation; ND, not detected; NK, normal karyotype; ns, not significant; W, week.
CD34 is a surrogate marker for normal HSPCs and LICs in some subtypes of AML.4,48 Because the transgenic human myeloid cytokines expressed by NSGS mice compromise the long-term engraftment of healthy CD34+ HSPCs,45-47 we next wanted to trace the CD34-expressing AML blasts before and after xenotransplantation as an indirect approach to gain insights into the differential long-term engraftment of AML cells according to the risk group (Figure 2D). Despite the heterogeneity of CD34 expression across AML patients, we found, for HR AML, that the proportion of CD34+ blasts observed in the primary samples is retained or increased after 18 weeks of expansion in NSGS mice (4.9% ± 2.6% vs 9.2% ± 2.9%; n = 9; P > .05; Figure 2D). In contrast, the CD34+ AML blasts were profoundly lost in PDXs transplanted with FR AML cells (18.5% ± 8.8% vs 4.4% ± 0.8%; P < .001) or IR AML cells (54.8% ± 9.3% vs 1.9% ± 0.3%; Figure 2D). We next performed LTC-IC assays using HR, IR, and FR primary AML samples (Figure 2E), and observed that HR AML patients showed the highest number of wells with CAs (Figure 2F) and the highest LIC estimation in a limiting dilution analysis using ELDA software (Figure 2G). Together, these data confirm the existence of AML-LICs in the CD34− leukemic fraction, regardless of the risk group, and reveal the presence of more (or fitter) LICs in HR AML samples than in FR or IR AML samples.
Orthotopic coadministration of patient-matched BM MSCs and primary blasts is dispensable for AML engraftment levels but favors peripheralization of engrafted AML cells
MSCs are an important cellular component of healthy and leukemic BM microenvironments. In fact, AML progression and chemoresistance are highly dependent on leukemia–BM stroma interactions.15,18 The impact of the coadministration of BM MSCs on the engraftment of primary human samples has been evaluated in the context of healthy CD34+ HSPCs, multiple myeloma, and MDS, with contradictory results.14,17,20-22 We next evaluated whether intra-BM coinjection of patient-matched BM MSCs affects the engraftment of risk-stratified AML samples. AML patient-matched MSCs were generated from passages 1 through 3, expanded, and functionally characterized in vitro and in vivo in previous studies from our group.24,25 A total of 105 bona fide AML-BM MSCs was IBM transplanted into NSGS mice together with 106 T-cell–depleted AML cells; the engraftment was monitored in BM and PB every 6 weeks until euthanization (week 18) (Figure 3A). The presence of patient-matched BM MSCs in the NSGS BM niche did not result in enhanced AML engraftment, regardless of the risk group (Figure 3B). Accordingly, follow-up of the animals confirmed an almost identical 18-week EFS for mice transplanted with risk-stratified AML samples in the presence or absence of patient-matched BM MSCs (P > .05; Figure 3C). We further assessed, in a limited number of NSGS mice, whether MSCs from healthy BM donors contribute to AML engraftment, but we did not find any enhanced engraftment associated with the coadministration of healthy donor BM MSCs (supplemental Figure 2).
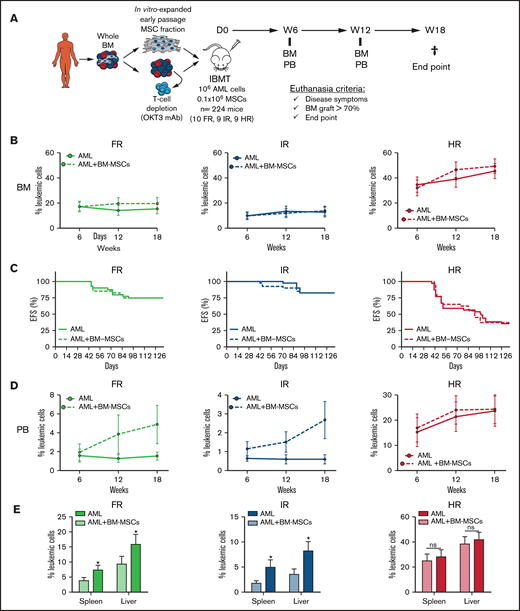
Orthotopic coadministration of patient-matched BM MSCs together with AML blasts does not enhance BM engraftment levels but favors peripheralization of engrafted AML cells. (A) Experimental design. (B) Kinetics of risk-stratified AML engraftment in BM at 6, 12, and 18 weeks in the presence or absence of patient-matched BM MSCs. Each point represents the mean (±SEM) of 36 to 40 mice. (C) Kaplan-Meier curves depicting 18-week EFS of mice transplanted with risk-stratified AML samples with or without BM MSCs (n = 28 AML samples; n = 224 mice). (D) Kinetics of risk-stratified AML engraftment in PB at 6, 12, and 18 weeks in the presence or absence of patient-matched BM MSCs. Each point represents the mean (±SEM) of 36 to 40 mice. (E) Blast infiltration in spleen and liver at euthanization for risk-stratified AML in the presence or absence of patient-matched BM MSCs. Data are mean ± SEM. *P < .05, unpaired Student t test. D, day; IBMT, intra-BM transplantation; ns, not significant; W, week.
Orthotopic coadministration of patient-matched BM MSCs together with AML blasts does not enhance BM engraftment levels but favors peripheralization of engrafted AML cells. (A) Experimental design. (B) Kinetics of risk-stratified AML engraftment in BM at 6, 12, and 18 weeks in the presence or absence of patient-matched BM MSCs. Each point represents the mean (±SEM) of 36 to 40 mice. (C) Kaplan-Meier curves depicting 18-week EFS of mice transplanted with risk-stratified AML samples with or without BM MSCs (n = 28 AML samples; n = 224 mice). (D) Kinetics of risk-stratified AML engraftment in PB at 6, 12, and 18 weeks in the presence or absence of patient-matched BM MSCs. Each point represents the mean (±SEM) of 36 to 40 mice. (E) Blast infiltration in spleen and liver at euthanization for risk-stratified AML in the presence or absence of patient-matched BM MSCs. Data are mean ± SEM. *P < .05, unpaired Student t test. D, day; IBMT, intra-BM transplantation; ns, not significant; W, week.
Of note, intra-BM coadministration of patient-matched BM MSCs favored the BM exit of FR and IR AML engrafted cells to the PB (Figure 3D) and spleen and liver (Figure 3E). However, such BM MSC–mediated peripheralization was not observed in NSGS mice transplanted with HR AML cells (Figure 3D-E). We hypothesized that the aggressiveness and high engraftment levels of HR AML cells may mask such a MSC-mediated effect. To address this, NSGS mice (n = 8) were IBM transplanted with a 10-fold lower dose of HR-AML cells (105 T-cell–depleted AML cells) together with 105 patient-matched BM MSCs. These dose-limiting in vivo assays revealed that, similar to FR and IR AML samples, the coadministration of BM MSCs also favors liver and spleen infiltration of HR AML cells (supplemental Figure 3). Together, the administration of patient-matched BM MSCs together with AML blasts is dispensable for BM engraftment levels but favors peripheralization of engrafted AML cells.
Discussion
AML is 1 of the most common hematopoietic cancers in adults; its incidence increases significantly in elderly people, and it is often associated with a dismal prognosis.1 AML is initiated by genetic and epigenetic lesions in HSPCs or myeloid progenitors, resulting in the accumulation of poorly differentiated myeloid cells in BM and infiltrating extramedullary tissues.49 The large cytogenetic and molecular heterogeneity observed in AML contributes to patient risk stratification, which has a major role in prognostication and contributes significantly to treatment decisions.2-4 AML is frequently associated with chemotherapy refractoriness and relapse, suggesting that a failure of current therapies to eradicate LICs is a major mechanism underlying AML progression/relapse.4,50,51 Moreover, BM MSCs, a major cellular component of the BM niche, have been shown to support the growth, survival, and chemoresistance of AML cells.15,18 Unfortunately, treatment of AML has not improved substantially in the last 2 or 3 decades; this is due, in part, to the lack of reliable in vivo models recapitulating the disease.2,3 In this sense, robust AML-PDX models represent a unique in vivo platform for studying AML pathogenesis and for testing novel targeted therapies.4-9 However, it has been challenging to anticipate the engraftment of primary samples derived from FR and IR AML patients in NSG mice, hindering the establishment of reliable PDX models covering AML heterogeneity.12 Here, we characterized the engraftment robustness/kinetics of cells from 28 risk-stratified AML patients in NSGS mice6,13,14 and assessed whether the orthotopic coadministration of patient-matched BM MSCs improves AML engraftment.
Our results show that the vast majority of NSGS mice were engrafted with primary AML samples independently of the risk group, with engraftment levels significantly superior for HR AML cells compared with FR and IR AML cells. Importantly, EFS of PDXs correlated well with the predictable prognosis of risk-stratified AML patients. Of note, AML PDXs consistently phenocopied the immunophenotype of the primary AMLs, and FISH and NGS studies confirmed that the engraftment stemmed from the original leukemic clone and not from residual healthy myeloid progenitor cells. Our results confirm recent studies suggesting the use of the NSGS mouse strain for robust engraftment of primary myeloid neoplasms6,14,45 and to reliably mimic human disease. Interestingly, IR AML PDXs also displayed multiple pathogenic mutations in myeloid genes that were absent in the diagnostic sample, likely reflecting minor leukemic clones that were undetectable at diagnosis or the acquisition of de novo environmental/niche pressure–mediated myeloid mutations. Further prospective experimental work should unravel the potential contribution of these de novo pathogenic myeloid mutations to leukemia initiation and maintenance in NSGS mice.
Importantly, the number of engrafted mice and the engraftment levels observed in NSGS mice by week 6 remained stable over time (up to week 18) regardless of the risk group, confirming that NSGS mice allow for a rapid and stable read-out of the engraftment of risk-stratified AML samples. This contrasts with previous studies reporting that the engraftment read-out of AML primary cells in NSG mice must be extended for several months. Of note, FR AML samples engraft poorly in NSG mice, likely as the result of a defective environmental support. Similar to the study by Ellegast et al,6 our data revealed that all (10/10) FR AML samples, including inv(16)-, PML-RARa–, AML1-ETO–, or NPM1-mutated samples, engrafted in 85% of NSGS mice. Of interest, cells from 1 NPM1/IDH comutated patient exhibited significantly lower engraftment levels than did cells from NPM1-mutated patients with germline IDH1. Although these data should be treated with caution because of the limited number of NPM1/IDH comutated patients assayed in this study, they provide biological clues about the impact of IDH mutations on the initiation and maintenance of AML.
MDS primary cells, but not healthy CD34+ HSPCs, have been shown to be serially transplanted in NSGS mice.45-47 In our cohort of risk-stratified AML patients, we found that HR AML samples, but not FR or IR AML samples, could be serially transplanted in NSGS mice. One exception were cells from an inv(16)+ AML patient that could be successfully retransplanted, in line with a previous study showing that inv(16)+ AML diagnostic samples may be serially transplanted in NSGS mice upon intrahepatic injection.6 Previous studies have shown that long-term exposure to the high levels of human cytokines occurring in NSGS mice could lead to the exhaustion of CD34+ HSPCs, affecting the long-term self-renewal activity of and impacting the establishment of secondary grafts.43-47 Therefore, we analyzed the dynamics of CD34+ AML blasts before and after transplantation into primary NSGS mice as an indirect approach to gain insights into the differential serial transplantation observed for AML cells according to the risk group. Despite the heterogeneity of CD34 expression across AML patients, the proportion of CD34+ blasts observed in HR AML samples was retained or slightly increased in primary NSGS mice, whereas CD34+ AML primary blasts were lost in PDXs transplanted with FR or IR AML samples. LTC-IC in vitro assays further confirmed the presence of more (or fitter) LICs in HR AML samples compared with FR or IR AML samples. Differences in sensitivity to specific niche cues involved in maintaining leukemic growth cannot be ruled out. Our data also suggest the presence of AML-LICs in the CD34− leukemic fraction, regardless of the risk group, which is in line with previous reports.48,52,53
The impact of the coadministration of BM MSCs in the engraftment of primary human samples has been evaluated in the context of normal hematopoiesis, multiple myeloma, and MDS, with inconsistent results.14,17,20-22 For example, Medyouf et al17 demonstrated that coinjection of MSCs from MDS patients enhanced engraftment in NSG mice. In contrast, Rouault-Pierre et al20 failed to find any improvement in the engraftment levels of MDS BM cells in mice coinjected with MSCs. Likewise, Krevvata et al14 reported that the coinjection of MSCs did not enhance human MDS engraftment. However, it remains elusive whether intra-BM coinjection of patient-matched BM MSCs impacts the engraftment of risk-stratified AML samples. In this study, we orthotopically IBM transplanted primary blasts with autologous BM MSCs from 28 risk-stratified AML patients and found that the coinfusion of patient-matched BM MSCs in the NSGS BM niche did not result in enhanced AML engraftment, regardless of the risk group. However, orthotopic cotransplantation of patient-matched BM MSCs favored peripheralization of engrafted AML cells, as revealed by the higher levels of leukemic cells in PB, spleen, and liver, especially in the FR and IR AML groups. Although the aggressiveness and high-engraftment levels of HR AML cells somehow masked such MSC-mediated effects, a potential greater resistance to external cues from stroma cannot be ruled out. The capacity of stroma to favor mobilization has been described in several studies focusing on different chemokine axes, such as CXCR4-SDF1.7,54 In summary, our study provides a comprehensive in vivo characterization of human AML in NSGS mice and reveals different inherent properties for each risk group with regard to engraftment ability, LICs, and interactions with the stroma. The contribution of these properties to the pathogenesis of AML warrants further study, which may help to improve the treatment and prognosis of AML patients.
Acknowledgments
The authors thank Paola Romecin and Virginia Rodriguez-Cortez for technical assistance.
This work was supported by the Spanish Ministry of Economy and Competitiveness (SAF2016-80481R, PID2019-108160RB-I00), the Obra Social La Caixa (LCF/PR/HR19/52160011), Interreg V-A programme (POCTEFA) 2014-2020 (grant PROTEOblood EFA360/19), Health Canada (H4080-144541), and Deutsche Josep Carreras Leukämie Stiftung (P.M.). Additional funding was provided by Consejería de Salud y Familia (PI-0119-2019) (R.D.d.l.G.), Health Institute Carlos III (ISCIII/FEDER, PI17/01028) and Asociación Española Contra el Cáncer (C.B.), Health Institute Carlos III/FEDER (CPII17/00032) (V.R.-M.), and Fundación Hay Esperanza (E.A.). CERCA/Generalitat de Catalunya and Fundació Josep Carreras-Obra Social la Caixa provided institutional support. B.L.-M. was supported by a Lady Tata Memorial Trust International Award and Asociación Española Contra el Cáncer (INVES20011LÓPE). O.M. and T.V.-H. were supported by Asociación Española Contra el Cáncer (INVES211226MOLI) and a Marie Sklodowska Curie Fellowship (792923), respectively. P.M. is an investigator in the Spanish Cell Therapy Network.
Authorship
Contribution: R.D.d.l.G. and B.L.-M. conceived the study, designed and performed experiments, analyzed data. and wrote the manuscript; T.V.-H., F.G.-A., H.R.-H., O.M., C.N.-A., L.Z., V.R.-M., J.C.R.-M., and C.B. performed experiments and analyzed data; A.B., J.L.F., E.A., S.V., J.N., M.T.G.-C., M.R.-O., and H.L. provided leukemia samples; and P.M conceived the study, designed experiments, wrote the manuscript, and financially supported the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pablo Menéndez, Josep Carreras Leukemia Research Institute, School of Medicine, University of Barcelona, Casanova 143, 08036 Barcelona, Spain; e-mail: pmenendez@carrerasresearch.org. Belen Lopez-Millan, Josep Carreras Leukemia Research Institute, School of Medicine, University of Barcelona, Casanova 143, 08036 Barcelona, Spain; e-mail: blopez@carrerasresearch.org; and Rafael Díaz de la Guardia, Josep Carreras Leukemia Research Institute, School of Medicine, University of Barcelona, Casanova 143, 08036 Barcelona, Spain; e-mail: rafael.diaz@genyo.es.