Abstract
Mucopolysaccharidoses (MPSs) are multiorgan devastating diseases for which hematopoietic cell transplantation (HCT) and, to a lesser extent, enzyme replacement therapy have substantially altered the course of the disease. Furthermore, they have resulted in increased overall survival, especially for Hurler disease (MPS-1). However, despite the identification of clinical predictors and harmonized transplantation protocols, disease progression still poses a significant burden to patients, although at a slower pace. To design better therapies, we need to understand why and where current therapies fail. In this review, we discuss important aspects of the underlying disease and the disease progression. We note that the majority of progressive symptoms that occur in “hard-to-treat” tissues are actually tissues that are difficult to reach, such as avascular connective tissue or tissues isolated from the circulation by a specific barrier (eg, blood-brain barrier, blood-retina barrier). Although easily reached tissues are effectively cured by HCT, disease progression is observed in these “hard-to-reach” tissues. We used these insights to critically appraise ongoing experimental endeavors with regard to their potential to overcome the encountered hurdles and improve long-term clinical outcomes in MPS patients treated with HCT.
Introduction
It has been >50 years since Fratantoni et al1 described that cocultured fibroblasts of patients with Hurler disease (mucopolysaccharidosis [MPS]-1) and Hunter disease (MPS-2) corrected each other, leading to a mutual reduction in the intracellular accumulation of glycosaminoglycans (GAGs). Hurler syndrome and Hunter syndrome are 2 of the 7 types of MPSs in which a deficiency in a specific lysosomal enzyme prevents proper degradation of specific metabolites, resulting in a devastating progressive multisystemic disease and, if severe, in premature death.2 In 1981, Hobbs et al3 reported the first hematopoietic cell transplantation (HCT) in a 1-year-old patient with MPS-1 based on the principle of cross-correction. HCT has become the standard of care in MPS-1, if diagnosed in a timely manner. Intense international collaboration during the last decade has identified predictors of clinical outcomes, including myeloablative conditioning, early timing of transplantation, and enzyme activity level in blood after HCT. This has resulted in optimized transplantation protocols and 5-year survival rates > 90%. Enzyme replacement therapy (ERT) for MPS-1 was introduced in 2003, followed by ERT for MPS-2, MPS-4, MPS-6, and MPS-7.4-6 Unfortunately, ERT comes with serious limitations (eg, neutralizing antibodies, lack of blood-brain barrier [BBB] passage, and huge costs).
Despite the greatly improved overall survival, current standard treatments still have their “weak spots,” because they are unable to completely halt the disease in specific tissues. Late outcome studies show significant residual disease burden.7-11 Although many researchers have been trying to develop new therapies to improve clinical outcomes, the primary outcomes in these (animal) studies are often enzyme activity and GAG concentration in receptive tissues: leukocytes in the circulation, brain, liver, spleen, and lungs. This is unfortunate because we are already capable of treating these tissues. In the medical field, Sutton’s Law, which is named after the infamous thief Willie Sutton who robbed banks because “that’s where the money is,” recommends that one should first consider the obvious. Therefore, we propose that, to evaluate whether new therapies are able to improve quality of life in MPS patients, the focus of the effect should be on the “hard-to-treat” tissues and how to improve this effect, because obviously “that’s where the money is.”
To design better strategies, we first need to understand why current therapies fail. Why are some tissues hard to treat? Therefore, a better understanding of disease pathogenesis and the mechanism of current treatments is necessary. In this review, we summarize important aspects of the underlying disease, establish which tissues are “hard to treat,” and define their unifying characteristics. Furthermore, we critically appraise experimental therapeutic endeavors with regard to their potential to overcome these hurdles and improve long-term clinical outcomes of MPS patients.
Understanding the underlying disease
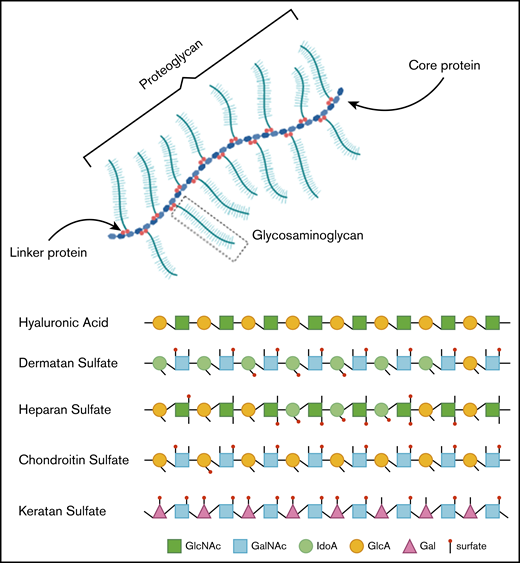
In healthy individuals, GAGs, formerly called mucopolysaccharides, represent complex sugar molecules that are degraded in a stepwise manner by enzymes in the lysosome (Figure 1).12 The 2 main groups are sulfated GAGs (heparan sulfate [HS], dermatan sulfate [DS], keratan sulfate [KS], and chondroitin sulfate) and nonsulfated GAGs (hyaluronic acid [HA]). All GAG chains, with the exception of HA, are linked to a core protein to form proteoglycans (PGs).12 PGs are important components of the extracellular matrix (ECM) of connective tissue and, furthermore, contribute to the ordering process of collagen fibers.13 During development, PGs are important in the assembly of ECM in tissue morphogenesis to determine form and function of a tissue.14 ECM is essential in the communication between cells. Communication is modulated via interactions between GAG chains and ECM components, such as collagen, fibronectin, and laminin.13,15 Some PGs are also known to bind and regulate a number of distinct proteins, including chemokines, cytokines, and growth factors.16
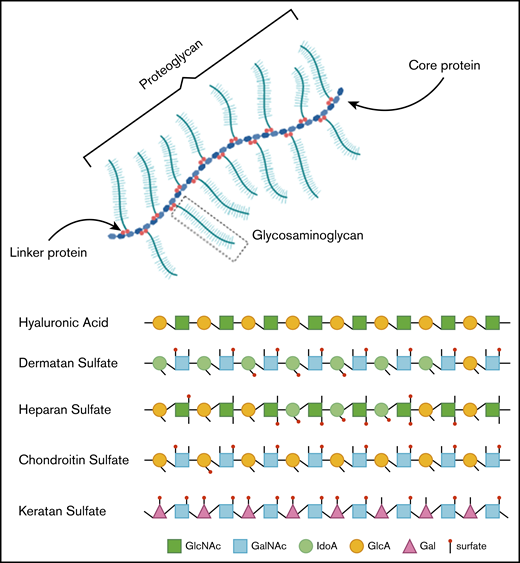
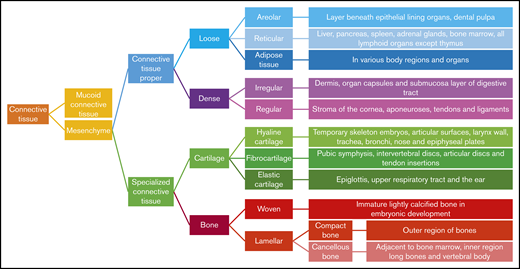
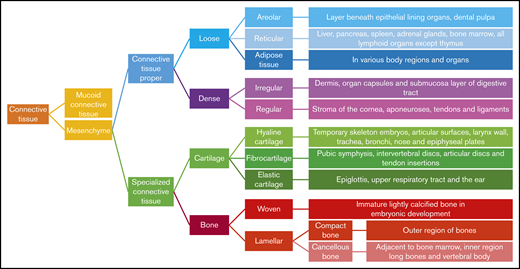
Hence, GAGs, either in chain form or as a part of larger PGs, are important components of connective tissue throughout the body, including the skeleton, cornea, cartilage, tendons, and, to a lesser extent, the brain (Figure 2).17 Free GAGs and proteoglycans are biologically active molecules that are involved in critical cellular pathways. They can isolate extracellular humoral factors, modulate the function of cell surface receptors in signal transduction and cross talk between cells. PGs are expressed during skeletal development, and they are spatially and temporally regulated in a highly defined pattern.18
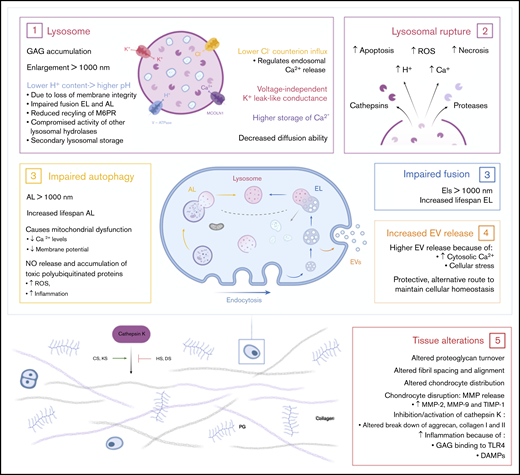
In MPS, deficiency of a lysosomal enzyme results in the accumulation of partially degraded GAGs. Primary GAG storage leads to perturbation of cellular, tissue, and organ homeostasis.19 Subsequently, the pathways in which they are involved will become affected. These secondary effects, such as impaired autophagy, increased apoptosis, and inflammation, are also important effectors of the disease symptoms (Figure 3).20 The main clinical symptoms are coarse facial features, hepato- and splenomegaly, dysostosis multiplex, joint stiffness, recurrent respiratory infections, sleep apnea, heart disease, and, in severe subtypes, cognitive impairment and a reduced life expectancy. The time of onset, rate of progression, and extent of disease manifestations vary, depending on the type of MPS.2 Details about the specific types of MPS can be found in Table 1.

Effects of accumulated lysosomal products on cellular and tissue homeostasis. (1) Accumulation of GAGs in the lysosome leads to enlargement of the lysosome, followed by a loss of membrane integrity. Leakage of H+ out of the lysosome results in a higher pH and may lead to compromised activity of lysosomal hydrolases and secondary lysosomal storage. Furthermore, the higher pH results in impaired fusion of endolysosomes and autolysosomes and a reduced recycling of the M6P receptor. Finally, enlarged lysosomes have decreased diffusion ability inside the cell. (2) Enlargement of the lysosome ultimately results in lysosomal rupture with the escape of cathepsins and proteases in the cell. Simultaneously, lysosomal rupture leads to increased concentration of H+ and Ca2+ in the cytosol. Together, this results in increased reactive oxygen species (ROS) production, necrosis, and apoptosis of cells. (3) The dysfunctional lysosome leads to impaired autophagy and fusion with endolysosomes. Therefore, increased lifespans are seen for autolysosomes and endolysosomes. Impaired autophagy subsequently results in mitochondrial dysfunction. (4) The increased concentration of cytosolic Ca2+ and cellular stress stimulates the release of extracellular vesicles (EVs). This is an alternative protective route for the cell to maintain cellular homeostasis. (5) The accumulation of GAGs in the cell and the secondary affected mechanisms result in tissue alterations and destruction. Connective tissue is the most affected tissue in MPS patients. Studies show altered proteoglycan turnover, altered fibril spacing and alignment, and altered chondrocyte distribution and disruption leading to increased concentrations of matrix metalloproteinase-2 (MMP-2), MMP-9, and tissue inhibitor of metalloproteinase-1 (TIMP-1). Depending on the type of GAG, undegraded GAGs lead to inhibition or activation of cathepsin K, which is necessary for the degradation of aggrecan and collagen I and II. Finally, HS and cellular stress lead to inflammation via activation of the Toll-like receptor 4 (TLR4) pathway. DAMPs, damage associated molecular patterns.
Effects of accumulated lysosomal products on cellular and tissue homeostasis. (1) Accumulation of GAGs in the lysosome leads to enlargement of the lysosome, followed by a loss of membrane integrity. Leakage of H+ out of the lysosome results in a higher pH and may lead to compromised activity of lysosomal hydrolases and secondary lysosomal storage. Furthermore, the higher pH results in impaired fusion of endolysosomes and autolysosomes and a reduced recycling of the M6P receptor. Finally, enlarged lysosomes have decreased diffusion ability inside the cell. (2) Enlargement of the lysosome ultimately results in lysosomal rupture with the escape of cathepsins and proteases in the cell. Simultaneously, lysosomal rupture leads to increased concentration of H+ and Ca2+ in the cytosol. Together, this results in increased reactive oxygen species (ROS) production, necrosis, and apoptosis of cells. (3) The dysfunctional lysosome leads to impaired autophagy and fusion with endolysosomes. Therefore, increased lifespans are seen for autolysosomes and endolysosomes. Impaired autophagy subsequently results in mitochondrial dysfunction. (4) The increased concentration of cytosolic Ca2+ and cellular stress stimulates the release of extracellular vesicles (EVs). This is an alternative protective route for the cell to maintain cellular homeostasis. (5) The accumulation of GAGs in the cell and the secondary affected mechanisms result in tissue alterations and destruction. Connective tissue is the most affected tissue in MPS patients. Studies show altered proteoglycan turnover, altered fibril spacing and alignment, and altered chondrocyte distribution and disruption leading to increased concentrations of matrix metalloproteinase-2 (MMP-2), MMP-9, and tissue inhibitor of metalloproteinase-1 (TIMP-1). Depending on the type of GAG, undegraded GAGs lead to inhibition or activation of cathepsin K, which is necessary for the degradation of aggrecan and collagen I and II. Finally, HS and cellular stress lead to inflammation via activation of the Toll-like receptor 4 (TLR4) pathway. DAMPs, damage associated molecular patterns.
Understanding disease progression in “hard-to-treat” tissues: in search of common themes
Disease progression related to connective tissue
Skeletal malformations.
Skeletal deformities are extremely common in MPS disorders, and the benefits of HCT or ERT are limited.5,21-23 Thoracolumbar kyphosis, hip dysplasia, and genu valgum progress in the majority of patients, albeit at a slower pace.24-26 Several factors are thought to contribute to skeletal malformations. First, ossification and mineralization depend on the distribution of growth factors (ie, insuline-like growth factors, fibroblast growth factors, parathryoid hormone–related protein, Indian Hedgehog), and GAGs play a major role in the regulation of these signaling pathways.27,28 Second, bone remodeling and growth are based on the ossification of cartilage, which is disturbed as a result of the malfunction of chondrocytes containing accumulated GAGs and altered cathepsin K activity (Figure 2).29 This is supported by the presence of nonossified cartilaginous precursors in bones of MPS patients.30,31 Cartilage is avascular and, thus, receives nutrients via diffusion.32 Enzyme delivery from the circulation will most likely also occur via diffusion. The distance that enzymes have to overcome in bone and articular cartilage is large; therefore, ossification continues to be impaired. Third, GAG accumulation stimulates secondary pathophysiological processes in ECM, like inflammation, contributing to osteopenia and the abnormal shape of bones seen in MPS patients.33 Finally, abnormal weight-bearing forces influence the morphology of bone structures.34,35 Malformations in these structures that are already present before HCT alter the forces between them and contribute to further progression of hip dysplasia. Progression of hip deformations also influences the progression of genu valgum.36,37 Importantly, many of these malformations already start to develop in utero.
In conclusion, many bone defects have already developed at the time of diagnosis and start of treatment as a result of abnormal ossification, lack of bone remodeling, inflammation, and abnormal forces. Posttreatment with HCT or ERT, enzyme availability continues to be insufficient in the avascular diffusion-dependent cartilage. Ongoing alterations in growth factor signaling pathways and inflammation may play additional roles in the changes in chondrogenesis and bone growth.38
Joints, tendons, and ligaments.
In all types of MPS, with the exception of MPS-4, GAG accumulation in ligaments, tendons, and joint capsules results in joint contractures.39-41 Studies on joint stiffness post-HCT or post-ERT show conflicting outcomes.5,24,42-45 The joint capsule is a dense connective tissue layer that enhances joint security and bone placement; it may also extend into the joint to form a fibrocartilaginous articular disc or meniscus. Normally, the capsule contains blood vessels, but altered forces can drive this tissue into a more fibrocartilage type that becomes much less vascularized. In this process, increases in collagen type II, KS, DS, and chondroitin sulfate are seen.46 As a consequence, changes in fibril arrangement, spacing, and size occur, which are destructive for tendons and ligaments; proper fibril arrangement is pivotal to create functional structures. In addition to joint contractions, weakening of the tendons is seen, which explains, in part, the thoracolumbar kyphosis.47 The skeletal malformations, in combination with weakening of the tendons, can also lead to neck instability and, together, complicate anesthetic airway management in patients.48 Furthermore, carpal tunnel syndrome, caused by median nerve constriction in the wrist due to GAG deposition in the transverse carpal ligament, occurs in 89% of patients, despite HCT. Also, trigger digits, caused by GAG deposition in the flexor tendons, are frequently observed, despite treatment.24,49-52
Thus, GAGs accumulate in ligaments, tendons, and joint capsules, despite current treatments. These tissues have in common that they are avascular connective tissues. Hence, they rely on diffusion of enzyme from the circulation after treatment with HCT or ERT, which is even more challenging in the case of ERT, because of the intermittent availability of enzyme with high peaks of enzyme in plasma for only a short duration. Moreover, most of these structures consist of dense connective tissue in which diffusion of nutrients is even more difficult, and the arrangement of the structure is highly important. This likely explains the poor efficacy of current systemic treatments.
Corneal clouding.
Progression of corneal clouding is observed posttreatment with HCT or ERT.5,53-55 The cornea, a transparent avascular connective tissue, consists of epithelial and endothelial cells, keratocytes, collagen, and PGs.56 GAG accumulation affects the size of keratocytes and disrupts the regular network of the stroma, resulting in abnormal arrangement, spacing, and size of collagen fibrils.57,58 Tear fluid could potentially be the supplier of donor enzyme after HCT. However, enzyme activity in tear fluid of MPS-1 patients after HCT remains low, despite normalization in the circulation, insinuating poor enzyme penetration.55
Heart valves.
Although disease manifestations in the coronary arteries and the heart muscle are effectively addressed following HCT, and partially for ERT, the mitral and aortic valve deformities, persist. In some, this may lead to progressive valvular dysfunction.5,59,60 The heart valves contain a highly specialized and organized ECM in which the alignment of collagen fibers is extremely important.61 GAG accumulation leads to abnormal fibril organization and weakening of valvular connective tissue as a result of decreased stiffness and increased extensibility, which have been associated with valvular prolapse.62-65 Similar to other avascular connective tissues, valvular disease progression is likely due to enzyme shortage.
Disease progression unrelated to connective tissue
Retinal degeneration.
Despite successful HCT, degeneration of the vascularized retina continues.53 This is likely explained by the blood-retina barrier (BRB), which hampers adequate enzyme delivery to the retina. Kaneko et al66 used enhanced green fluorescent protein–positive bone marrow cells in mice to study whether donor cells end up in the retina after HCT. They compared damaged retinas (induced by injection of N-methyl-N-nitrosourea or creation of retinal detachment) with normal retinas and found that only a minute number of donor cells end up in uninjured retinas of mice up to 12 months following HCT. However, damaged retinas showed massive recruitment of donor-derived cells into the retina.66 Whether retinas of MPS patients show similar damage and, thus, recruit donor-derived macrophages after HCT to produce enzyme locally warrants further investigation.
Taken together, we propose that “hard-to-treat” tissues in MPS are actually “hard-to-reach” tissues for which 2 major hurdles need to be addressed: overcoming avascular zones and blood-tissue barriers. Poorly vascularized tissues are dependent on diffusion of the enzyme from the circulation, which can pose a significant hurdle if the distance is too large or the tissue is very dense. An advantage of HCT is its ability to allow monocytes to migrate to the brain and, thus, pass the BBB, suggesting that this physiological barrier is overcome. However, MPS-3 patients, in whom central nervous system symptoms predominate, deteriorate despite HCT.67 The predominant accumulating GAG in MPS-3 is HS, which plays an important role in the brain. Speculatively, the central nervous system of these patients may require an amount of enzyme that, apparently, is not provided by HCT. Furthermore, progression of retinal degeneration insinuates that the effect of therapy is still inhibited by the BRB. Therefore, we propose that additional local therapies or therapies that enhance transport of systemic available enzyme to connective tissues or tissues isolated from the circulation because of a specific barrier could help to improve long-term outcomes in MPS patients.
Toward treating “hard-to-reach” tissues
Based on the general considerations above, alternative approaches for enzyme delivery to “hard-to-reach” tissues have been proposed. These include, in order of their proximity to clinical use, gene therapy (GT), direct local administration, small molecule therapies, targeted enzyme-delivery systems, mesenchymal stem cell therapy, and focused ultrasound therapy.
Gene therapy
GT is currently the most advanced experimental therapy. Viral GT has been studied widely; however, nonviral strategies have been explored as well.
Viral GTs.
Viral GT in MPS has been explored using retroviruses, lentiviruses, and adeno-associated viruses.68 Because of the risk of mutagenesis when using retroviruses, research now focuses mainly on lentiviruses and adeno-associated viruses. Ex vivo LV GT is used in combination with autologous stem cell therapy, aiming for supraphysiological enzyme activity levels in the circulation, with the rationale to improve penetration in poor responding tissues, of which animal studies have shown better correction of the bones and brain.69-71 Autologous gene-modified HCT is being studied in a clinical trial for MPS-1 (NCT03488394) and for MPS-3A (NCT04201405) and is being developed for MPS-3B. An important advantage of autologous stem cell GT is reduced morbidity and mortality as a result of the absence of graft failure and graft-versus-host-disease, as well as the reduced toxicity of immunosuppressive medication.72 However, it is still unknown whether patients develop antibodies against the enzyme that could subsequently reduce the effect of therapy. Although the initial results are promising, it is still an open question whether greater hematologically available enzyme is able to overcome the diffusion distances in poorly responding avascular tissues and completely cure MPS. In addition to gene-modified autologous HCT, direct administration of GT is being studied and is addressed in "Direct local administration."
Nonviral GTs.
A nonviral GT strategy is the Sleeping Beauty (SB) transposons.73 SB transposons consist of a system using 2 plasmids: 1 containing the internal sequence for the system, the inverted terminal repeat (ITR) and the gene of interest, and the other 1 containing the SB transposase that recognizes the ITR to ensure translocation of the gene into the genome. The SB strategy was studied in MPS-1 mice and showed efficacy in most, but not all (ie, kidney, heart, and aorta), somatic organs. The central nervous system also was not treated with the SB transposons. Therefore, the SB strategy is not yet sufficient for the treatment of MPS.74
Direct local administration
To overcome avascularity of tissues and blood-tissue barriers, local (intra-articular) administration of recombinant enzyme has been studied in MPS-1 and MPS-6 animal models.75-77 By injecting the recombinant enzyme directly into the joint cavity of MPS-6 cats, obvious histological improvements were consistently detectable. Following treatment, clearance of lysosomal storage in synovial cells and chondrocytes was seen. Joints that received recombinant enzyme had improved thickness of cartilage, fewer cartilage lesions, better overall joint shape, healthy well-vascularized subchondral bone, and improved synovium compared with the contralateral buffer-treated joints. These encouraging results have led to the treatment of 2 MPS-6 patients with monthly intra-articular injections in the hips for 2 years.76 However, the results have yet to be published. Wang et al77 found similar results for intra-articular recombinant enzyme in MPS-1 dogs. Administration into intra-articular spaces resulted in reduction of GAG storage in the synovium and chondrocytes in the articular surface of the joint cartilage, as well as in chondrocytes further away from the articular surface.
Major disadvantages of injecting recombinant enzyme are the need for repeated injections, and, possibly, the risk of antibody formation. Vance et al78 explored AAV-mediated GT for enzyme delivery by intrastromal injection as an effective treatment for MPS-1–related corneal clouding. Seven days after injection of an AAV8G9-optimized enzyme construct in human corneas, the enzyme was overproduced in the corneal stroma with widespread distribution in multiple cell types, as well as a 10-fold increase in enzyme activity without any indications of toxicity.78 Furthermore, AAV-mediated GT directly administered into the brain was successful in animals79-81 and resulted in clinical trials that are currently open for MPS-1 (NCT03580083), MPS-2 (NCT03566043), and MPS-3A (NCT03612869 and NCT02716246).
Small-molecule therapies
Alternative strategies that overcome the transport obstacles aim to improve enzyme activity locally or decrease the workload of the faulty enzyme.
An estimated 70% of severe MPS phenotypes are caused by premature stop-codon mutations.82 Therefore, stop-codon read-through therapy could be a promising approach; however, most of the well-known drugs with read-through effects, such as gentamycin (an aminoglycoside) and chloramphenicol, are toxic. Therefore, a less toxic derivate was constructed (NB84). The results of a phase 1 clinical trial with the synthetic version of NB84, ELX-02, showed an acceptable safety profile in healthy adults83 ; however, clinical efficacy in MPS patients has yet to be proven. PTC-124 is a nonaminoglycoside stop-codon read-through therapy compound. Although promising results for PTC-124 were seen in MPS mouse models, an open-label clinical phase 2 trial was ended early because of low enrolment (European Union Drug Regulating Authorities Clinical Trials Database number 2014-002596-28). No conclusions could be drawn about its effect.
Substrate reduction or optimization therapy.
Molecules that reduce the synthesis of GAGs, such as genistein and rhodamine B, are a subject of research in MPS-1, MPS-2, and MPS-3.84-86 A pilot study in MPS-3 patients showed that genistein was safe, reduced urine GAG concentration, and arrested behavioral and cognitive deficits.87 A phase 3 study of genistein in MPS-3 patients was completed in 2018; however, the results have yet to be published (European Union Drug Regulating Authorities Clinical Trials Database number 2013-001479-18). In MPS-2 patients, genistein improved joint and connective tissue elasticity.88 In addition to substrate-reduction therapies, substrate optimization therapy has been applied. Substrate optimization therapy includes compounds that alter the accumulating substrate to make it more amenable to degradation by other enzymes. In vitro studies showed that modified GAGs were indeed more susceptible to degradation in fibroblasts, and the therapy is currently being developed for MPS-1.89,90
Chaperone therapy.
Chaperones are small proteins that aid in the correct folding of enzymes, leading to increased residual enzyme activity.91 In vitro experiments on several MPS 3B– and 3C-causing mutations indicated that pharmaceutical chaperones can bind and stabilize mutant enzymes and improve their enzymatic activity. Thus, the use of pharmaceutical chaperones to alter enzyme activity might be plausible, especially for those mutations that cause abnormal glycosylation patterns.91
Small-molecule therapies show promising results in vitro; however, their in vivo efficacy has yet to be proven in clinical trials. Because most compounds are smaller than 50 kilodaltons, they are able to cross the BBB and, possibly, the BRB. Although not suitable as general standard therapies, they could be helpful in achieving better clinical outcomes in specific mutations seen in MPS patients.
Targeted enzyme delivery systems
Targeted drug delivery seeks to concentrate the medication in the tissues of interest and can potentially improve drug delivery in avascular tissues or isolated tissues because of a blood-tissue barrier. Two types of targeted enzyme delivery systems have been studied in recent years: a so-called “Trojan horse strategy” and a nano-targeted delivery route.
Trojan horse strategy.
The Trojan horse strategy entails the use of fusion proteins to cross a therapeutic over a specific barrier.92 The therapeutic part of the fusion protein can be conjugated to a monoclonal antibody against a tissue-specific target-receptor or to an endogenous ligand for natural binding to the receptor.93 In MPS, fusion proteins have mainly been studied to cross the BBB via the receptors of transferrin, insulin, low-density lipoprotein, or lectin and were demonstrated to create higher yields of enzyme in the brain.94-98 Targeted delivery of drugs to bones with the use of a hydroxyapatite-binding site or alendronate (a drug with a high affinity for bone) has been investigated for several bone diseases and shows promising results in vitro.99 In vivo efficacy has yet to be proven. This approach has not been studied for MPS, but it could be a strategy to get the deficient enzyme into the cartilage and bone and possibly achieve a better clinical response.
Nano-targeted delivery.
Nano-therapy has been proposed for gene products and direct enzyme delivery and offers promising therapeutic methods in lysosomal storage diseases.100,101 It uses different strategies, including micelles, liposomes, nano-emulsions, or nanoparticles. The choice of which biomaterial is used depends on the biochemicals of the desired drug.100 Nano-targeted delivery of enzyme might be specifically interesting for the treatment of bone and cartilage disease in MPS patients. Wang et al102 created a liposome specific for hydroxyapatite, which binds with a high affinity to collagen-hydroxyapatite compositions in vitro. This suggests the possibility of using mineral-targeted ligands in nano-particles for the delivery of lysosomal enzymes to mineralized tissues. Bidone et al103 studied intra-articular administration of cationic nano-emulsions complexed with a plasmid encoding for the IDUA protein. They demonstrated the localization of the nano-emulsion in synovial joints, cellular uptake by synoviocytes, high cell viability, and a 10-fold increase in IDUA expression in the joint 48 hours after injection. IDUA activity was increased fivefold in synovial fluid and 20-fold in synovial joints.103
Mesenchymal stem cell therapy and extracellular vesicles
Bone marrow, in addition to HSCs, contains mesenchymal stem cells (MSCs). MSCs are multipotent stromal cells that are characterized by their intrinsic self-renewal capacity and ability to differentiate into cells of the mesenchymal lineage, including osteoblasts and chondrocytes.104,105 They are important in tissue homeostasis and regeneration because of their immunomodulatory potential and release of trophic factors;106 however, direct infusion of expanded MSCs leads to entrapment in the lungs and rapid clearance by the spleen.107 Furthermore, several studies have shown that MSCs do not engraft after infusion; thus, the effect is only temporary.107 However, recent studies indicate that extracellular vesicles (EVs) are mainly responsible for the positive effects of MSCs.108 EVs are small endosome-derived lipid nanoparticles (40-500 nm) that play an important role in intercellular trafficking.109 Because of their very small size, they are interesting for the delivery of enzyme to avascular or blood-tissue barrier–isolated tissues. EVs can carry macromolecules, including enzymes, to adjacent cells.110 Although literature on EVs in metabolic diseases is scarce, EVs are known to contain lysosomal proteins.111 Furthermore, transplantation of MSCs in the cornea of MPS-3 and MPS-7 mice showed that cross-correction of the deficient cells was indeed mediated by EVs and corrected GAG accumulation.111,112 Thus, EVs from MSCs could potentially deliver enzyme to deficient cells.
Focused ultrasound therapy
In the past years, ultrasound contrast agents have been used in diagnostic ultrasound imaging.113 They can also carry drugs or genes to select targeted tissues and cells for treatment or transfection.114 The mechanism behind (focused) ultrasound therapy relies on the administration of microbubbles. These microbubbles are lipid- or protein-containing shells filled with a heavy gas. The use of ultrasonic pressure waves on microbubbles causes them to collapse, resulting in a local widening of endothelial tight junctions, called sonoporation.115 Sonoporation causes temporary disruption of, for example, the BBB.116 As a consequence, larger molecules up to 2000 kilodalton are able to pass through the BBB.117 The disturbance in the BBB leads to an increased drug concentration in the brain, as already demonstrated in several disease models, including the delivery of recombinant human IDUA in the case of MPS-1 mice.118
Ultrasound therapy has also been investigated to deliver drugs past the BRB. Using a rat model, Park et al119 demonstrated that burst ultrasound, together with a microbubble agent, was able to induce transient increases in retinal vascular permeability for retinal drug delivery in rats without any significant side effects. The BRB appeared to be restored within a few hours, which provided a suitable time window for ocular pharmaceutical agent delivery while avoiding undesired effects that may result from long-term BRB disruption. In light of this knowledge, focused ultrasound therapy could be especially interesting for cell therapies and GTs targeting the brain and retina, because it would create a 1-time temporary disruption of the BBB and BRB, which allows the product to enter the tissue of interest and, possibly, increase biodistribution.
Conclusions
ERT and HCT are not sufficient to cure MPS disorders. Therefore, the management of MPSs will most likely evolve from monotherapy into combination therapy. A combination of HCT or autologous gene-modified HCT with additional therapies, as mentioned above, could allow for successful and complete correction of the tissue deficiencies seen in MPS (Figure 4). We have summarized 2 important hurdles that explain disease progression in “hard-to-reach” tissues in MPS patients, avascularity and tissue-specific barriers; both lead to unavailability of enzyme. The majority of disease progression occurs in connective tissue because of its avascularity. Additionally, the presence of the BRB and BBB prevent current standard therapies from providing optimal correction. Because the 2 major hurdles presented in this review, avascularity and tissue-specific barriers, are not MPS specific, they can also be applied to other metabolic diseases in which systemic treatments are not sufficient to completely cure the disease. For example, HCT is attempted in, among other diseases, leukodystrophies, α-mannosidosis, fucosidosis, Niemann-Pick disease, Tay-Sachs disease, Pompe disease, and Gaucher disease with variable success.120 The experimental therapies mentioned in this review that try to overcome these hurdles might also be explored in these metabolic diseases to achieve a complete treatment of the underlying disease. For MPS, autologous gene-modified HCT appears to be the most promising; although it is likely to lower disease burden, is unlikely to result in a complete cure for MPS. Based on the nature and characteristics of the different MPS subtypes, each might require a different and unique treatment strategy for successful outcomes. Of note, because part of the damage already occurs in utero, some tissues might even be “out-of-reach.” Thus, newborn screening programs are extremely important to identify patients in an early disease state to prevent further irreversible damage. Finally, biomarkers related to the extent of residual disease or the secondary manifestations, such as impaired autophagy, increased apoptosis, and inflammation, are necessary to evaluate the effect of (new) therapies on “hard-to-reach” tissues, because in the end, “that’s where the money is.”
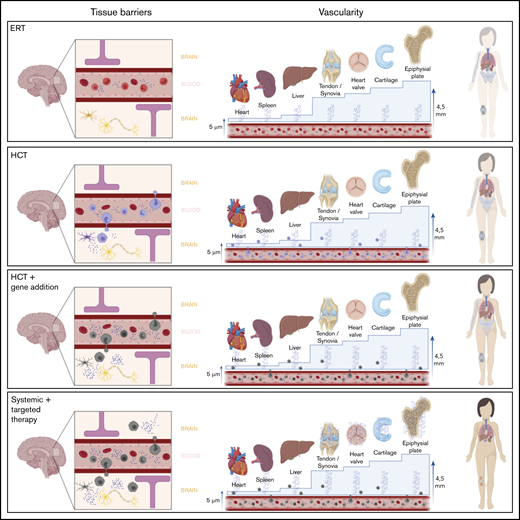
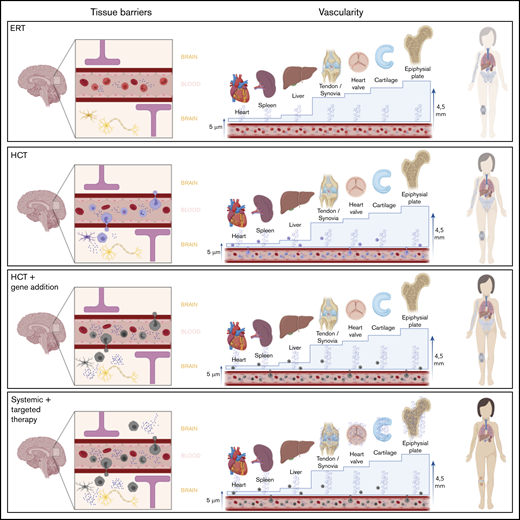
Overview of different therapy strategies on the 2 major hurdles in MPS. Patients treated with current standard treatments, ERT and HCT, still experience significant disease progression. Two major hurdles in the failure of these treatments in specific tissues are the presence of a physical barrier, such as the BBB or the BRB, and the vascularity of the tissue. Connective tissues, in which most disease progression is seen and which are avascular, are isolated from the circulation. Thus, they completely depend on diffusion of monocytes and free enzyme. For the epiphyseal plate for instance, the distance to overcome is 4.5 mm, which is too far; therefore, enzyme availability in the epiphyseal plate is still zero, despite treatment. Only increasing the available amount of enzyme in the circulation, like in the GT strategies, will most likely not solve this problem. Furthermore, it is unknown whether monocytes migrate past the BRB into the retina like it is assumed in the central nervous system. Progression of retinal degeneration insinuates that the available amount of enzyme is insufficient, at the least. Therefore, the route to cure MPS disease is to focus on increasing enzyme availability in these “hard-to-reach” tissues.
Overview of different therapy strategies on the 2 major hurdles in MPS. Patients treated with current standard treatments, ERT and HCT, still experience significant disease progression. Two major hurdles in the failure of these treatments in specific tissues are the presence of a physical barrier, such as the BBB or the BRB, and the vascularity of the tissue. Connective tissues, in which most disease progression is seen and which are avascular, are isolated from the circulation. Thus, they completely depend on diffusion of monocytes and free enzyme. For the epiphyseal plate for instance, the distance to overcome is 4.5 mm, which is too far; therefore, enzyme availability in the epiphyseal plate is still zero, despite treatment. Only increasing the available amount of enzyme in the circulation, like in the GT strategies, will most likely not solve this problem. Furthermore, it is unknown whether monocytes migrate past the BRB into the retina like it is assumed in the central nervous system. Progression of retinal degeneration insinuates that the available amount of enzyme is insufficient, at the least. Therefore, the route to cure MPS disease is to focus on increasing enzyme availability in these “hard-to-reach” tissues.
Data sharing requests should be sent to Peter M. van Hasselt (p.vanhasselt@umcutrecht.nl).
Acknowledgments
All figures were created with BioRender.com.
B.T.A.v.d.B. was supported by a research grant from the Sylvia Toth Charity Foundation (The Hague, The Netherlands) while working on this study.
B.T.A.v.d.B. is a PhD candidate at Utrecht University and this work is submitted in partial fulfillment of the requirements for the PhD.
The sponsors of this study are public or nonprofit organizations that support science in general. They had no role in gathering, analyzing, or interpreting the data.
Authorship
Contribution: B.T.A.v.d.B. conceptualized and designed the review and wrote the manuscript; J.v.D. and C.V.H. wrote and critically reviewed the manuscript; S.N. and J.J.B. designed the review and critically reviewed the manuscript; C.A.L. and N.V.-D. critically reviewed the manuscript; and P.M.v.H. conceptualized and designed the review, wrote the manuscript, and critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Peter M. van Hasselt, Department of Pediatric Metabolic Diseases, Wilhelmina Children’s Hospital, Internal mail number: KE.04.133.1, Lundlaan 6, 3584 EA Utrecht, The Netherlands; e-mail: p.vanhasselt@umcutrecht.nl.