Key Points
The adverse prognostic value of RUNX1 mutations in the ELN 2017 classification seems to be limited to their co-occurrence with SF mutations.
SF3B1 mutations conferred relatively poor prognosis upon patients with AML classified as having favorable or intermediate risk.

Abstract
Splicing factor (SF) mutations are important contributors to the pathogenesis of hematological malignancies; however, their relevance in risk classification of acute myeloid leukemia (AML) warrants further investigation. To gain more insight into the characteristics of patients with AML carrying SF mutations, we studied their association with clinical features, cytogenetic and molecular abnormalities, and clinical outcome in a large cohort of 1447 patients with AML and high-risk myelodysplastic syndrome. SF mutations were identified in 22% of patients and were associated with multiple unfavorable clinical features, such as older age, antecedent myeloid disorders, and adverse risk factors (mutations in RUNX1 and ASXL1). Furthermore, they had significantly shorter event-free and overall survival. Notably, in European LeukemiaNet (ELN) 2017 favorable- and intermediate-risk groups, SF3B1 mutations were indicative of relatively poor prognosis. In addition, patients carrying concomitant SF mutations and RUNX1 mutations had a particularly adverse prognosis. In patients without any of the 4 most common SF mutations, RUNX1 mutations were associated with relatively good outcome, which was comparable to that of intermediate-risk patients. In this study, we propose that SF mutations be considered for incorporation into prognostic classification systems. First, SF3B1 mutations could be considered an intermediate prognostic factor when co-occurring with favorable risk features and as an adverse prognostic factor for patients currently categorized as having intermediate risk, according to the ELN 2017 classification. Second, the prognostic value of the current adverse factor RUNX1 mutations seems to be limited to its co-occurrence with SF mutations.
Introduction
Recurrent mutations in genes regulating splicing (splicing factors [SFs]) were first discovered in hematological malignancies.1-3 The most commonly mutated genes from this novel class include splicing factor 3B subunit 1 (SF3B1), serine and arginine rich splicing factor 2 (SRSF2), U2 small nuclear RNA auxiliary factor 1 (U2AF1) and zinc finger, CCCH type, RNA-binding motif and serine and arginine rich 2 (ZRSR2), all of which are believed to act during the early stages of spliceosome assembly.4 Several studies documented the contribution of SF mutations to the pathogenesis of myeloid malignancies. Mutations in SRSF2, SF3B1, and U2AF1 were demonstrated to result in widespread changes in the transcriptome accompanied by altered hematopoiesis,5 and they are generally considered to be early leukemogenic events.1,5
Interestingly, the prognostic impact of SF mutations was shown to differ between hematological malignancies.6 The association of SF3B1 mutations with a better prognosis in the context of low-risk myelodysplastic syndrome (MDS) is already well established.2 In 2016, an updated version of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia incorporated SF3B1 mutations as a diagnostic criterion for MDS with ring sideroblasts.7 In contrast, 2 recent whole-genome investigations, as well as a small study focused on SF mutations, suggest that SF mutations are associated with inferior treatment outcome in acute myeloid leukemia (AML); in particular. SRSF2 mutations could be considered for incorporation into prognostic guidelines.8-10 However, an in-depth study focused specifically on SF mutations in a large well-annotated cohort is lacking.
The most widely accepted classification and prognostic schemes for AML include cytogenetic lesions together with multiple genetic mutations, including those in NPM1, FLT3, and CEBPA.7 Importantly, European LeukemiaNet (ELN) endorsed mutations in TP53, RUNX1, and ASXL1 as adverse risk factors, whereas AML with mutations in RUNX1 is considered a new provisional entity according to WHO.11 Interestingly, mutations in RUNX1 and ASXL1 were previously shown to co-occur with SF mutations.8,9 The high frequency of SF mutations in AML, together with their co-occurrence with RUNX1 and ASXL1 mutations, suggests that the prognostic impact of each of these factors could be examined in more detail.
Here, we report a comprehensive study of mutations in SF genes in a large well-annotated cohort of patients with AML (N = 1447). This cohort provides the unique opportunity to explore the significance of mutations in SF genes individually and cumulatively as an entity, as well as to investigate clinical and biological features of AML with SF mutations. In the present study, we assessed the frequency, genetic background, and prognostic value of SF mutations. To the best of our knowledge, this is the largest study that specifically focuses on this emerging class of molecular aberrations.
Methods
Patients
This study included a total of 1447 samples taken at the time of diagnosis from patients with AML/refractory anemia with excess blasts (RAEB), including 1253 de novo AML, 70 secondary AML (sAML), 72 MDS (RAEB), and 52 therapy-related AML (tAML) patients. The patients were included in clinical trials of the Dutch-Belgian Cooperative Trial Group for Hematology-Oncology (HOVON; n = 889) and treated according to 1 of the 3 clinical protocols (HOVON-42A, n = 133; HOVON-92, n = 43; or HOVON-102, n = 713)12,13 or treated according to standard protocols in Germany (collected by the Munich Leukemia Laboratory [MLL] between 2005 and 2016; n = 558). Despite the heterogenous origin of our cohort, no incremental differences were found in the genetic landscape, and no significant differences in overall survival (OS) were found between HOVON- and intensively treated patients with MLL (supplemental Figure 9A). More details regarding treatment protocols and study design can be found in the supplemental Methods. In total, the cohort included 1223 patients treated with intensive chemotherapy (1047 de novo AML, 61 sAML, 72 MDS, and 43 patients with tAML; Table 1, supplemental Table 1). All patients provided written informed consent. The study was approved by the internal review board of the MLL and local ethics committee of Amsterdam University Medical Center and was conducted in accordance with the Declaration of Helsinki.
Genetic profile
The mutational profile of each patient from the HOVON dataset was defined based on molecular diagnostics, as described previously.12,13 Also, for 430 patients (all of whom reached complete remission [CR]) additional data on mutations in 54 genes, based on targeted sequencing using an Illumina TruSight Myeloid Panel (Illumina, San Diego, CA), were generated previously, as described by Jongen-Lavrencic et al.14 The mutational profiles of patients from the MLL dataset were based on routine molecular diagnostics (including a combination of gene scan analysis, melting curve analysis, Sanger sequencing, and next-generation amplicon sequencing, as described previously),15-18 complemented by whole-genome sequencing (supplemental Methods). The cytogenetics were determined as described previously for patients included in the HOVON trials12,13 and in the MLL dataset following International Cancer Screening Network guidelines (2013).16,19,20
Statistical analysis
The associations between SF mutations and other genetic abnormalities, as well as categorical clinical and biological variables, were analyzed using Fisher’s exact test with the Benjamini-Hochberg correction for multiple testing. Associations between continuous variables and the presence of SF mutations were assessed using the Mann-Whitney U test. Survival analyses were performed in a subset of patients with AML treated with an intensive chemotherapy regimen (N = 1223). Samples for which data on the mutational status for a particular SF were missing were excluded from the analyses (for SFmut4: patients for whom data were missing for 1 of the 4 major SF factors were excluded from the analysis). These analyses included univariable testing (Kaplan-Meier analysis with log-rank test) and multivariable Cox proportional hazards model of the association between SF mutations and primary end points: OS and event-free survival (EFS). Further details about the statistical analyses used can be found in supplemental Methods.
Results
Recurrent SF mutations in AML
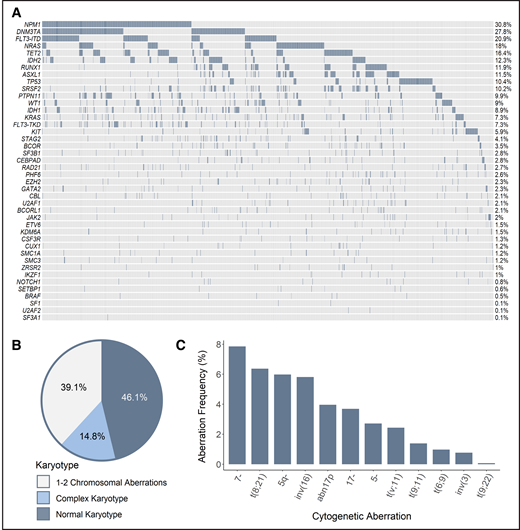
Recurrent SF mutations were assessed in a total of 1447 patients, including data gathered in multicenter clinical trials of the HOVON group (n = 889), as well as data collected by the MLL (n = 558). Although all 3 HOVON studies included patients aged 18 to 65 years, the MLL dataset contains data from aged at least 18 years, without an upper age restriction. The combination of the different cohorts of patients with AML resulted in a large and potentially heterogeneous population of interest. However, comprehensive analysis of genetic and clinical features, as well as treatment outcome, revealed comparability of characteristics that allowed combined analyses (supplemental Study design). Accordingly, the genetic landscape of our cohort was typical of AML; frequencies of cytogenetic and molecular aberrations were consistent with those reported in previous studies (Figure 1).8,9
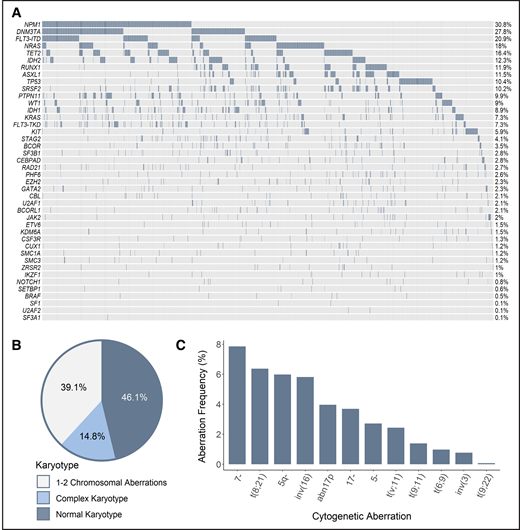
Genetic make-up of the total AML cohort. (A) Frequency of recurrent mutations. Proportion of patients based on the number of karyotypic aberrations (B) and frequency of recurring cytogenetic abnormalities (C). The assignment of patients to particular cytogenetic subgroups was not hierarchical; therefore, a single patient could be assigned to multiple subgroups when appropriate (ie, when >1 cytogenetic aberration was found).
Genetic make-up of the total AML cohort. (A) Frequency of recurrent mutations. Proportion of patients based on the number of karyotypic aberrations (B) and frequency of recurring cytogenetic abnormalities (C). The assignment of patients to particular cytogenetic subgroups was not hierarchical; therefore, a single patient could be assigned to multiple subgroups when appropriate (ie, when >1 cytogenetic aberration was found).
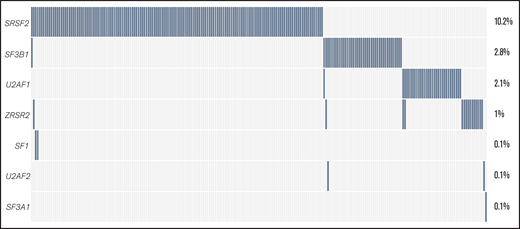
Data about the mutational status of 7 splicing regulators were available for a variable number of cases: SF3B1 (n = 1447), SRSF2 (n = 1447), U2AF1 (n = 988), ZRSR2 (n = 988), and SF1, SF3A1, and U2AF2 (n = 558 each). The 4 most common SF mutations (SRSF2, SF3B1, U2AF1, and ZRSR2) were identified in 22% of all of the assessed cases (Figure 2; see supplemental Table 2 for percentages of mutated cases per gene and supplemental Table 3 for mutated cases separated per disease type). The majority of identified mutations were hotspot mutations (supplemental Table 4). Although SF mutations were mostly mutually exclusive, 9 of 231 patients had coinciding mutations in 2 SF genes at diagnosis (supplemental Table 5, supplemental Results).

The frequency of SF mutations in the total AML cohort. The percentage of patients with SF mutations among the total number of AML cases in this study (N = 1447) is shown. The number of cases evaluated for each gene is shown in supplemental Table 2. Cumulatively, SF mutations were identified in 22% of evaluated cases, including 148 individuals with an SRSF2 mutation, 41 patients with an SF3B1 mutation, 31 individuals with a U2AF1 mutation, and 15 patients with a ZRSR2 mutation. Furthermore, 2 patients with an SF1 mutation and 1 patient with SF3A1 and U2AF2 mutations were found.
The frequency of SF mutations in the total AML cohort. The percentage of patients with SF mutations among the total number of AML cases in this study (N = 1447) is shown. The number of cases evaluated for each gene is shown in supplemental Table 2. Cumulatively, SF mutations were identified in 22% of evaluated cases, including 148 individuals with an SRSF2 mutation, 41 patients with an SF3B1 mutation, 31 individuals with a U2AF1 mutation, and 15 patients with a ZRSR2 mutation. Furthermore, 2 patients with an SF1 mutation and 1 patient with SF3A1 and U2AF2 mutations were found.
The genetic landscape and clinical features of SF-mutated AML
To gain more insight into the characteristics of patients with AML carrying SF mutations, we studied their associations with other recurrent gene mutations, cytogenetics, and clinical characteristics (Figure 3). Mutations in any single SF gene are relatively infrequent, and patients with mutations in SFs have not been elaborately studied as a subgroup. Therefore, associations were examined for mutations in each individual SF gene separately, as well as collectively. The analysis combining multiple SFs included 2 variables: mutations in at least 1 of the 4 most commonly mutated SFs (SF3B1, SRSF2, U2AF1 and ZRSR2; SFmut4) and mutations in at least 1 of all of the evaluated SF genes, including sporadic mutations in SF1, SF3A1, and U2AF2 (SFmut7). Because the latter variable included a limited number of cases (because the sporadic SF mutations were only assessed in 558 patients), we primarily focused our analyses on SFmut4.
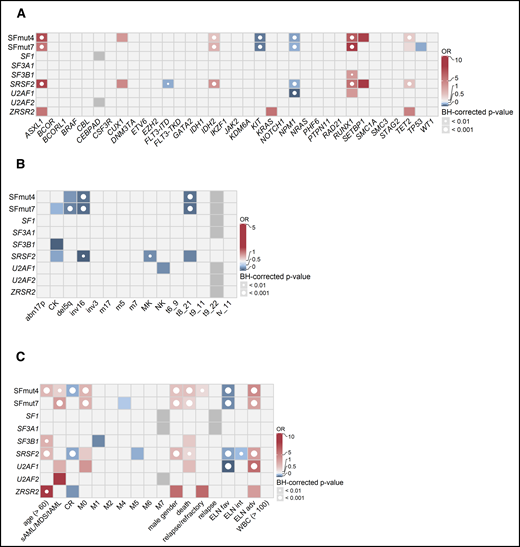
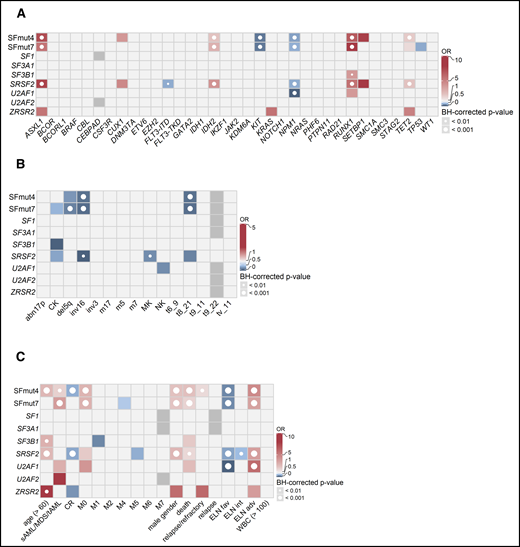
Associations between SF mutations and other recurrent gene mutations, cytogenetics, and clinical variables. Associations were determined using Fisher’s exact test with the Benjamini-Hochberg (BH) correction for multiple testing. The odds ratios (ORs) for significant associations are color coded. Red colors indicate a positive association (co-occurrence), whereas blue colors indicate a negative association (mutual exclusivity). Gray indicates missing data. The significance levels are indicated by the presence and size of the circle in each cell. (A) Co-occurrence and mutual exclusivity of SF mutations and other gene mutations. (B) Associations between SF mutations and cytogenetics. (C) Associations between gene mutations and clinical patient characteristics. adv, adverse; fav, favorable; int, intermediate.
Associations between SF mutations and other recurrent gene mutations, cytogenetics, and clinical variables. Associations were determined using Fisher’s exact test with the Benjamini-Hochberg (BH) correction for multiple testing. The odds ratios (ORs) for significant associations are color coded. Red colors indicate a positive association (co-occurrence), whereas blue colors indicate a negative association (mutual exclusivity). Gray indicates missing data. The significance levels are indicated by the presence and size of the circle in each cell. (A) Co-occurrence and mutual exclusivity of SF mutations and other gene mutations. (B) Associations between SF mutations and cytogenetics. (C) Associations between gene mutations and clinical patient characteristics. adv, adverse; fav, favorable; int, intermediate.
When associations with other commonly mutated genes were investigated, SF mutations frequently coincided with RUNX1, ASXL1, IDH2, and TET2 mutations and were mutually exclusive with KIT and NPM1 mutations, as well as FLT3-internal tandem duplication (Figure 3A). Furthermore, although we did not find any cytogenetic aberrations or chromosomal abnormalities co-occurring with SF mutations, we did find the core-binding factor aberrations and del5q (regardless of other molecular features) to be mutually exclusive with SF mutations (Figure 3B).
With respect to clinical characteristics, SFmut4 was correlated with older age (median, 64.5 vs 56.0 years; P < .001) and lower white blood cell (WBC) counts (median, 10.8 × 109/L; range: 0.3-264.3 × 109/L vs median, 16.3 × 109/L; range, 0.3-351.0 × 109/L; P < .001), both of which are common features of AML with antecedent myeloid disorders (Figure 3C). Accordingly, SFmut4 was more common in the sAML/MDS/tAML subgroup (34.2% of cases compared with 20.5% of de novo AML samples). Furthermore, 83% of all SF mutations affected patients classified as having adverse or intermediate risk according to the ELN 2017 criteria. We also examined the relationship between SF mutations and the differentiation status of the leukemia cells, as reflected by the French–American–British (FAB) classification, even though it is no longer used in risk stratification. We observed a strong association between SFmut4 and the M0 (immature) FAB subtype, which indicates the undifferentiated state of cells carrying these mutations. Finally, SF mutations were associated with male sex (SFmut4: 28.8% of males vs 14.3% of females; Figure 3C). The majority of the associations with clinical characteristics, cytogenetics, and recurrent gene mutations held true when examined in de novo AML, excluding all sAML/MDS/tAML cases (supplemental Table 6).
SF mutations are associated with inferior survival
All analyses of OS and EFS were performed in a subset of the cohort including patients treated with an intensive chemotherapy regimen only (n = 1223, Table 1; supplemental Table 1; also see "Methods"). Inferior survival was observed when the relationship between SF3B1, SRSF2, U2AF1, or ZRSR2 mutations and OS and EFS was analyzed individually for each gene in the total cohort (supplemental Figures 1 and 2). In addition, we found that SFmut4 was associated with shorter EFS, as well as OS, in Kaplan-Meier analyses (Figure 4A). The same associations were apparent within de novo AML, as well as within the sAML/MDS/tAML patient subset (data not shown). Furthermore, comparable trends were observed when the association between SFmut4 and OS or EFS was analyzed in separate ELN 2017 risk groups (supplemental Figure 3). Notably, mutations in SF3B1 were related to significantly inferior EFS and OS in the intermediate-risk group, whereas the association was of borderline significance for OS (P = .058) in favorable-risk patients (Figure 4B-C; supplemental Table 7). Survival rates of SF3B1-mutated patients in the intermediate-risk group were comparable to those of adverse-risk patients (5-year OS, 20.0% vs 23.8%, respectively; 5-year EFS, 10.0% vs 17.0%, respectively). In the favorable-risk group, 5-year OS of SF3B1-mutated patients was similar to that of adverse-risk patients (28.6% vs 23.8%, respectively), whereas 5-year EFS resembled that of intermediate-risk patients (28.6% vs 36.4%, respectively). SRSF2 mutations were associated with a slightly shorter EFS only within the adverse-risk group (supplemental Figure 3B). SRSF2-mutated patients were classified into this subgroup predominantly as a result of the co-occurrence with RUNX1 or ASXL1 mutations; the majority had a normal karyotype (data not shown). This suggests that co-occurrence of SRSF2 mutations with RUNX1 or ASXL1 mutations could be associated with a particularly adverse outcome.
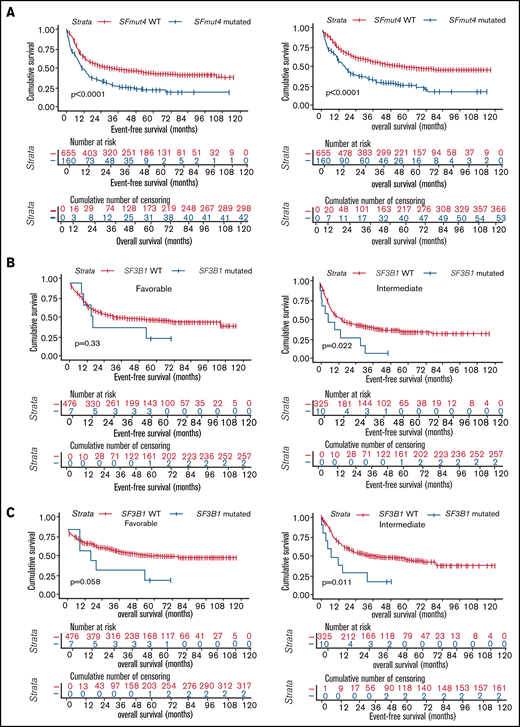
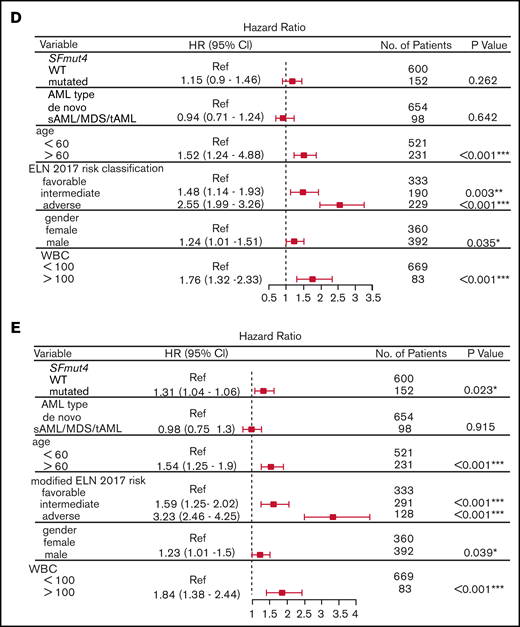
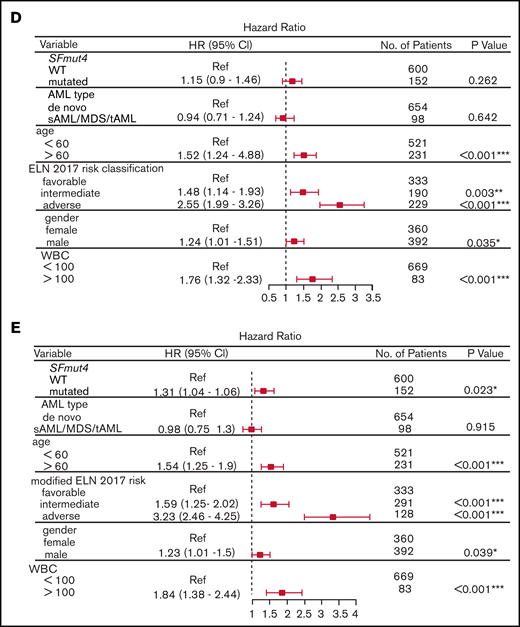
Survival of AML patients in relation to the presence of SF mutations. (A) Kaplan-Meier analyses of EFS (left) and OS (right) in the total AML cohort in relation to SFmut4. Multivariable Cox regression analysis of EFS in relation to SFmut4 with complete (C) and modified (D) ELN 2017 classification. In the modified ELN 2017 classification RUNX1 and ASXL1 mutations were excluded, so that patients carrying RUNX1 or ASXL1 mutations were reclassified based on the presence of the rest of aberrations in this classification system. The type of stem cell transplantation violated the proportional hazard assumption; therefore, it was used as strata variable in all of the multivariable Cox regression models (for variables used in the model as strata, the statistics were not calculated and do not appear in the results). Kaplan-Meier curves for EFS (D) and OS (E) in relation to the mutation status of SF3B1 within the favorable- or intermediate-risk groups, as defined by the ELN 2017 classification. HR, hazard ratio; Ref, reference; WT, wild-type; 95% CI, 95% confidence interval.
Survival of AML patients in relation to the presence of SF mutations. (A) Kaplan-Meier analyses of EFS (left) and OS (right) in the total AML cohort in relation to SFmut4. Multivariable Cox regression analysis of EFS in relation to SFmut4 with complete (C) and modified (D) ELN 2017 classification. In the modified ELN 2017 classification RUNX1 and ASXL1 mutations were excluded, so that patients carrying RUNX1 or ASXL1 mutations were reclassified based on the presence of the rest of aberrations in this classification system. The type of stem cell transplantation violated the proportional hazard assumption; therefore, it was used as strata variable in all of the multivariable Cox regression models (for variables used in the model as strata, the statistics were not calculated and do not appear in the results). Kaplan-Meier curves for EFS (D) and OS (E) in relation to the mutation status of SF3B1 within the favorable- or intermediate-risk groups, as defined by the ELN 2017 classification. HR, hazard ratio; Ref, reference; WT, wild-type; 95% CI, 95% confidence interval.
Finally, to assess the independent prognostic value of SF mutations, we performed a multivariable Cox-regression analysis accounting for known risk factors and potential confounders (which were also significantly associated with OS and EFS in univariable analysis, supplemental Table 8). These included age, sex, WBC count, disease entity, type of stem cell transplantation (none vs autologous or allogeneic stem cell transplantation) and ELN 2017 risk classification. We found that adding SFmut4 does not improve the baseline model of ES and OS when the current ELN 2017 classification is included (Figure 4D; supplemental Figure 4A). Because SF mutations often co-occur with RUNX1 and ASXL1 mutations, which are currently part of the ELN 2017 classification, we created a modified ELN 2017 classification by excluding RUNX1 and ASXL1 mutations (Figure 4E; supplemental Figure 4B). In the model constructed with this modified ELN classification, SFmut4 was independently associated with OS and EFS. Furthermore, addition of SFmut4 improved the model fit (Akaike Information Criterion [AIC] 4368.7 for baseline model vs 4365.7 for SF-including model of EFS; AIC 3626.1 for baseline model vs 3622.6 for SF-including model of OS; supplemental Tables 9-13). In addition, the AIC values for models including SFmut4 and modified ELN 2017 classification were superior to AIC values for the baseline models with the current ELN 2017 classification (AIC 4376.2 for EFS and 3634.5 for OS). The effects of SFmut4 were stronger in comparison with mutations in individual SFs separately, none of which showed significant independent predictive value. Strikingly, in contrast to SFmut4, addition of RUNX1 or ASXL1 mutation to the baseline model with modified ELN 2017 classification did not improve its predictive value (supplemental Figure 9). Thus, RUNX1 and ASXL1 mutations on their own are not strong independent predictors of the treatment outcome. Taken together, these findings highlight the importance of SF mutations for prognostication of AML treatment outcome.
Influence of gene interactions on survival
Next, we further explored the prognostic value of the interactions between SFmut4 and RUNX1 and ASXL1 mutations. Interestingly, Kaplan-Meier analysis of EFS and OS showed that the co-occurrence of SFmut4 with RUNX1 mutation was associated with a particularly adverse outcome (Figure 5A). This effect was largely driven by the interaction of SRSF2 mutations with RUNX1 mutations (Figure 5B). Moreover, the presence of RUNX1 mutations without SFmut4 no longer predicted inferior survival (Figure 5A-B; supplemental Figure 6A); this suggests that mutated RUNX1 is not the driver behind the independent prognostic value of SFmut4 in the context of the modified ELN 2017 classification. In addition, RUNX1 mutations were previously shown to be significantly associated with adverse outcome in patients with AML younger than 60 years21; therefore, we also analyzed the interaction between SFmut4 and RUNX1 mutations in this group separately and confirmed our findings from the total cohort (supplemental Results). The lack of an adverse prognostic value for RUNX1 mutations without co-occurring SFmut4 was further supported by analysis within the ELN 2017 adverse-risk group; patients with a mutation in RUNX1 without any of the 4 most common SF mutations (SRSF2, SF3B1, U2AF1, and ZRSR2) had a relatively good outcome (supplemental Figure 10). The OS and EFS of these patients were comparable to the intermediate-risk group: 5-year OS of 43.6% (vs 45.6% for intermediate-risk patients) and 5-year EFS of 38.0% (36.4% for intermediate-risk patients). In addition, double-mutated patients showed worse OS and EFS within the chromatin-spliceosome subgroup defined by Papaemanuil et al (data not shown).8 Similar associations, although somewhat less pronounced, were found for the combination of SFmut4 and ASXL1 mutations (supplemental Figure 6B).

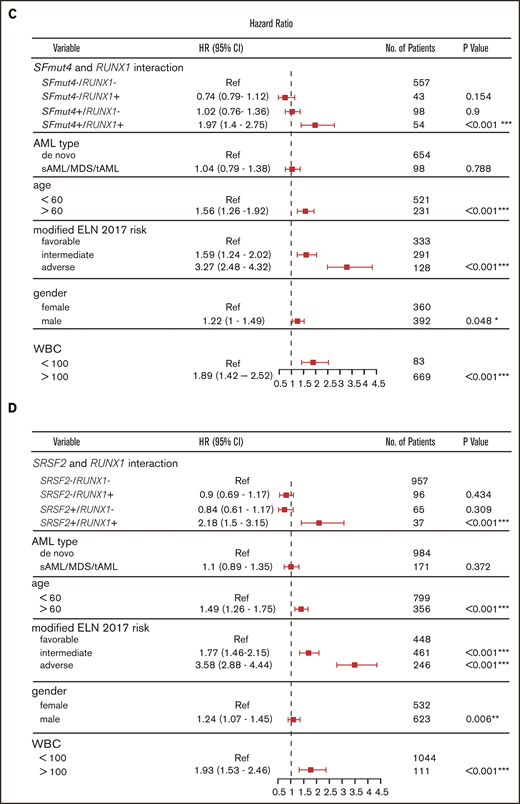
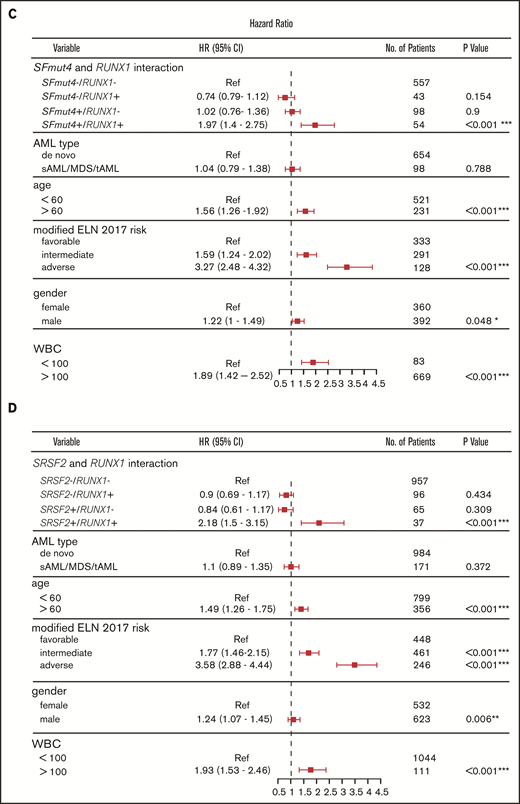
Influence of SF mutations on survival of RUNX1- and ASXL1-mutated patients. (A) Kaplan-Meier curves for EFS in relation to SFmut4 in combination with the mutation status of RUNX1. (B) Kaplan-Meier curves for EFS in relation to the mutation status of SRSF2 in combination with RUNX1. Multivariable Cox regression analysis of EFS in relation to the mutation status of SRSF2 (C) or SFmut4 (D) with RUNX1 mutations and modified ELN 2017 classification. In the modified ELN 2017 classification, RUNX1 and ASXL1 mutations were excluded; patients carrying a RUNX1 or ASXL1 mutation were reclassified based on the presence of the rest of the aberrations in this classification system. Type of stem cell transplantation violated the proportional hazard assumption; therefore, it was used as strata variable in all of the multivariable Cox regression models (for variables used in the model as strata, the statistics were not calculated and do not appear in the results). Because the number of cases for which data on all 4 of the most common SF mutations (included in the SFmut4 variable) were available was smaller than the number of cases for which SRSF2 alone was available, the patient numbers used in the analyses differ in panel B and panel C differ. *P < .05; **P < .01; ***P < .001. HR, hazard ratio; Ref, reference; sAML, sAML/tAML/MDS; 95% CI, 95% confidence interval.
Influence of SF mutations on survival of RUNX1- and ASXL1-mutated patients. (A) Kaplan-Meier curves for EFS in relation to SFmut4 in combination with the mutation status of RUNX1. (B) Kaplan-Meier curves for EFS in relation to the mutation status of SRSF2 in combination with RUNX1. Multivariable Cox regression analysis of EFS in relation to the mutation status of SRSF2 (C) or SFmut4 (D) with RUNX1 mutations and modified ELN 2017 classification. In the modified ELN 2017 classification, RUNX1 and ASXL1 mutations were excluded; patients carrying a RUNX1 or ASXL1 mutation were reclassified based on the presence of the rest of the aberrations in this classification system. Type of stem cell transplantation violated the proportional hazard assumption; therefore, it was used as strata variable in all of the multivariable Cox regression models (for variables used in the model as strata, the statistics were not calculated and do not appear in the results). Because the number of cases for which data on all 4 of the most common SF mutations (included in the SFmut4 variable) were available was smaller than the number of cases for which SRSF2 alone was available, the patient numbers used in the analyses differ in panel B and panel C differ. *P < .05; **P < .01; ***P < .001. HR, hazard ratio; Ref, reference; sAML, sAML/tAML/MDS; 95% CI, 95% confidence interval.
Our multivariable analysis including the modified ELN 2017 risk classification further supports the observation that co-occurrence of RUNX1 mutations with SFmut4 and, in particular, co-occurrence of RUNX1 mutations with SRSF2 mutations, is independently associated with a particularly poor EFS and OS (Figure 5C-D; supplemental Figure 8A). In the absence of any of the 4 most recurrent SF mutations, RUNX1 was not indicative of adverse risk in the multivariable model. The interactions of SFmut4 with ASXL1 mutations (supplemental Figure 8B) was not indicative of inferior EFS or OS when examined in the multivariable model (supplemental Tables 9-12).
Discussion
In this study, we focused specifically on characterization of the genetic background and clinical relevance of recurrent SF mutations in a large cohort of AML patients. For the purpose of our analyses, we combined several independent AML patient cohorts with confirmed compatibility (and limited heterogeneity) with respect to genetic and clinical features. The heterogeneity between cohorts was further limited by inclusion of only intensively treated patients in our survival analyses.
We demonstrated that SF mutations as combined groups affect 22% or 35% of patients with AML for SFmut4 and SFmut7, respectively. In addition, the vast majority of SF mutations were found to be mutually exclusive.5 The presence of SF mutations in AML and the mutual exclusivity of these mutations, together with the effectivity of recently developed splicing modulators, indicate that splicing deregulation in AML is key and cooperates with other lesions to promote leukemogenesis.22 Therefore, it is essential to explore the involvement of SF mutations in relation to other mutations and their (combined) relevance for patient outcome.
In this respect, the 2 most commonly comutated genes together with SFs include 2 transcription factors (RUNX1 and ASXL1). Because splicing occurs cotranscriptionally, it is conceivable that mutations in this class of gene expression regulators work together to initiate leukemia.23 Accordingly, mutations in RUNX1 were previously reported to be associated with increased intron retention in AML.24 In line with these observations, a recent study demonstrated that mutated RUNX1 knockout alone resulted in splicing alterations that were further broadened in the concomitant presence of SRSF2 mutation.9,10
Exploration of associations between SF mutations and specific AML features showed that any of the 4 common SF mutations (also coinciding with RUNX1 mutations) were strongly associated with a very immature cell state (FAB-M0). In addition, an intriguing association was noted between SF mutations (particularly SRSF2 and ZRSR2) and male sex. Several previous studies observed the same association in MDS and AML; however, it did not reach statistical significance in some cases because of small sample numbers.25,26 Furthermore, it was suggested that sex has an impact on splicing profiles of various tissues, and the degree of global aberrant splicing was reported to be higher in men with MDS compared with women with the same disorder.8,9 Future studies that are better designed to examine sex-specific genetic backgrounds and clinical associations with SF mutations, including treatment outcome, should be conducted.
We found that SF mutations were associated with older age, lower WBC count, and sAML, which is in agreement with previous observations.27 In parallel to these clinical features, SF mutations in our cohort were correlated with shorter OS and EFS, which can be explained by an inferior response to therapy, as reflected in the lower rates of CR and higher rates of disease relapse or refractoriness and death. Recently, Lindsley et al postulated that mutations in the chromatin-spliceosome subtype, including mutations in SRSF2, SF3B1, U2AF1, and ZRSR2, are characteristic of sAML, and they identified a subtype with poor prognosis in elderly de novo AML.27,28 Although this study also highlights the importance of cohesin mutations in this context, we did not identify a significant co-occurrence of this type of aberrations with SF mutations. In addition, another recent report suggests that the chromatin-spliceosome mutational signature in de novo AML identifies a disease subtype with a poor treatment outcome resembling that of secondary AML.9,29 This is further supported by the gene interactions observed in our study. SF mutations frequently co-occurred with mutations in ASXL1, RUNX1, and TET2, which constitute aberrations commonly found in older patients and sAML/MDS.9,29 At the same time, SF mutations were mutually exclusive with CBF translocations, which typically show a lower incidence in older patients.9 Taken together, these results support the notion that SF mutations identify a subtype of de novo AML with more sAML-like features. However, even in our subgroup analysis of patients with sAML/MDS/tAML, SF mutations were associated with worse OS and EFS (data not shown).
Overall, SFmut4 was associated with shorter OS and EFS in univariable and multivariable models. Just as reported by Papaemanuil et al, this suggests that splicing deregulation in general, via mutation in any SF, confers a poor prognosis.8 In our study, we identified 2 factors that could drive further improvements in the prognostication of AML. First, SF3B1 mutations among patients classified as having favorable risk and, in particular, intermediate risk, marked individuals with worse OS and EFS, suggesting that these patients would benefit from more intensive treatment or innovative therapies, such as splicing modulation. Second, we found that patients carrying concomitant SFmut4 and RUNX1 mutations had a particularly poor prognosis in univariable and multivariable analyses. This effect was largely due to the particularly strong interaction of SRSF2 and RUNX1 mutations. Strikingly, patients with RUNX1 mutations without any of the 4 common SF mutations had longer OS and EFS compared with the rest of patients within the adverse-risk group according to the ELN 2017 classification. Hence, RUNX1 mutations may not be relevant for risk assessment without the co-occurrence of any of the 4 most common SF mutations. Importantly, exclusion of patients carrying any of the 4 most common SF mutations from the RUNX1-mutated subgroup had a stronger effect than did exclusion of only SRSF2-mutated patients, highlighting the importance of considering mutations in any SF as a subgroup. Further underscoring the prognostic value of SFmut4, the multivariable model including SFmut4 and modified ELN 2017 classification, excluding ASXL1 and RUNX1 mutations, was superior at predicting survival than was a multivariable model without SFmut4 but including the current ELN 2017 classification.
Finally, it was suggested previously that RUNX1 mutations are predictive of inferior outcome only in AML patients younger than aged 60 years.8 Strikingly, the association of concomitant SFmut4 and RUNX1 mutations with inferior survival was even more pronounced in AML patients within this age group. The fact that this association was weaker in patients with AML older than 60 years seems to stem, at least in part, from the fact that the survival of older patients is, in general, relatively short. As a result, the impact of genetic lesions is thought to be less pronounced in this subgroup. The interaction of SFmut4 with mutations in ASXL1 showed similar, but less pronounced, associations with survival in Kaplan-Meier analysis and was not associated with survival in multivariable analysis, although a previous study suggested that co-occurrence of any 2 genes within the chromatin-spliceosome subgroup typically confers particularly short survival.14 Accordingly, the prognostic value of ASXL1 mutations without SF mutations should be explored further.
In summary, we propose that SF mutations should be considered for incorporation into diagnostic and prognostic guidelines, because the adverse prognostic value of RUNX1 mutations may not be valid without co-occurrence of any of the 4 most common SF mutations. Furthermore, mutations in SF3B1 should be considered a prognostic marker in individuals currently classified as having favorable or intermediate risk. SF3B1-mutated patients in those subgroups have relatively short OS and EFS, suggesting that they could benefit from more intensive treatment or innovative therapies, such as splicing modulation. Although our study included several independent cohorts, future studies should validate our proposed refinements and extend the analysis of SF mutations in relation to minimal residual disease.
Acknowledgments
I.v.d.W. was supported by the Oncology Graduate School Amsterdam (OOA) Diamond Program. A.W. was supported by the Torsten Haferlach Leukämiediagnostik Stiftung.
Authorship
Contribution: I.v.d.W. designed research, analyzed data, and wrote the manuscript; A.W. designed research, analyzed data, performed statistical analyses, and wrote the manuscript; M.M., S.H., C.B., and P.J.M.V. generated and analyzed data and edited the manuscript; M.H. supported statistical analyses; W.K., C.H., J.J.W.M.J., and G.J.O. supervised the analyses and edited the manuscript; and J.C. and T.H. designed the study, supervised analyses, and edited the manuscript.
Conflict-of-interest disclosure: T.H., C.H., and W.K. are part owners of MLL. M.M., S.H., and C.B. are employees of MLL. The remaining authors declare no competing financial interests.
Correspondence: Anna Wojtuszkiewicz, Department of Hematology Amsterdam UMC, Location VU University Medical Center, Cancer Center Amsterdam, De Boelelaan 1117, 1081HV, Amsterdam, The Netherlands; e-mail: a.wojtuszkiewicz@amsterdamumc.nl.