Key Points
Molecular profiling of MPN-SVT may identify high-risk patients as candidates for disease-modifying therapy.
JAK2V617F allele burden ≥50% or presence of chromatin/spliceosome/TP53 mutations is associated with adverse MPN-SVT hematologic outcome.

Abstract
Myeloproliferative neoplasms (MPNs) are the most frequent underlying causes of splanchnic vein thromboses (SVTs). MPN patients with SVTs (MPN-SVT) often have a unique presentation including younger age, female predominance, and low Janus kinase 2 (JAK2) mutation allele burden. This study aimed at identifying risk factors for adverse hematologic outcomes in MPN-SVT patients. We performed a retrospective study of a fully characterized cohort of MPN-SVT patients. The primary outcome was the incidence of evolution to myelofibrosis, acute leukemia, or death. Eighty patients were included in the testing cohort. Median follow-up was 11 years. Most of the patients were women with a mean age of 42 years and a diagnosis of polycythemia vera. The primary outcome was met in 13% of the patients and was associated with a JAK2V617F allele burden ≥50% (odds ratio [OR], 14.7) and presence of additional mutations in genes affecting chromatin/spliceosome (OR, 9). We identified high-risk patients (29% of the cohort) as those harboring at least 1 molecular risk factor: JAK2-mutant allele burden ≥50%, presence of chromatin/spliceosome/TP53 mutation. High-risk patients had worse event-free survival (81% vs 100%; P = .001) and overall survival at 10 years (89% vs 100%; P = .01) than low-risk patients. These results were confirmed in an independent validation cohort of 30 MPN-SVT patients. In conclusion, molecular profiling identified MPN-SVT patients with dismal outcome. In this high-risk population, a disease-modifying therapy should be taken into consideration to minimize the probability of transformation.
Introduction
Splanchnic vein thromboses (SVTs), including Budd-Chiari syndrome (BCS) and portal vein thrombosis (PVT), are severe vascular events.1 The pathogenesis of SVTs is mostly dependent on the presence of systemic prothrombotic conditions like inherited or acquired thrombophilia, paroxysmal nocturnal hemoglobinuria, or myeloproliferative neoplasms (MPNs). Indeed, MPNs represent 30% to 40% of the etiologies of BCS and PVT.2-4 However, the subgroup of MPN patients with SVT (MPN-SVT) has been shown to have peculiar clinical (young age, female predominance)5 and molecular features (large predominance of Janus kinase 2 [JAK2] gene V617F mutation6 ). During SVT, portal hypertension leads to hypersplenism and hemodilution, which may mask features of MPNs (high blood counts and splenomegaly) and blur the usual diagnostic criteria, the most reliable of which remains the presence of an MPN-related driver mutation. JAK2V617F6 and calreticulin (CALR)7,8 mutations are found in 80% and 4% of MPN-SVT, respectively,9-11 whereas JAK2 exon 12 or MPL gene mutations are very rarely identified.9 Similarly to classical MPN patients, MPN-SVT patients may harbor additional nondriver mutations targeting genes involved in the regulation of various intracellular pathways like epigenetics, messenger RNA splicing, signalization, and transcription. The impact of such additional mutations on the outcome of MPN-SVT patients has never been studied.
MPN-SVT is a unique presentation of MPNs due to particular disease features: the majority of patients are women of young age (<45 years old), mostly diagnosed with polycythemia vera (PV) with a low JAK2V617F-mutant allele burden. These peculiarities of MPN-SVT have already been described, but the mechanisms underlying such occurrences of thromboses in young MPN patients with low JAK2V617F allele burden remain unknown.9,12,13 Interestingly, a large study in the Danish population found that very low JAK2-mutant allele burdens were indeed associated with a higher risk of venous thrombosis, although these patients did not have an MPN phenotype, further strengthening the apparent paradox of MPN-SVT.14 However, the individual disease course is heterogeneous and difficult to predict. Some patients have an indolent disease for many years, and others experience multiple complications and disease progression. Transformation to secondary myelofibrosis and evolution to acute leukemia occur with similar frequencies in young patients at diagnosis (<45 years) compared with older ones (>60 years), but clear prognostic factors allowing prediction of long-term evolution are lacking.12 Also, better risk stratification in MPN-SVT could help to select the best treatment strategy when potential modifying therapies are available. Molecular profiling for MPN patients could offer personalized risk stratification in MPNs.15 The main objective of this study was to find predictive factors associated with adverse hematologic outcomes in patients with MPN-SVT.
Methods
Patients
This retrospective study was performed at the hospital group Assistance Publique–Hôpitaux de Paris (AP-HP) North–the University of Paris (Hôpital Saint-Louis and Hôpital Beaujon, Paris, France) for the testing cohort, and at the University of Insubria (Varese, Italy) for the validation cohort. Inclusion criteria were: (1) diagnosis of PVT or BCS3 ; (2) MPN diagnosis (2008 World Health Organization [WHO] classification16 ); (3) presence of a driver mutation (JAK2, CALR, MPL); and (4) available next-generation sequencing (NGS) result or archived DNA for NGS analysis. The database of SVT patients referred for MPN suspicion at Hôpital Saint-Louis (n = 284) and the NGS database of MPN patients routinely followed (n = 1371) were cross-checked to identify these patients. Clinical data were extracted from medical records and the French Network for Vascular Disorders of the Liver database, approved by the institutional review board (Comité de Protection des Personnes [CPP] Ile de France IV, Paris, France; number 2003/21). Demographic data including age, sex, MPN diagnosis, SVT type, blood counts at diagnosis, spleen size at diagnosis, associated thrombophilia (including factor V, factor II, C677T methylenetetrahydrofolate reductase mutations, protein C, protein S, and antithrombin deficiencies, antiphospholipid antibodies, and paroxysmal nocturnal hemoglobinuria determined by flow cytometry), and cytoreductive and anticoagulant therapy were recorded. The study was performed in accordance with the ethical guidelines of the Declaration of Helsinki. All patients gave their written consent for the registration of clinical and biological data. Data were collected and processed anonymously in a dedicated study after authorization from the French National Commission for Data Protection and Liberties (CNIL; no. 2215642).
Molecular analysis
JAK2V617F-mutant allele burden was measured as previously described.9 Molecular profiling of patients was performed on 200 ng of DNA extracted from peripheral blood (QIAcube instrument; Qiagen) using a capture-based custom NGS panel provided by SOPHiA Genetics (Lausanne, Switzerland) targeting 36 myeloid genes involved in MPNs. Genes tested in the panel are as follows: ABL1, ASXL1, BRAF, CALR, CBL, CCND2, CEBPA, CSF3R, CUX1, DNMT3A, ETNK1, ETV6, EZH2, FLT3, HRAS, IDH1, IDH2, IKZF1, JAK2, KIT, KRAS, MPL, NFE2, NPM1, NRAS, PTPN11, RUNX1, SETBP1, SF3B1, SH2B3, SRSF2, TET2, TP53, U2AF1, WT1, and ZRSR2. Bioinformatics analysis was performed by SOPHiA Genetics, and significant variants were identified and retained using SOPHiA DDM software with a detection limit of variants set at 1% for additional mutation. For the validation cohort, NGS analysis was performed on 20 to 50 ng of DNA extracted from polymorphonuclear cells using an automatic extractor (Maxwell RSC Buccal Swab DNA kit). The library preparation used the capture-based Myeloid Solution (MYS) panel by SOPHiA Genetics which covers 30 relevant myeloid genes, identical to those sequenced in the testing cohort except CCND2, CUX1, ETNK1, IKZF1, NFE2 and SH2B3. The NGS libraries were paired-end sequenced (2 × 150 bp) on an Illumina MiniSeq system.
Statistics
Quantitative variables were described using medians (range), whereas qualitative variables were described by frequencies (percentage). Categorical variables were compared using the χ2 or Fisher's exact test as appropriate. Continuous variables were compared using the Student t or Wilcoxon test, as appropriate.
The primary outcome was the incidence of transformation to secondary myelofibrosis, acute leukemia, or death. Event-free survival (EFS) was computed as the interval between diagnosis (SVT or MPN, the first to occur) and death of any cause or evolution into secondary myelofibrosis, acute leukemia, or last follow-up. Overall survival (OS) was computed as the interval between diagnosis (SVT or MPN, the first event to occur) and death or last follow-up. The cumulative probability of OS and EFS was estimated using the Kaplan-Meier method. For score development, the Paris cohort was used as the learning set and the Varese cohort as the validation set. All statistical tests were performed using R v3.4.4 (https://www.r-project.org).
Results
Patient characteristics
A total of 110 patients with MPN-SVT were analyzed in this study. For the testing cohort, 80 of 195 consecutive SVT patients (41%) referred to Hôpital Saint-Louis for the diagnostic workup of MPNs fulfilled inclusion criteria (Figure 1), mostly due to the availability of full molecular profile using NGS. Of note, no difference in initial characteristics and outcomes was observed between the 80 patients with NGS and the 115 patients without NGS analysis except for follow-up duration (11 years vs 8.3 years; P = .003) (supplemental Table 1).

Study profile. Of 284 patients with SVT and suspicion of MPNs, 195 patients had a confirmed MPN diagnosis. We excluded 8 patients without driver mutation and 107 patients without available NGS data. Eighty patients had an NGS analysis and were included in the testing cohort. Among them, 43 patients exhibited at least 1 additional mutation.
Study profile. Of 284 patients with SVT and suspicion of MPNs, 195 patients had a confirmed MPN diagnosis. We excluded 8 patients without driver mutation and 107 patients without available NGS data. Eighty patients had an NGS analysis and were included in the testing cohort. Among them, 43 patients exhibited at least 1 additional mutation.
For the testing cohort, the mean age at diagnosis was 42 years (range, 17-73 years). Median follow-up was 11 years (0.3-37 years) (Table 1). Localization of SVT was PVT (n = 52; 65%) and BCS (n = 28; 35%). Among PVT, associated thrombosis of splenic and mesenteric veins was present in 12 patients (23%) and 11 patients (21%), respectively. The driver mutation was JAK2V617F in 95%, and CALR type 1 in 5% of patients, respectively. Most of the patients were women (n = 50; 63%) with PV (n = 52; 65%). Patients with BCS were younger than those with PVT (mean, 35 vs 45 years old; P = .002), with a greater female predominance (85% vs 50%; P = .001). In 63 patients (79%), MPN and SVT diagnoses were coincident; in 9 patients (11%), the SVT preceded MPN diagnosis by a median time of 10 years (range, 1-29 years), and in 8 patients (10%), SVT occurred after a median of 5.5 years from the diagnosis of MPN (range, 1-12 years). In 17 patients with normal blood counts at diagnosis of SVT, a formal phenotype could not be assigned, and they were initially diagnosed as MPN unclassifiable. In all of them, a diagnosis of PV (in 11 patients) or essential thrombocythemia (ET; in 6 patients) could eventually be made due to changes in blood cell counts during follow-up. In all, as shown in Table 1, there were no distinguishing features at presentation between patients with additional mutations vs driver mutations alone. Ninety-eight percent of the patients received anticoagulants mostly with vitamin K antagonist (94%). Ninety-four percent of patients were treated by cytoreductive therapy mostly with hydroxyurea (84%). The transjugular intrahepatic portosystemic shunt procedure and orthoptic liver transplantation were performed in 11% of patients each. Two patients (3%) whose condition evolved into acute leukemia received an allogeneic stem cell transplantation.
MPN-SVT patients were younger at diagnosis than the global cohort of 1291 MPN patients with NGS data routinely followed in Hôpital Saint-Louis (42 vs 61 years; P < 10−5). At least 1 additional mutation was found in 37 patients (46%). Clinical characteristics and treatment were not significantly different between patients with and without additional mutations (Table 1).
Patients with additional mutations had a significantly higher JAK2V617F allele burden than those with only JAK2 mutation (median 31% vs 11%; P = .00003). A JAK2V617F allele burden above 50% was also more frequent in patients with than those without additional mutations (35% vs 7%; P = .0001).
Ten patients (13%) experienced an event of the primary outcome during follow-up (Table 1). Patients with additional mutations had more events than patients without (22% vs 5%; P = .04), mainly related to an increased incidence of transformation into secondary myelofibrosis (21% vs 2%; P = .04). No difference for acute leukemia or death was found between patients with and without additional mutations.
Molecular profiling
The most frequent additional mutations were found in genes involved in DNA methylation (31%) and chromatin or spliceosome pathways (15%) (Figure 2), including mutations in TET2 (21%), DNMT3A (11%), and ASXL1 (8%). Three patients (4%) had a TP53 mutation. No difference was observed in MPN-SVT patients when compared with NGS data derived from the 1291 MPN patients without SVT except for a lower incidence of ASXL1 mutations (8% vs 23%; P = .02).

Molecular profiling. Mutations found using a 36-gene NGS panel in the testing cohort, represented according to the presence or absence of clinical events of the primary outcome. Each column represents 1 sequenced patient. Eighty percent of patients with events had additional mutations vs 41% of patients without events. MF, myelofibrosis; ND, not determined.
Molecular profiling. Mutations found using a 36-gene NGS panel in the testing cohort, represented according to the presence or absence of clinical events of the primary outcome. Each column represents 1 sequenced patient. Eighty percent of patients with events had additional mutations vs 41% of patients without events. MF, myelofibrosis; ND, not determined.
Construction of the model and validation
Clinical characteristics and type of driver mutation were not associated with primary outcomes (Table 2). In contrast, 2 molecular features were associated with the primary outcome: a JAK2V617F allele burden ≥50% (odds ratio [OR], 14.7; 95% confidence interval [CI], 3.2-67.9; P = .0005) and the presence of additional mutations (OR, 5.7; 95% CI, 1.11-28.6; P = .03). Among additional mutations, those affecting genes involved in chromatin spliceosome (ie, targeting ASXL1, EZH2, SF3B1, SRSR2, U2AF1, or ZRSR2) had a very high effect on the primary outcome (OR, 9.0; 95% CI, 2.1-39; P = .003). TP53 mutations were not associated with the primary outcome (OR, 4.3; 95% CI, 0.4-53; P = .25) probably due to the low number of patients with mutations (n = 3) but were associated with the risk of transformation to acute leukemia (OR, 38.0; 95% CI, 1.7-850; P = .02). The number of mutations was not associated with the primary outcome (P = .1). A multivariate analysis could not be performed due to the low incidence of events.
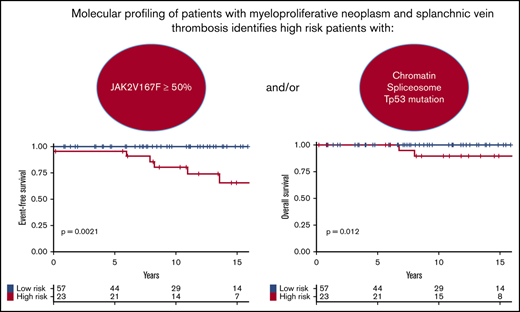
Based on these results, we developed a prognostic stratification allowing us to identify a group of high-risk patients harboring 1 or both of the following molecular risk factors: JAK2V617F allele burden ≥50% or presence of chromatin/spliceosome/TP53 mutation. This high-risk group represented 29% of the cohort. According to this stratification, at 10 years, high-risk patients had poorer EFS (81% vs 100%; P = .001; Figure 3A) and poorer OS (89% vs 100%; P = .01; Figure 3B) than low-risk patients.
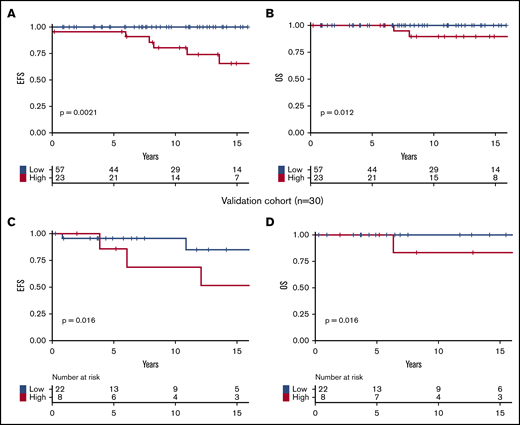
Survival of patients. Survival curves are represented in yellow for high-risk patients and in blue for patients with low-risk score. (A) EFS in the testing cohort. (B) OS in the testing cohort. (C) EFS in the validation cohort. (D) OS in the validation cohort.
Survival of patients. Survival curves are represented in yellow for high-risk patients and in blue for patients with low-risk score. (A) EFS in the testing cohort. (B) OS in the testing cohort. (C) EFS in the validation cohort. (D) OS in the validation cohort.
The Varese validation cohort included 30 MPN-SVT patients with NGS data (Table 3). Median follow-up was 7 years (range, 0.3-39 years), not significantly different than that of the testing cohort (P = .34). Clinical characteristics and treatments were also not significantly different between the testing and validation cohorts, except for the distribution of MPN subtypes (more ET patients in the validation cohort; P = .04) and the rate of transformation into secondary myelofibrosis (more frequent in the validation cohort; P = .04). In 15 cases (50%), MPN and SVT diagnosis were coincident; in 5 patients (17%), SVT preceded MPN diagnosis by a median time of 4 years (range, 2-35 years); and in 10 (33%), SVT occurred after a median of 10 years post-MPN diagnosis (range, 4-27 years). Nine patients (30%) had an additional mutation. Eight patients (27%) had the high-risk profile as defined in the testing cohort. In the validation cohort, we confirmed that patients belonging to the high-risk category had inferior EFS and OS than those within the low-risk category (Figure 3C-D). Because the time between MPN diagnosis and NGS analyses was variable in both cohorts, we verified that high-molecular-risk profile remained significantly associated with worse outcomes in subgroups of patients in whom NGS was performed before or after 11 years of follow-up (the median follow-up of our cohort). The 10-year EFS of high-risk vs low-risk patients was 65% vs 100% (P = .0008), and 62% vs 94% (P = .03) among the shorter interval group of the testing and validation cohorts, respectively (supplemental Figure 2A,C). The 10-year EFS of high- vs low-risk patients was 59% vs 100% (P = .017) among the longer interval group of the testing cohort (supplemental Figure 2) (not possible in the validation cohort due to insufficient numbers [n = 3]).
Thrombosis recurrence
In the initial cohort, 12 patients (15%) had a thrombosis recurrence: SVT recurrence for 6 patients and thrombosis in another site for 6 patients (2 ischemic strokes, 2 cerebral venous thromboses, and 2 coronary thromboses). Thrombophilia was not associated with thrombosis recurrence (29% vs 8%; P = .2); neither was the presence of an additional mutation (P = .76).
Discussion
To the best of our knowledge, this is the first study showing a prognostic impact of NGS molecular analysis in MPN-SVT patients. We found that the presence of a JAK2V617F allele burden >50%, or of chromatin, spliceosome, or TP53 mutations, was associated with adverse hematological outcomes. These findings were reproducible in an external independent validation cohort for patients followed at a different hospital with NGS analysis performed on a different molecular platform, suggesting that our proposed risk-stratification algorithm could be easily and widely applicable.
One-half of the patients had additional mutations, mostly affecting TET2, DNMT3A, and ASXL1 genes, as previously reported in MPNs.15,17,18 Our results are in agreement with the worse outcomes of patients harboring high-risk mutations in genes affecting chromatin or spliceosome.15 Median JAK2V617F allele burden was low in our study as expected in MPN-SVT patients.4 However, patients with a high JAK2V617F allele burden had adverse hematologic outcomes (OR, 14.7), in agreement with previous studies in patients with homozygous JAK2 mutation.15,19,20 We included in the high-risk molecular criteria the presence of TP53 mutation (4%) due to its well-known adverse impact on the prognosis of MPN patients, in particular the strong association with transformation into acute leukemia.15,17,18 These TP53 mutations were present in the cells of our patients during the chronic phase of MPNs at a low allelic burden, but in 1 of them, an expansion of the clone and acute leukemia eventually occurred, as described usually after the loss of the wild-type allele.18 In contrast, there was no negative impact of mutations in genes affecting DNA methylation, for example, TET2 and DNMT3A, suggesting that these mutations could be related to clonal hematopoiesis18,21 rather than MPNs per se in this particular context. Indeed, patients with TET2 and DNMT3A mutations were significantly older compared with patients without such mutations (median age of 47 vs 40 years; P = .04).
Patients with MPN-SVT are often considered as affected by an early stage of MPNs, and indeed the majority of patients in this study had an indolent evolution during the median 11 years of follow-up. However, in the long-term, 13% of the patients suffered a hematological progression or died (mostly related to hematological complications), a proportion higher than usually expected in this young population but close to that reported by Stein et al in a study focusing on the long-term evolution of younger PV patients.12 Our approach using NGS analysis for molecular profiling was able to detect such patients with a high risk of poor outcomes. This stratification is needed because the actual prognostic scores for PV and ET may not be relevant in this particular group of patients with SVT.22 Although the presentation of MPN-SVT is unique, our findings are in agreement with previous studies of the impact of molecular abnormalities in MPN patients in general, validating their importance in this particular context. The vast majority of MPN-SVT patients in this study received cytoreductive therapy (mostly hydroxyurea) with the primary objective of decreasing thrombosis recurrence, and it was therefore impossible to assess the impact of cytoreduction on the primary outcome. With the PROUD-PV and CONTINUATION-PV studies, pegylated-interferon α showed promising results in PV patients with good tolerability and the potential to decrease not only the JAK2-mutant allele burden but also that of additional mutation.23,24 Ruxolitinib, a JAK1-JAK2 inhibitor, also demonstrated capacity to reduce the JAK2-mutant allele burden.25,26 Longer follow-up of these studies will provide important information on the clinical impact of these molecular responses on long-term outcomes and the ability to change MPN natural history.
The retrospective nature of our study is a limit, but, otherwise, we would not have had any prospective possibility of capturing information in the long-term in such a rare disease. In addition, the clinical picture of patients we selected based on DNA availability is superimposable to that of the whole series of SVT patients we collected in 10 years. Hence, we can generalize the results we found to the whole population of SVT. On the other hand, we may have overlooked some patients with severe acute hepatic injury and early death, eventually determining the low rate of SVT-associated death observed (only 1 event).
In summary, the addition of NGS data to clinical variables allowed the design of a new prognostic score able to predict MPN-SVT patients with a poor long-term outcome. High-risk features included a high JAK2-mutant allele burden or the presence of chromatin/spliceosome/TP53 mutations, associated with shorter EFS and OS. A disease-modifying therapy could be proposed for high-risk patients, when possible, to minimize the risk of hematological transformation. On the other hand, low-risk patients did not have any evolution after 10 years and cytoreductive therapy with hydroxyurea seems adequate when needed on top of anticoagulation to reduce the risk of thrombosis recurrence. However, confirmation of these findings in a prospective study would be important, but it will necessarily require international collaboration and enough follow-up.
Data-sharing requests may be e-mailed to the corresponding author, Jean-Jacques Kiladjian, at jean-jacques.kiladjian@aphp.fr.
Acknowledgments
The authors thank the French Intergroup of Myeloproliferative Neoplasms (FIM) and the French National Network for Vascular Liver Diseases for helpful discussion; in particular, the authors thank Kamal Zekrini for help in database management.
N.M. was supported by a Poste d’accueil grant of the Institut National Du Cancer (INCa). The Varese group was supported by grants from the Fondazione Regionale Ricerca Biomedica (Milan, Italy; FRRB project no. 2015-0042); the Ministero della Salute (Rome, Italy; Finalizzata 2018, NET-2018-12365935); the Ministero dell’Istruzione, dell’Università e della Ricerca (Roma, Italy; PRIN 2017, 2017WXR7ZT); Fondazione Matarelli (Milan, Italy); and AIL-Varese ONLUS (Varese, Italy).
Authorship
Contribution: P.-E.D., B.C., S.G., and J.-J.K. designed research; P.-E.D., B.C., S.G., F.P., and J.-J.K. performed research and analyzed and interpreted data; B.C., B.M., E.V., N.M., and C.S. contributed analytical tools; P.-E.D. performed statistical analysis; P.-E.D., F.P., and J.-J.K. wrote the manuscript; and all authors participated in collecting, analyzing, and interpreting the data, checked the final version of the manuscript, and are fully responsible for the content and editorial decisions pertaining to this manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jean-Jacques Kiladjian, Centre d’Investigations Cliniques, Hôpital Saint-Louis, 1 Ave Claude Vellefaux, Paris, France; e-mail: jean-jacques.kiladjian@aphp.fr; or Francesco Passamonti, Department of Medicine and Surgery, University of Insubria, Via Guicciardini, 21100 Varese, Italy; e-mail: francesco.passamonti@uninsubria.it.