

Key Point
NUDT15 variants may confer a risk of second cancers after treatment with 6-MP in patients with ALL.

Abstract
The effect of genetic variation on second malignant neoplasms (SMNs) remains unclear. First, we identified the pathogenic germline variants in cancer-predisposing genes among 15 children with SMNs after childhood leukemia/lymphoma using whole-exome sequencing. Because the prevalence was low, we focused on the association between SMNs and NUDT15 in primary acute lymphoblastic leukemia (ALL) cases. NUDT15 is one of the 6-mercaptopurine (6-MP) metabolic genes, and its variants are common in East Asian individuals. The prevalence of NUDT15 hypomorphic variants was higher in patients with SMNs (n = 14; 42.9%) than in the general population in the gnomAD database (19.7%; P = .042). In the validation study with a cohort of 438 unselected patients with ALL, the cumulative incidence of SMNs was significantly higher among those with (3.0%; 95% confidence interval [CI], 0.6% to 9.4%) than among those without NUDT15 variants (0.3%; 95% CI, 0.0% to 1.5%; P = .045). The 6-MP dose administered to patients with ALL with a NUDT15 variant was higher than that given to those without SMNs (P = .045). The 6-MP–related mutational signature was observed in SMN specimens after 6-MP exposure. In cells exposed to 6-MP, a higher level of 6-MP induced DNA damage in NUDT15-knockdown induced pluripotent stem cells. Our study indicates that NUDT15 variants may confer a risk of SMNs after treatment with 6-MP in patients with ALL.
Introduction
Second malignant neoplasms (SMNs) are one of the most serious late effects in children and adolescents who have been treated for cancer. The cumulative incidence of SMNs increases substantially with time, even after 10 years, approaching 10%.1,2 SMNs are a leading cause of mortality among long-term survivors of childhood cancer.3
Previous studies have reported that radiation therapy and certain anticancer drugs (eg, etoposide) increase the risk of SMNs.4,5 Additionally, several reports have demonstrated the possible contribution of host genetics to SMN development. Recent comprehensive analyses have revealed a high prevalence of germline variants in cancer-predisposing genes (CPGs) in children with cancer.6,7 Because a family history of cancer is one of the risk factors for SMNs,8 mutations in CPGs are thought to be associated with SMN development after childhood cancer. For example, recent studies report that TP53 variants are overexpressed in pediatric SMN cases after the treatment of ALL and solid tumors.9,10 A large-scale comprehensive study of pediatric cancer survivors revealed the prevalence of germline variants of CPGs in SMN cases (6.4%).11 However, the contribution of germline variants in CPGs among SMNs is still not fully understood.
Polymorphisms of thiopurine methyltransferase (TPMT), one of the 6-mercaptopurine (6-MP)–metabolizing genes, is considered another potential genetic risk factor for SMNs, because the DNA-damaging effect of 6-MP during treatment of acute lymphoblastic leukemia (ALL) increased in patients with low TPMT activity.12-14 However, the mechanism of 6-MP–induced SMNs remains unclear. Additionally, TPMT polymorphisms are rare in East Asian individuals; instead, polymorphisms of nucleoside diphosphate–linked moiety X-type motif 15 (NUDT15), another 6-MP–metabolizing gene, are common.15,16 However, there are no reports showing an association between NUDT15 and SMNs.
We investigated the genetic basis of SMNs among children with SMNs after childhood leukemia/lymphoma. We report an association between NUDT15 variants and SMNs after 6-MP exposure.
Patients and methods
Enrollment of the patients
We first sent a questionnaire to the collaborating institutions and retrospectively collected the available samples that fulfilled the eligibility criteria: (1) development of SMNs after treatment of childhood leukemia/lymphoma, (2) availability of specimens, and (3) obtainment of informed consent. An SMN was defined as a new independent cancer occurring in a person who has had cancer in the past. Germline samples (n = 15) with available somatic samples (n = 6) meeting these criteria were obtained from 13 institutions belonging to the Tokyo Children’s Cancer Study Group (TCCSG) and other collaborating institutions. Moreover, for four patients who developed SMNs after ALL treatment, only tumor samples of primary ALL were obtained.
Identification of germ line pathogenic variants in CPGs
We designed this retrospective study to examine the genetic risk factors for SMNs after treatment of childhood leukemia/lymphoma. First, to identify potential pathogenic variants in SMN cases, we performed whole-exome sequencing (WES) in 15 samples. Complete details of sample preparation and WES are provided in the supplemental Methods. Variants of 162 known CPGs listed in previous reports7,17 were extracted in each sample. Pathogenicity of the variants was evaluated using the guideline of the American College of Medical Genetics and Genomics18 and online databases, such as National Center for Biotechnology Information ClinVar (detailed in the supplemental Methods).
Subgroup analysis in patients with primary ALL
NUDT15 variant analysis.
The prevalence of the major NUDT15 nonfunctional allele (rs116855232)19 was evaluated using WES data for 10 patients with ALL as the primary cancer. We also evaluated the NUDT15 genotype using direct sequencing in 4 samples for which germline samples were unavailable, under the assumption that the genotype does not change in tumor samples. Because all our patients were East Asian, the prevalence of NUDT15 variants in this SMN cohort was compared with that in the general population of East Asia registered in the gnomAD database (https://gnomad.broadinstitute.org/). To confirm the significance of NUDT15 variants, the cumulative incidence of SMNs with or without NUDT15 nonfunctional alleles in an unselected cohort of 438 patients with ALL studied in TCCSG L04-1620 was evaluated using cmprsk (https://CRAN.R-project.org/package=cmprsk). The average 6-MP dose during primary ALL treatment was compared with that administered to 98 Japanese children with the same NUDT15 genotype without SMNs after 6-MP–containing therapy, as reported in a previous study.21
Mutational signature analysis.
To assess the impact of 6-MP on SMN development, we performed mutational signature analysis. First, to extract somatic mutations in SMN specimens, available SMN samples from six patients after 6-MP treatment were analyzed using WES. Somatic mutations were extracted using tumor-normal pairs from WES data. Mutational signature analysis was performed to extract known mutational signatures (detailed in the supplemental Methods). Known mutational signatures were obtained from the COSMIC database (mutational signatures version 3; synapse.org ID syn12009743) and Li et al22 (6-MP–related signatures A and B).
hPSC maintenance cultures
We performed in vitro analysis using hematopoietic cells derived from NUDT15-knockdown induced pluripotent stem cells to assess the effect of NUDT15 expression on the extent of 6-MP–induced DNA damage. This study used two lines of human pluripotent stem cells (hPSCs): human ES cells (cell line KhES1) and human induced pluripotent stem cells (cell line 409B7). KhES1 was provided by Dr Norio Nakatsuji (Kyoto University, Kyoto, Japan). 409B7 was provided by Dr Shinya Yamanaka (Kyoto University). These hPSCs were cultured in tissue culture dishes coated with 0.25 μg/cm2 of iMatrix 511 (catalog #892011; Nippi, Osaka, Japan) in AK02N medium (catalog #AJ100; Ajinomoto, Tokyo, Japan).
hPSC transduction of piggyBac-based short hairpin RNA expression vectors
Short hairpin RNA sequences targeting two different loci in the NUDT15 gene (NM_018283.4), ACAAGGCTATGATCCATTTAA (sh01) and TTTCATTGAGAAGGAGAATTA (sh02), were designed and cloned into piggyBac-based short hairpin RNA expression vectors containing the puromycin-resistant cassette. hPSCs were electroporated with the vectors using a Nepa21 electroporater (Nepa Gene Co., Chiba, Japan).
Differentiation of hematopoietic cells from hPSCs
We modified our previously reported protocol23 to induce hematopoietic cells from hPSCs. The medium of PSC colonies that grew to a diameter of 750 to 1000 μm was replaced with Essential 8 (catalog #A1517001; Thermo Fisher Scientific, Waltham, MA) containing 2 μM of GSK-3 inhibitor CHIR-99021 (catalog #038-23101; Wako, Osaka, Japan), 80 ng/mL of BMP4 (catalog #314-BP-010; R&D Systems, Minneapolis, MN), and 80 ng/mL of vascular endothelial growth factor (catalog #293-VE-010; R&D Systems) on days 0 to 2 to induce cells equivalent to those of the primitive streak. Next, the medium was replaced with Essential 6 (catalog #A1516401; Thermo Fisher Scientific) containing 2 μM of ALK5 inhibitor SB431542 (catalog #031-24291; Wako), 50 ng/mL of stem cell factor (catalog #255-SC-010; R&D Systems), and 80 ng/mL of vascular endothelial growth factor on days 2 to 4 to induce hemangioblast-like bipotent progenitor cells. To induce hematopoietic progenitor cells,24 the medium was replaced with Stemline-II medium (catalog #S0192; Sigma-Aldrich, St Louis, MO) containing 50 ng/mL of stem cell factor, 10 μM of thrombopoietin, and 50 ng/mL of Flt-3 ligand (catalog #308-FK-005; R&D Systems) on days 4 to 15.
Flow cytometry and cell sorting
On days 15 to 17 of differentiation, blood cells were harvested from culture and stained with APC-conjugated anti-human CD45 and Qdot605-conjugated anti-human CD34 antibodies (BioLegend, San Diego, CA). Cells positive for both CD45 and CD34 were sorted using the BD FACSAria II (BD Biosciences, San Jose, CA) for subsequent culture. To detect the phosphorylated histone H2AX (pH2AX), cells exposed to 6-MP were fixed and permeabilized using BD Cytofix/Cytoperm solution (BD Biosciences) following the manufacturer’s procedure. Intracellular staining was then performed using fluorescein isothiocyanate antiphosphorylated (ser139) H2AX (clone 2F3; BioLegend). pH2AX levels are presented as the median fluorescence intensity.
Statistical analysis
The prevalence of NUDT15 variants was compared between groups using Fisher’s exact test. The 6-MP dose was compared between groups using the Mann-Whitney U test. Cumulative incidences were determined using the Gray test. A P value <.05 was considered statistically significant. In the experiments using PSCs, the Kruskal-Wallis test was first performed. For lines with a significant difference of P < .05, multiple comparisons (Dan-Bonferroni method) were performed as post hoc comparisons.
Ethics
This study was approved by the institutional ethics board of the National Center for Child Health and Development (#1025 and #1035; Tokyo, Japan), and informed consent was obtained from the patients and/or guardians. The use of human embryonic stem cells was approved by the Japanese Ministry of Education, Culture, Sports, Science and Technology.
Results
Patient characteristics
Characteristics of the 19 patients with SMNs are presented in Figure 1, Table 1, and supplemental Table 2. The diagnoses of primary cancer included ALL (n = 14), lymphoma (n = 4), and AML (n = 1). As for subsequent cancers, 14 patients had subsequent leukemia (n = 13) and lymphoma (n = 1), and five patients had subsequent solid tumors, including brain tumor (n = 1) and nonbrain solid tumors (n = 4). The median ages at diagnosis of primary cancer and SMNs were 5 years (range, 1-14 years) and 13.5 years (range, 5-44 years), respectively. The median time from diagnosis of primary malignancy to SMN was 6.5 years (range, 2-30 years). Radiation therapy was administered in seven cases. Three patients developed a solid tumor within the radiation field, whereas one developed a solid SMN outside of the field; the remaining SMNs were AML/myelodysplastic syndrome (Figure 1; supplemental Table 2). In the 14 patients whose primary cancer was ALL, the risk at diagnosis was classified as high in 7 patients, intermediate in 3 patients, and standard in 4 patients (Table 2). All patients were treated according to the Berlin-Frankfurt-Munich–type ALL protocol.
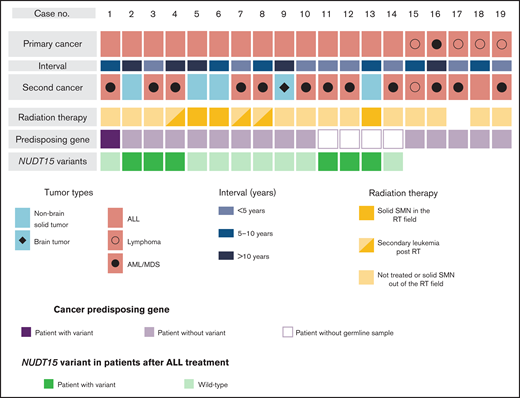
Overview of patient characteristics and genetic variations in SMNs. The subtypes of primary cancers and SMNs, intervals between primary cancers and SMNs, cancer-predisposing gene variants, and NUDT15 variants in patients with SMNs after ALL treatment are shown. NUDT15 genotype was evaluated using WES data (n = 10) and direct sequencing (n = 4). AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; RT, radiation therapy.
Overview of patient characteristics and genetic variations in SMNs. The subtypes of primary cancers and SMNs, intervals between primary cancers and SMNs, cancer-predisposing gene variants, and NUDT15 variants in patients with SMNs after ALL treatment are shown. NUDT15 genotype was evaluated using WES data (n = 10) and direct sequencing (n = 4). AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; RT, radiation therapy.
Clinical characteristics of SMN patient cohort
| Characteristic | n |
|---|---|
| Total patients | 19 |
| Primary cancer | |
| Median age at diagnosis, y (range) | 4 (0-14) |
| ALL | 14 |
| AML/MDS | 1 |
| Lymphoma | 4 |
| SMN | |
| Median age at diagnosis, y (range) | 13.5 (5-44) |
| Median interval from primary cancer, y (range) | 6.5 (2-30) |
| Hematologic malignancies | 14 |
| ALL | 1 |
| AML/MDS | 12 |
| Lymphoma | 1 |
| Solid tumor | 5 |
| CNS tumor | 1 |
| Non-CNS tumor | 4 |
| Characteristic | n |
|---|---|
| Total patients | 19 |
| Primary cancer | |
| Median age at diagnosis, y (range) | 4 (0-14) |
| ALL | 14 |
| AML/MDS | 1 |
| Lymphoma | 4 |
| SMN | |
| Median age at diagnosis, y (range) | 13.5 (5-44) |
| Median interval from primary cancer, y (range) | 6.5 (2-30) |
| Hematologic malignancies | 14 |
| ALL | 1 |
| AML/MDS | 12 |
| Lymphoma | 1 |
| Solid tumor | 5 |
| CNS tumor | 1 |
| Non-CNS tumor | 4 |
CNS, central nervous system; MDS, myelodysplastic syndrome.
Clinical characteristics and NUDT15 variants in primary ALL cases
| Case n | Primary cancer | Risk group | Genetic subtypes of primary ALL | SMN subtype | Interval, y* | Cytogenetics of tMDS/AML | NUDT15 genotype† | 6-MP dose, mg/m2 |
|---|---|---|---|---|---|---|---|---|
| 1 | BCP-ALL | HR | — | MDS | 10 | Normal karyotype | WT | 48 |
| 2 | BCP-ALL | IR | — | Bladder cancer | 24 | — | HT | 60 |
| 3 | BCP-ALL | HR | ETV6-RUNX1 | AML | 3 | MLL rearrangement | HT | 32 |
| 4 | BCP-ALL | HR | — | AML | 30 | 45,X,-X, t(8;16;21)(q22;p11.2;q22) | HT | 70 |
| 5 | BCP-ALL | IR | t(1;19) | BCC | 25 | — | WT | 47 |
| 6 | BCP-ALL | HR | — | ES | 9 | — | WT | NA |
| 7 | BCP-ALL | HR | FUS-ERG | AML | 3 | NA | WT | 16.6 |
| 8 | BCP-ALL | SR | — | AML | 8 | MLL rearrangement | WT | 85 |
| 9 | BCP-ALL | SR | — | Neurilemmoma | 12 | — | WT | 68 |
| 10 | T-ALL | SR | — | AML | 2 | Normal karyotype | WT | 115 |
| 11 | BCP-ALL | IR | — | MDS | 4 | 46,XX, add(3)(q26), −7, +mar | HT | 60 |
| 12 | BCP-ALL | HR | — | AML | 4 | Normal karyotype | HT | 38 |
| 13 | BCP-ALL | HR | MLL-AF4 | OS | 8 | — | HT | 37 |
| 14 | BCP-ALL | SR | ETV6-RUNX1 | MDS | 1 | 46,XX t(5;12;17)(q35;p13;q12) | WT | 27 |
| Case n | Primary cancer | Risk group | Genetic subtypes of primary ALL | SMN subtype | Interval, y* | Cytogenetics of tMDS/AML | NUDT15 genotype† | 6-MP dose, mg/m2 |
|---|---|---|---|---|---|---|---|---|
| 1 | BCP-ALL | HR | — | MDS | 10 | Normal karyotype | WT | 48 |
| 2 | BCP-ALL | IR | — | Bladder cancer | 24 | — | HT | 60 |
| 3 | BCP-ALL | HR | ETV6-RUNX1 | AML | 3 | MLL rearrangement | HT | 32 |
| 4 | BCP-ALL | HR | — | AML | 30 | 45,X,-X, t(8;16;21)(q22;p11.2;q22) | HT | 70 |
| 5 | BCP-ALL | IR | t(1;19) | BCC | 25 | — | WT | 47 |
| 6 | BCP-ALL | HR | — | ES | 9 | — | WT | NA |
| 7 | BCP-ALL | HR | FUS-ERG | AML | 3 | NA | WT | 16.6 |
| 8 | BCP-ALL | SR | — | AML | 8 | MLL rearrangement | WT | 85 |
| 9 | BCP-ALL | SR | — | Neurilemmoma | 12 | — | WT | 68 |
| 10 | T-ALL | SR | — | AML | 2 | Normal karyotype | WT | 115 |
| 11 | BCP-ALL | IR | — | MDS | 4 | 46,XX, add(3)(q26), −7, +mar | HT | 60 |
| 12 | BCP-ALL | HR | — | AML | 4 | Normal karyotype | HT | 38 |
| 13 | BCP-ALL | HR | MLL-AF4 | OS | 8 | — | HT | 37 |
| 14 | BCP-ALL | SR | ETV6-RUNX1 | MDS | 1 | 46,XX t(5;12;17)(q35;p13;q12) | WT | 27 |
BCP-ALL, B-cell precursor ALL; BCC, basal cell carcinoma; ES, Ewing sarcoma; HR, high risk; HT, heterozygous type; IR, intermediate risk; MDS, myelodysplastic syndrome; NA, not available; OS, osteosarcoma; SR, standard risk; T-ALL, T-cell precursor ALL; WT, wild type.
Time from primary cancer to SMN.
HT indicates heterozygous for rs116855232.
Germ line pathogenic variants in CPGs
WES identified 23 nonsilent germ line variants in the 110 cancer-associated genes listed as having a dominant inheritance pattern. Among them, only two LZTR1 variants (1 frameshift and 1 nonsense variant) were considered pathogenic; they were detected in the same patient (case 1; Figure 1; supplemental Table 3). Of the 52 genes with a recessive inheritance pattern, none were detected as homozygous or compound heterozygous mutations.
NUDT15 variants in patients with SMNs after ALL
Because there was only one CPG pathogenic variant case among the SMN cases, we considered the possibility of other potential SMN-causing genetic factors. Most cases in the primary hematological malignancy group were ALL (n = 14 of 19); therefore, we suspect that the genetic factors were related to the ALL treatment. Based on previous reports that polymorphism in TPMT, one of the metabolism-related genes in 6-MP, is associated with a risk of SMN development in American and European populations,13,14 we hypothesized that NUDT15 polymorphism is a risk factor of SMNs in our East Asian SMN cohort. Therefore, we evaluated the frequency of an altered nucleotide in rs116855232 of NUDT15 (p.R139C), which is an important determinant of sensitivity to 6-MP,15,16,19 using WES data in 10 cases and direct sequencing in 4 cases where only somatic samples were available (Figure 1). Strikingly, 6 of the 14 patients with ALL (42.9%) had NUDT15 heterozygous variants, although its prevalence in the general East Asian population in the gnomAD database was 19.7% (P = .042; Figure 2).
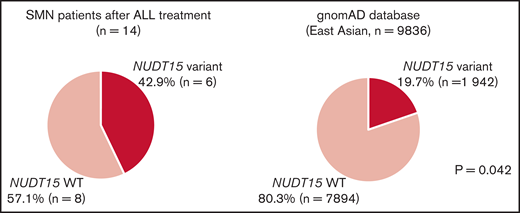
NUDT15 variant frequency in patients with SMNs after ALL treatment. Comparisons of the frequency of NUDT15 variant cases between the SMN cohort and gnomAD database are shown. Six (42.9%) of 14 patients with SMNs after ALL treatment had NUDT15 heterozygous variants, whereas prevalence in the general East Asian population in the gnomAD database was 19.7%. (P = .042). WT, wild type.
NUDT15 variant frequency in patients with SMNs after ALL treatment. Comparisons of the frequency of NUDT15 variant cases between the SMN cohort and gnomAD database are shown. Six (42.9%) of 14 patients with SMNs after ALL treatment had NUDT15 heterozygous variants, whereas prevalence in the general East Asian population in the gnomAD database was 19.7%. (P = .042). WT, wild type.
Because the sample size of our SMN cohort was small, we additionally investigated the presence of NUDT15 variants in a cohort of 438 unselected patients with ALL in the TCCSG L04-16 study to validate the potential association between NUDT15 hypomorphic variants and tumorigenesis. Among the 1033 patients in the original TCCSG L04-16 study, genomic DNA was available in 438 cases. All of these patients were treated according to the TCCSG protocol, which was based on the Berlin-Frankfurt-Munich–type ALL protocol. Among them, six patients developed SMNs. The clinical characteristics of these cases are presented in Table 3. Consequently, the cumulative incidence of SMNs was significantly higher among cases with the NUDT15 variant (3.0% at 7 years; 95% CI, 0.6% to 9.4%) than among those without (0.3%; 95% CI, 0.0% to 1.5%; P = .045; Figure 3A).
Clinical characteristics of patients with SMNs in the TCCSG L04-16 cohort
| TCCSG n | Primary cancer subtype | Primary risk | Day-8 risk | SMN subtype | NUDT15 genotype* |
|---|---|---|---|---|---|
| 404 | BCP-ALL | HR | HR | AML | HT |
| 105 | BCP-ALL | HR | HR | AML | WT |
| 014-008 | BCP-ALL | SR | SR | AML | WT |
| 0616084 | BCP-ALL | HR | HR | AML | HT |
| 0616086 | BCP-ALL | HR | HR-SCT | OS | HT |
| 568 | BCP-ALL | SR | SR | MDS | WT |
| TCCSG n | Primary cancer subtype | Primary risk | Day-8 risk | SMN subtype | NUDT15 genotype* |
|---|---|---|---|---|---|
| 404 | BCP-ALL | HR | HR | AML | HT |
| 105 | BCP-ALL | HR | HR | AML | WT |
| 014-008 | BCP-ALL | SR | SR | AML | WT |
| 0616084 | BCP-ALL | HR | HR | AML | HT |
| 0616086 | BCP-ALL | HR | HR-SCT | OS | HT |
| 568 | BCP-ALL | SR | SR | MDS | WT |
BCP-ALL, B-cell precursor ALL; HR, high risk; HR-SCT, high-risk stem cell transplantation; HT, heterozygous type; MDS, myelodysplastic syndrome; OS, osteosarcoma; SR, standard risk; WT, wild type.
HT indicates heterozygous for rs116855232.
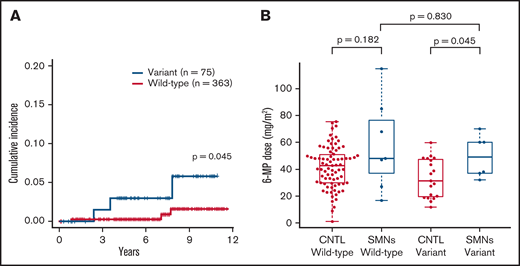
Cumulative incidence and comparison of 6-MP dose in patients with SMNs after ALL. (A) Cumulative incidence of SMNs in 438 children in TCCSG L04-16 who received ALL therapy was significantly higher in patients with NUDT15 variants (3.0% at 7 years) than in those with wild-type alleles (0.3%; P = .045). P value was calculated using the Gray test. (B) Average daily dose of 6-MP in maintenance therapy for patients with SMNs with a NUDT15 heterozygous variant was significantly higher than the dose administered to control (CNTL) patients with the same NUDT15 genotype, but they did not develop SMNs after 6-MP–containing therapy in a previous study (P = .045). P value was calculated using the Mann-Whitney U test.
Cumulative incidence and comparison of 6-MP dose in patients with SMNs after ALL. (A) Cumulative incidence of SMNs in 438 children in TCCSG L04-16 who received ALL therapy was significantly higher in patients with NUDT15 variants (3.0% at 7 years) than in those with wild-type alleles (0.3%; P = .045). P value was calculated using the Gray test. (B) Average daily dose of 6-MP in maintenance therapy for patients with SMNs with a NUDT15 heterozygous variant was significantly higher than the dose administered to control (CNTL) patients with the same NUDT15 genotype, but they did not develop SMNs after 6-MP–containing therapy in a previous study (P = .045). P value was calculated using the Mann-Whitney U test.
Clinical characteristics of patients with ALL with NUDT15 variants
Of the six cases with a NUDT15 heterozygous variant, the median ages at diagnosis of ALL and SMN were 10 and 17.5 years, respectively. The median time from ALL diagnosis to SMN was 6 years in variant cases and 8.5 years in wild-type cases (Figure 1; Table 2). In six SMN cases with a NUDT15 variant, the SMN subtypes were AML/myelodysplastic syndrome (n = 4), bladder cancer (n = 1), and osteosarcoma (n = 1). Radiation therapy was performed in three of the six patients with variants. The bladder cancer developed outside of the radiation field, whereas the osteosarcoma developed within the radiation field. Notably, the dose of 6-MP administered to patients with SMNs with a NUDT15 heterozygous variant was significantly higher than that administered to those with the NUDT15 genotype but who did not develop SMNs after 6-MP–containing therapy in the previous cohort study21 (P = .045; Figure 3B; Table 2).
Identification of 6-MP-induced signature
All 6 patients with available tumor (SMN)–normal pair samples had been treated with 6-MP for primary malignancies (ALL, n = 4; lymphoblastic lymphoma, n = 2). These six cases included one and five cases of NUDT15 variant and wild type, respectively. To evaluate the contribution of 6-MP–induced DNA damage to SMNs, mutational signature analysis was performed (supplemental Methods). Of the six cases, signature B, reported as the 6-MP–related signature,22,25 was present in four, including one variant case (66.7%; Figure 4).
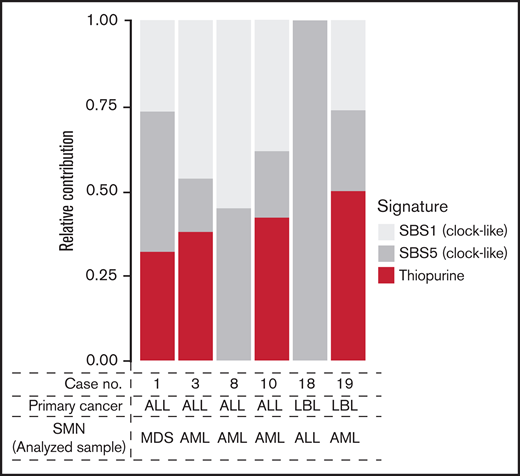
Relative contribution of therapy-related signatures in the 6 cases with available SMN samples. AML, acute myeloid leukemia; LBL, lymphoblastic lymphoma; MDS, myelodysplastic syndrome; SBS, single base substitution.
Relative contribution of therapy-related signatures in the 6 cases with available SMN samples. AML, acute myeloid leukemia; LBL, lymphoblastic lymphoma; MDS, myelodysplastic syndrome; SBS, single base substitution.
Reduced NUDT15 expression increased DNA sensitivity to damage by 6-MP
To evaluate the relationship between NUDT15 expression and sensitivity to 6-MP–induced DNA damage in hematopoietic stem/progenitor cells, we assessed the accumulation of double-strand DNA breaks using flow cytometry targeting pH2AX (Figure 5A). Cells with NUDT15 gene knockdown were significantly less viable after treatment with 6-MP (0.39 μM) for 1 week and did not survive treatment at higher doses (1.56 and 6.25 μM; Figure 5B-C). The viability of the scramble control cells was not affected by 6-MP exposure at these concentrations. Consistent with these observations, pH2AX levels were significantly higher in NUDT15-knockdown cells than in controls after treatment with 6-MP (0.39 μM; Figure 5D-E). These results indicate that reduced NUDT15 expression results in increased DNA damage induced by 6-MP.
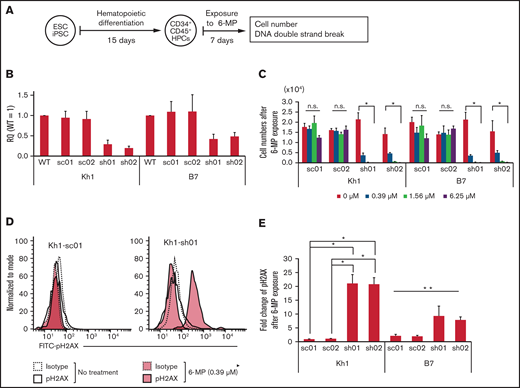
Reduced NUDT15 expression increased sensitivity to 6-MP–induced DNA damage. (A) Schematic diagram of experiments. (B) Relative expression of the NUDT15 gene in PSC-derived hematopoietic progenitor cells (HPCs). Relative quantity (RQ) values were calculated via the ΔΔCt method using multiple control genes in wild-type (WT) cells as an internal control in Kh1 and B7 strains. (C) Cell numbers after exposure to 6-MP. (D-E) Representative fluorescence-activated cell sorting panel and median fluorescence intensity of pH2AX levels after exposure to 6-MP. *Significant difference (P < .05) in both the Kruskal-Wallis test and post hoc comparison, **significant difference only in the Kruskal-Wallis test. ESC, embryonic stem cell; iPSC, induced pluripotent stem cell; n.s., no significant difference in Kruskal-Wallis test; sc, scrambled; sh, short hairpin.
Reduced NUDT15 expression increased sensitivity to 6-MP–induced DNA damage. (A) Schematic diagram of experiments. (B) Relative expression of the NUDT15 gene in PSC-derived hematopoietic progenitor cells (HPCs). Relative quantity (RQ) values were calculated via the ΔΔCt method using multiple control genes in wild-type (WT) cells as an internal control in Kh1 and B7 strains. (C) Cell numbers after exposure to 6-MP. (D-E) Representative fluorescence-activated cell sorting panel and median fluorescence intensity of pH2AX levels after exposure to 6-MP. *Significant difference (P < .05) in both the Kruskal-Wallis test and post hoc comparison, **significant difference only in the Kruskal-Wallis test. ESC, embryonic stem cell; iPSC, induced pluripotent stem cell; n.s., no significant difference in Kruskal-Wallis test; sc, scrambled; sh, short hairpin.
Discussion
Our most striking finding is that hypomorphic NUDT15 alleles may be a risk factor for developing SMNs after 6-MP exposure. The potential carcinogenic effect of thiopurines has been demonstrated in several studies showing an increased risk of therapy-related cancers in inflammatory bowel disease and autoimmune disease.26-29 NUDT15 is the most potent determinant of 6-MP sensitivity in the Asian population, and TPMT is the counterpart of NUDT15 in White populations. Thus, our results are consistent with previous studies reporting that TPMT polymorphisms confer a risk of SMNs after 6-MP treatment.12-14 This hypothesis is reinforced by our observation that patients who developed SMNs had been administered a higher average dose of 6-MP and that the 6-MP–related signature B was present in SMN specimens after 6-MP exposure. The 6-MP–related signature has also been identified in NUDT15 wild-type cases. A higher percentage of signature B is likely detected in NUDT15 variant cases; however, this could not be demonstrated because of the lack of available specimens. The results of in vitro experiments with PSC-derived hematopoietic progenitor cells may also suggest a relationship between insufficient NUDT15 activity and increased sensitivity to DNA damage by 6-MP. These findings demonstrate that high doses of 6-MP lead to an increased risk of SMNs, particularly in patients with NUDT15 variants.
A recent germline analysis of SMN cases after pediatric ALL treatment revealed that the relative prevalence of germline pathogenic variants was not high.30 In our study, mutations in LZTR1, which have been shown to be associated with B-cell ALL in recent studies, were identified in only one case.7 For children with ALL, variants of thiopurine-metabolizing genes such as TMPT and NUDT15 might be the most potent and frequent germline determinants of the risk of developing SMNs.
Among the patients with SMNs with a NUDT15 variant, there was one each who developed osteosarcoma within the radiation field and bladder cancer outside the radiation field. Because the expression of NUDT15 in each organ is not precisely known, it is unclear how 6-MP was associated with these SMNs. Additional studies are required to address how 6-MP–induced DNA damage confers each SMN type.
With regard to SMN prevention, determining the dose of 6-MP during maintenance therapy is an important issue. There are cases with very low thiopurine tolerance despite the absence of TPMT and NUDT15 polymorphisms.21 Finding a specific indicator, such as measuring thiopurine metabolites, as opposed to the current regulation that uses white blood cell counts, is a future challenge.
Our results have several limitations. First, the sample size of our study was small because of the rarity of SMN development. Furthermore, a validation study with a larger number of cases, ideally with a cohort, including other SMN phenotypes common in survivors, such as breast cancer and soft tissue sarcoma, is needed. Also, our in vitro experimental model corresponds to a NUDT15 homozygous variant. Ideally, a heterozygous variant model should be constructed to compare the level of DNA damage. Moreover, because our study was retrospective, we could not evaluate 6-MP metabolites during ALL treatment. Additional studies are required to determine the best method for optimizing the 6-MP dose for each genetic variant.
In conclusion, we found that NUDT15 variants may confer a risk of SMNs after treatment with 6-MP in patients with ALL. Our study underscores the importance of genetic background in childhood cancers, both in primary disease and in the development of SMNs.
Acknowledgments
The authors thank the staff at the Center for Clinical Research and Development at the National Center for Child Health and Development for their editorial support. The authors are grateful to Siqiaozhi Li, Takafumi Mano, and Etsuko Mochizuki for their technical assistance.
This work was supported by the Japan Society for the Promotion of Science through a Grant-in-Aid for Scientific Research (19K22608 and 16K10026), Japan Agency for Medical Research and Development (JP20ck0106467 and JP20kk0305014), National Center for Child Health and Development of Japan (2020A-1 and 2019A-4), and the US National Institutes of Health (R01GM118578 and R35GM141947).
Authorship
Contribution: J.J.Y. and M.K. planned, designed, and coordinated the study; M.Y., K.N., W.Y., A.N., J.J.Y., and M.K. contributed to the writing of the manuscript; M.Y., S.T., K.Y., R.S., T.O., C.K., Y.S., K.T., S. Ishimura, Y.Y., M.T., H.T., Y.A., A.K., M.H., T.I., D.H., Y.N., M.O., H.K., N.T., A.I., M.S., T.Y., D.T., K.K., K.M., N.K., A.O., A.M., and A.N. contributed to data gathering; M.Y., K.N., A.S.-O., W.Y., S.T., H.O.-K., T.K., K.I., M.S., K.O., K.Y., R.S., T.M., R.N., M.S., S.O., A.N., and K.H. contributed to data analysis; M.Y., K.N., A.S.-O., M.S., A.N., J.J.Y., and M.K. interpreted the data; S. Ito and N.K. discussed the results and provided comments and feedback; and all authors critically reviewed the manuscript and agreed to submit the manuscript for publication.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Motohiro Kato, Department of Pediatric Hematology and Oncology Research, National Center for Child Health and Development, 2-10-1, Okura, Setagaya, Tokyo 157-8535, Japan; e-mail: katom-tky@umin.ac.jp.

