Key Points
Virus-specific IgG levels were stable for up to 1 year after CD19-CARTx in adults with durable complete remission.
Preexisting humoral immunity may be preserved in adult recipients of CD19-CARTx.

Abstract
The long-term effects of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy (CD19-CARTx) for B-cell malignancies on humoral immunity are unclear. We examined antiviral humoral immunity in 39 adults with B-cell malignancies who achieved durable complete remission without additional therapy for >6 months after CD19-CARTx. Despite CD19+ B-cell aplasia in all patients, the incidence of viral infections occurring >90 days post–CD19-CARTx was low (0.91 infections per person-year). Because long-lived plasma cells are CD19− and should not be direct targets of CD19-targeted chimeric antigen receptor T cells, we tested the hypothesis that humoral immunity was preserved after CD19-CARTx based on linear mixed-effects models of changes in serum total immunoglobulin G (IgG) concentration, measles IgG concentration, and the number of viruses or viral epitopes to which serum IgG was directed (the “antivirome”) using the novel VirScan assay. Samples were tested pre–CD19-CARTx and ∼1, 6, and 12 months post–CD19-CARTx. Although total IgG concentration was lower post–CD19-CARTx (mean change, −17.5%), measles IgG concentration was similar (mean change, 1.2%). Only 1 participant lost measles seroprotection post–CD19-CARTx but had undergone allogeneic hematopoietic cell transplantation before CD19-CARTx. The antivirome was also preserved, with mean absolute losses of 0.3 viruses and 6 viral epitopes detected between pre- and post–CD19-CARTx samples. Most participants gained IgG to ≥2 epitopes for ≥2 viruses, suggesting that humoral immunity to some viruses may be maintained or recover after successful CD19-CARTx. These findings may differ in children. Studies of immunoglobulin replacement and vaccination after CARTx are warranted.
Introduction
CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy (CD19-CARTx) is a novel treatment for relapsed or refractory B-cell malignancies, including acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and non-Hodgkin lymphoma (NHL).1-12 CD19-CARTx targets cells expressing the B-cell lineage antigen CD19, which is present on malignant and nonmalignant B cells.6,7 However, terminally differentiated B cells, such as long-lived plasma cells (LLPCs), have low CD19 expression and may survive after lymphodepletion chemotherapy and CD19-CARTx.13-17 CD19-CARTx products are now commercially available for treatment of ALL and diffuse large B-cell lymphomas in children and adults,18-20 but the long-term effects of CD19-CARTx on humoral immunity are not well understood.
CD19-directed chimeric antigen receptor (CAR) T cells can persist in the recipient for years and cause prolonged depletion of normal CD19+ B cells.4-8,21 In light of the association between B-cell depletion and hypogammaglobulinemia,22,23 monthly monitoring of serum immunoglobulin G (IgG) with prophylactic IV immunoglobulin (IVIG) replacement for patients with IgG < 400 to 600 mg/dL is often recommended after CD19-CARTx.24 However, the effects of CD19-CARTx on serum IgG concentration and infection risk due to CD19+ B-cell depletion are unclear. LLPCs that produce antibodies to previously encountered pathogens may not be affected by CD19-CARTx.13,25,26 One study demonstrated stable pathogen-specific antibody titers in 2 participants and bone marrow LLPCs in 8 participants in complete remission (CR) for 1 to 2 years after CD19-CARTx.27 In persons treated with rituximab alone, a monoclonal antibody directed against CD20 that results in depletion of normal B-cell subsets similar to subsets expressing CD19, total and pathogen-specific IgG concentrations are preserved, and infection rates are not increased.25 However, the half-life of rituximab is ∼1 month, and hypogammaglobulinemia and increased infections may occur after rituximab treatment in the context of other risk factors.22,23 Prophylactic IVIG is approved in some immunocompromised populations based on studies demonstrating reduced rates of serious bacterial infections.28,29 However, IVIG is associated with substantial costs and toxicities, has not been shown to improve overall mortality, and is not routinely recommended after other cancer therapies.28-33 Thus, the role of prophylactic IVIG after CD19-CARTx is unclear.
We sought to examine the effects of CD19-CARTx on humoral immunity by evaluating the incidence of viral infections and changes in serum total IgG concentrations, measles-specific IgG concentrations, and the antiviral antibody repertoire in CD19-CARTx recipients who achieved a CR ≥6 months duration without additional antitumor therapy.
Methods
Participants and treatment characteristics
Participants in this study were adults ≥18 years old with relapsed or refractory CD19+ ALL, CLL, or NHL who achieved a CR >6 months duration in the absence of additional antitumor treatment after lymphodepletion chemotherapy and CD19-directed CAR T-cell infusion in a single institution clinical trial (https://clinicaltrials.gov/ct2/show/NCT01865617; Table 1; supplemental Methods).4 This study was approved by the Fred Hutchinson Cancer Research Center institutional review board, and all participants provided informed consent in accordance with the Declaration of Helsinki.
Supportive care and monitoring
The total serum IgG concentration was evaluated prior to lymphodepletion and approximately monthly after CAR T-cell infusion. IVIG (400 mg/kg) was recommended if the serum IgG concentration was <400 mg/dL. Neurotoxicity and cytokine release syndrome (CRS) were graded as previously described.34-36 Antimicrobial prophylaxis is detailed in supplemental Methods.
Infection categorization
We identified definite (microbiologically diagnosed) or clinical (nonmicrobiologically diagnosed) viral infections occurring >90 days post–CD19-CARTx, after the acute toxicities of CARTx waned. We categorized infections as nonserious (grades 1-2) or serious (grade 3), according to the Blood and Marrow Transplant Clinical Trials Network Manual of Procedures (version 3.0).37
CAR T-cell and CD19+ B-lymphocyte enumeration
We quantitated CAR T cells in peripheral blood using polymerase chain reaction (PCR) targeting a flap and elongation factor-1α (FlapEF1α) sequence in the vector. We determined the percentage of normal CD19+ B cells in the peripheral blood using immunophenotyping by flow cytometry after lysis of the erythroid cells.
Measles antibody testing
We measured measles IgG as a marker of humoral immunity given the high population prevalence of seropositivity, the stability of measles IgG over time in healthy individuals, and the low likelihood of boosting due to reactivation or a new infection.38 Serum samples collected prior to lymphodepletion and at ∼1, 6, and 12 months after CD19-CARTx were retrospectively batch tested for measles IgG using a US Food and Drug Administration–cleared indirect fluorescent antibody assay (Zeus Scientific Inc., Branchburg, NJ; supplemental Methods).
Systematic viral epitope scanning (VirScan) testing
We used a novel systematic viral epitope scanning method (VirScan) to perform a comprehensive serosurvey for IgG to viruses, as previously described.39,40 VirScan combines DNA microarray synthesis and bacteriophage display to create a synthetic representation of peptide epitopes covering >1000 strains of 206 viruses. We determined the number of unique viruses to which IgG was directed above a predefined seropositivity threshold and the number of viral epitopes recognized by IgG for these viruses (supplemental Methods).
Data collection and statistical considerations
We collected data from up to 90 days preceding CD19-CARTx until the time of new antitumor therapy for relapse or new malignancy, death, or last clinical contact post–CD19-CARTx. For most analyses, total or pathogen-specific IgG results were only included from samples obtained ≥16 weeks after IVIG (>4 half-lives of blood IgG), unless otherwise indicated.41,42
We computed cumulative incidences for the following end points after CD19-CARTx: (1) peripheral blood CD19+ B-cell detection (≥0.01% CD19+ normal B cells in blood leukocytes), (2) loss of CD19-directed CAR T cells (≤10 FlapEF1α copies per microgram of DNA in 2 consecutive tests), (3) IgG < 400 mg/dL after CD19-CARTx among participants with IgG ≥ 400 mg/dL pre–CD19-CARTx, and (4) viral infections occurring >90 days after CD19-CARTx (new antitumor therapy for relapse or new malignancy treated as competing risks). Additional details are in supplemental Methods.
We used a noninferiority design to test the hypotheses that post–CD19-CARTx metrics of humoral immunity were not lower than pre–CD19-CARTx metrics for the following outcomes: total serum IgG concentration, measles serum IgG concentration, and number of viruses or viral epitopes to which serum IgG antibodies were detected by VirScan. Based on power calculations (supplemental Methods; supplemental Table 1) and clinical relevance, we used the following noninferiority margins for each outcome: 20% mean change for total or measles IgG concentrations, and 2 viruses or 30 epitopes mean change (∼20%) for the number of viruses or viral epitopes to which IgG was detected by VirScan. We used linear mixed-effects models to compare changes in outcomes after CD19-CARTx and estimated changes within subgroups of age, underlying disease, and prior hematopoietic cell transplantation (HCT). Noninferiority (ie, the outcome was no lower in the post–CD19-CARTx period than in the pre–CD19-CARTx period) was recognized when the lower limit of the 95% confidence interval (CI) for the change estimates remained above the negative specified margin.
Results
Participant and treatment characteristics
Among 166 adults with CD19+ B-cell malignancies who received CD19-CARTx through March 2017, we identified 40 participants with CR ≥6 months without additional antitumor therapy; 1 participant was excluded because of insufficient records (Table 1). The median duration of follow-up prior to censoring was 508 days (range, 251-1067) after CD19-CARTx.
Serious viral infections were infrequent despite prolonged CD19+ B-cell aplasia after CD19-CARTx
In this cohort of 39 participants, 38 were tested for blood CD19+ B cells pre–CD19-CARTx; 20 (53%) of these participants had B-cell depletion (<0.01% CD19+ B cells) prior to CD19-CARTx. Among 35 participants tested in the first 28 days post–CD19-CARTx, 100% had CD19+ B-cell depletion. Among 38 participants with testing after day 28 post–CD19-CARTx, 17 (45%) had an increase in blood CD19+ B cells to levels ≥0.01%, which occurred at a median of 89 days (range, 57-275; Figure 1A). In all 39 participants, we detected blood CD19-directed CAR T cells by PCR within 28 days after infusion. Among 38 participants with subsequent testing, CD19-directed CAR T cells became undetectable in only 9 (24%) participants, which occurred at a median of 136 days (range, 71-655; Figure 1B). Additional details can be found in supplemental Table 2.
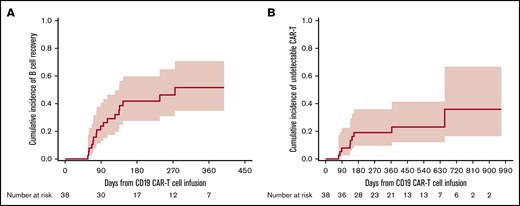
Cumulative incidence curves of time to CD19+normal B-cell detection and loss of CD19-directed CAR T-cell detection. (A) Cumulative incidence of peripheral blood normal CD19+ B-cell detection at a level ≥0.01% CD19+ normal B cells in blood leukocytes after day 28 post–CD19-CARTx. (B) Cumulative incidence of loss of peripheral blood CD19-directed CAR T-cell detection, defined as ≤10 FlapEF1α per microgram of DNA on 2 consecutive tests, as detected by quantitative PCR after day 28 post–CD19-CARTx. Participants were censored at the last peripheral blood measurement. The shaded area represents the upper and lower limits of the 95% CIs.
Cumulative incidence curves of time to CD19+normal B-cell detection and loss of CD19-directed CAR T-cell detection. (A) Cumulative incidence of peripheral blood normal CD19+ B-cell detection at a level ≥0.01% CD19+ normal B cells in blood leukocytes after day 28 post–CD19-CARTx. (B) Cumulative incidence of loss of peripheral blood CD19-directed CAR T-cell detection, defined as ≤10 FlapEF1α per microgram of DNA on 2 consecutive tests, as detected by quantitative PCR after day 28 post–CD19-CARTx. Participants were censored at the last peripheral blood measurement. The shaded area represents the upper and lower limits of the 95% CIs.
Despite prolonged CD19+ B-cell depletion and CD19-directed CAR T-cell persistence in most participants, the incidence of serious viral infections >90 days after CD19-CARTx in all 39 participants was low. There were only 2 serious viral infections in 1 participant; rhinovirus was detected in the lower respiratory tract in the context of 2 episodes of bacterial pneumonia, but neither required hospitalization. The median number of any clinically or microbiologically confirmed viral infection (supplemental Table 3) was 1 per participant (interquartile range, 0-2), with an incidence rate of 0.91 viral infections per person-year. Significant cytopenias beyond day 90 occurred in 4 participants, and 1 of these individuals was the participant with serious viral infections of the lower respiratory tract (supplemental Results). Viral infection rates were similar prior to and after CD19+ B-cell detection. Furthermore, serum IgG levels were similar in participants with and without viral infections, and the incidence of viral infections was not higher in patients not receiving IVIG (supplemental Results; supplemental Figure 1).
Total serum IgG concentration was lower after CD19-CARTx
Based on our observations of few serious viral infections, we were interested in understanding the direct effect of CARTx on serum total IgG. In 38 participants with an IgG result within 90 days pre–CD19-CARTx, 11 (29%) had a total IgG concentration <400 mg/dL. Among all 39 participants, 22 (56%) had a total IgG concentration <400 mg/dL at any time post–CD19-CARTx. Eighteen participants (46%) never received IVIG post–CD19-CARTx, 7 of whom had ≥1 IgG level <400 mg/dL. Among 65 instances of IgG measurements < 400 mg/dL in 23 subjects, IVIG was administered within 28 days in 55% of instances in 14 (61%) participants. After excluding IgG levels obtained within 16 weeks of IVIG administration, 162 IgG levels were available from 35 participants. Only 4 participants were excluded using this strategy, and the timing of included vs excluded samples was similar (supplemental Table 2). An evaluation of the temporal effect of IVIG on IgG levels demonstrated that our approach effectively mitigated the impact of IVIG on total IgG (supplemental Figure 2). Total serum IgG levels over time are shown in Figure 2A (excluding values ≤16 weeks after IVIG) and supplemental Figure 3 (all results).

Change in serum total IgG concentrations after CD19-CARTx. (A) Spaghetti plot of serum total IgG concentrations before and after CD19-CARTx; 162 total IgG measurements were obtained for 35 participants. Each line represents 1 participant. A dashed reference line is shown at 400 mg/dL, the concentration below which IVIG was recommended. Data from samples obtained ≤16 weeks after IVIG administration were excluded. (B) Forest plot showing linear mixed-effects model estimates for the mean change in serum total IgG concentrations. Exploratory subgroup estimates were computed from separate models, including the subgroup variable, the time period (post- vs pre–CD19-CARTx), and their interaction. For underlying disease, interaction P = .04. Filled squares represent the change estimates, and bars indicate the 95% CI. Dashed vertical reference lines are shown at our prespecified noninferiority margin and at no change. LCL, lower confidence limit of the 95% CI; UCL, upper confidence limit of the 95% CI.
Change in serum total IgG concentrations after CD19-CARTx. (A) Spaghetti plot of serum total IgG concentrations before and after CD19-CARTx; 162 total IgG measurements were obtained for 35 participants. Each line represents 1 participant. A dashed reference line is shown at 400 mg/dL, the concentration below which IVIG was recommended. Data from samples obtained ≤16 weeks after IVIG administration were excluded. (B) Forest plot showing linear mixed-effects model estimates for the mean change in serum total IgG concentrations. Exploratory subgroup estimates were computed from separate models, including the subgroup variable, the time period (post- vs pre–CD19-CARTx), and their interaction. For underlying disease, interaction P = .04. Filled squares represent the change estimates, and bars indicate the 95% CI. Dashed vertical reference lines are shown at our prespecified noninferiority margin and at no change. LCL, lower confidence limit of the 95% CI; UCL, upper confidence limit of the 95% CI.
In linear mixed-effects models including 35 participants with IgG results >16 weeks after IVIG, mean concentrations of IgG post–CD19-CARTx (469 mg/dL) were lower than mean concentrations of IgG pre–CD19-CARTx (569 mg/dL; mean difference, −17.5%; 95% CI, −25.2 to −9.1; Figure 2B). These findings were similar among subgroups defined by age and prior HCT. The largest decrease in IgG concentrations occurred in participants with ALL, and the smallest decrease was noted in participants with CLL (Figure 2B), although participants with CLL had the lowest mean IgG levels pre–CD19-CARTx (Table 1). Among 22 participants with IgG concentrations ≥400 mg/dL pre–CD19-CARTx, the cumulative incidence of an IgG concentration <400 mg/dL by 3 and 12 months post–CD19-CARTx was 36% and 60%, respectively (Figure 3A), with a mean decrease of 272 mg/dL (range, 40-1175 mg/dL) from pre–CD19-CARTx to first IgG <400 mg/dL. These findings were similar by age and prior HCT but may vary by underlying disease (Figure 3B-D).

Cumulative incidence of time to serum total IgG <400 mg/dL post–CD19-CARTx among participants with IgG ≥400 mg/dL pre–CD19-CARTx. This figure includes 22 participants with IgG concentrations ≥400 mg/dL pre–CD19-CARTx who also had IgG measurements post–CD19-CARTx. (A) The overall cumulative incidence of IgG <400 mg/dL by day 365 was 60% (95% CI, 38-82). The shaded area represents the upper and lower limits of the 95% CIs. The cumulative incidence of IgG <400 mg/dL varied by underlying disease (P = .06) (B) but was similar by age (P = .8) (C) and prior HCT (P = .9) (D).
Cumulative incidence of time to serum total IgG <400 mg/dL post–CD19-CARTx among participants with IgG ≥400 mg/dL pre–CD19-CARTx. This figure includes 22 participants with IgG concentrations ≥400 mg/dL pre–CD19-CARTx who also had IgG measurements post–CD19-CARTx. (A) The overall cumulative incidence of IgG <400 mg/dL by day 365 was 60% (95% CI, 38-82). The shaded area represents the upper and lower limits of the 95% CIs. The cumulative incidence of IgG <400 mg/dL varied by underlying disease (P = .06) (B) but was similar by age (P = .8) (C) and prior HCT (P = .9) (D).
We considered that a decline in IgG might correlate with the duration of CD19+ B-cell aplasia. We compared the decrease in mean total IgG concentrations in the early (1-100 days) or late (>100 days) time periods after CD19-CARTx, which revealed an initial decline that was subsequently stable (supplemental Table 4). There was no evidence of an effect of CD19+ B-cell aplasia duration on mean total IgG (supplemental Figure 4A), and mean serum total IgG levels were similar before and after blood CD19+ B-cell detection ≥0.01% (supplemental Figure 5). Furthermore, there were no clear differences between mean total IgG levels pre–CD19-CARTx and after peak blood CD19-directed CAR T-cell counts (supplemental Figure 6A).
These data demonstrate that hypogammaglobulinemia is common pre–CD19-CARTx, there is a modest decline (100 mg/dL) in mean serum total IgG concentrations after CD19-CARTx occurring within the first 100 days, and this does not vary by duration of CD19+ B-cell depletion.
Measles IgG concentrations were stable after CD19-CARTx
Given that we found a modest decline in total serum IgG levels after CD19-CARTx, we were interested in changes in pathogen-specific IgG levels after CD19-CARTx. We analyzed measles IgG concentrations in 87 samples from 30 participants (supplemental Table 2). Eight (27%) participants had a negative (n = 7) or indeterminate (n = 1) measles IgG level pre–CD19-CARTx; 7 of these participants had undergone HCT before CD19-CARTx (allogeneic, n = 4; autologous, n = 2; both, n = 1). Among the remaining 22 participants with a seroprotective measles IgG level pre–CD19-CARTx (Figure 4A), only 1 (4.5%) had a negative measles IgG level ∼1-year post–CD19-CARTx. This participant had received an autologous and allogeneic HCT before CD19-CARTx without revaccination. One participant had decreasing antibody concentrations after CD19-CARTx, suggesting possible impending seroreversion. An additional participant who received a prior autologous HCT without measles revaccination had a low-positive measles IgG concentration pre–CD19-CARTx that decreased to an indeterminate level post–CD19-CARTx.

Change in serum measles IgG concentrations after CD19-CARTx. (A) Spaghetti plot of serum measles IgG concentrations before and after CD19-CARTx; 63 measles IgG measurements were obtained for 22 participants. Each line represents 1 participant. A dashed reference line is shown at 110 mIU/mL, the concentration above which an individual is considered seroprotected for measles. Negative and indeterminate results were based on values ≤90 mIU/mL and values between 91 and 109 mIU/mL, respectively. (B) Forest plot showing linear mixed-effects model estimates for the mean change in serum measles IgG concentrations. Exploratory subgroup estimates were computed from separate models, including the subgroup variable, the time period (post– vs pre–CD19-CARTx), and their interaction. Underlying disease was not included because of the limited sample size for each disease group. Additional data are presented from models excluding 1 outlier who seroreverted (blue boxes). In the model excluding the participant who seroreverted, participants without a prior HCT had a greater decrement in measles IgG concentrations than did those with a prior HCT (interaction P = .009). Squares represent the change estimates, and bars indicate the 95% CIs. Dashed vertical reference lines are shown at our prespecified noninferiority margin and at no change. For both panels, we excluded data from samples obtained ≤16 weeks after IVIG administration and from participants lacking seroprotective measles IgG pre–CD19-CARTx (n = 8). LCL, lower confidence limit of the 95% CI; UCL, upper confidence limit of the 95% CI.
Change in serum measles IgG concentrations after CD19-CARTx. (A) Spaghetti plot of serum measles IgG concentrations before and after CD19-CARTx; 63 measles IgG measurements were obtained for 22 participants. Each line represents 1 participant. A dashed reference line is shown at 110 mIU/mL, the concentration above which an individual is considered seroprotected for measles. Negative and indeterminate results were based on values ≤90 mIU/mL and values between 91 and 109 mIU/mL, respectively. (B) Forest plot showing linear mixed-effects model estimates for the mean change in serum measles IgG concentrations. Exploratory subgroup estimates were computed from separate models, including the subgroup variable, the time period (post– vs pre–CD19-CARTx), and their interaction. Underlying disease was not included because of the limited sample size for each disease group. Additional data are presented from models excluding 1 outlier who seroreverted (blue boxes). In the model excluding the participant who seroreverted, participants without a prior HCT had a greater decrement in measles IgG concentrations than did those with a prior HCT (interaction P = .009). Squares represent the change estimates, and bars indicate the 95% CIs. Dashed vertical reference lines are shown at our prespecified noninferiority margin and at no change. For both panels, we excluded data from samples obtained ≤16 weeks after IVIG administration and from participants lacking seroprotective measles IgG pre–CD19-CARTx (n = 8). LCL, lower confidence limit of the 95% CI; UCL, upper confidence limit of the 95% CI.
The mean measles IgG concentration post–CD19-CARTx (335 mIU/mL) was similar to the mean concentration pre–CD19-CARTx (353 mIU/mL) in linear mixed-effects models including all patients with seroprotection pre–CD19-CARTx (mean difference, −7.4%; 95% CI, −26.0 to 16.0%; Figure 4B). The 95% CIs included our noninferiority margin and a difference of 0%, indicating inconclusive results (neither lower nor noninferior). In an exploratory analysis excluding the participant who seroreverted after CD19-CARTx but had prior autologous and allogeneic HCTs, the estimated mean change was 1.2% (95% CI, −7.9 to 11.1; Figure 4B), which met the noninferiority criteria. These findings were similar among participants stratified by age, but participants without a prior HCT had a greater decrement in measles IgG concentrations compared with those with a prior HCT (Figure 4B). When stratified by early and late time periods post–CD19-CARTx, no significant mean change in measles IgG was seen in either time period (supplemental Table 4). There was no evidence of an effect of CD19+ B-cell depletion duration on mean measles IgG levels (supplemental Figure 4B), and levels were similar after CD19+ B-cell detection (supplemental Figure 5), after peak blood CD19-directed CAR T-cell counts (supplemental Figure 6B), and in individuals with hypogammaglobulinemia (supplemental Figure 7).
These data demonstrate that pathogen-specific immunity, using measles IgG for proof of concept, may be preserved after CD19-CARTx. However, patients who underwent HCT before CD19-CARTx may have substantial deficits in preexisting immunity.
Antibody reactivity to the human virome was preserved after CD19-CARTx
The preservation of measles IgG levels in most participants after CD19-CARTx suggested that preexisting humoral IgG-mediated immunity to other viruses might also remain intact after CD19-CARTx. We used the VirScan assay to evaluate the number of unique viruses to which antibody responses were directed above a predefined seropositivity threshold, along with the number of viral epitopes for these viruses, in serum samples from the 30 participants who underwent measles IgG testing (supplemental Table 2). In pre– and post–CD19-CARTx samples, we detected qualifying antibody responses to 34 viruses, primarily herpesviruses and respiratory viruses (Figure 5A). Pre–CD19-CARTx, we detected a median of 10 viruses above the seropositivity threshold (range, 1-19) and 144 epitopes for these viruses (range, 49-241). These findings were similar in post–CD19-CARTx samples, with a median of 10 viruses (range, 1-17) and 139 epitopes (range, 32-258; Figure 5B).

Viruses and viral epitopes to which serum IgG antibodies were directed based on the VirScan assay. This figure includes data for 28 participants with serum sample results from both the pre– and post–CD19-CARTx time periods; 2 participants who did not have data for both periods were excluded. (A) Each column depicts whether a participant had IgG for a given virus (indicated in rows) in the pre–CD19-CARTx (light shading) or last post–CD19-CARTx (dark shading) sample; participants are grouped by prior HCT status. (B) Each column depicts the number of epitopes to which IgG antibodies were directed in a participant for a given virus (indicated in rows) in the pre–CD19-CARTx or last post–CD19-CARTx sample; shading is based on a heat map for the number of epitopes, and participants are grouped by prior HCT status. Forest plots showing linear mixed-effects model estimates for the mean change in the total number of viruses (C) and epitopes (D) recognized by IgG. Exploratory subgroup estimates were computed from separate models, including the subgroup variable, the time period (post– vs pre–CD19-CARTx), and their interaction. Participants without a prior HCT had a greater decrement in the total virus and epitope IgG (C-D) compared with participants with a prior HCT, although the differences did not reach statistical significance (interaction P = .07 and .12, respectively). Squares represent the change estimates, and bars indicate the 95% CIs. Dashed vertical reference lines are shown at our prespecified noninferiority margins and at no change.
Viruses and viral epitopes to which serum IgG antibodies were directed based on the VirScan assay. This figure includes data for 28 participants with serum sample results from both the pre– and post–CD19-CARTx time periods; 2 participants who did not have data for both periods were excluded. (A) Each column depicts whether a participant had IgG for a given virus (indicated in rows) in the pre–CD19-CARTx (light shading) or last post–CD19-CARTx (dark shading) sample; participants are grouped by prior HCT status. (B) Each column depicts the number of epitopes to which IgG antibodies were directed in a participant for a given virus (indicated in rows) in the pre–CD19-CARTx or last post–CD19-CARTx sample; shading is based on a heat map for the number of epitopes, and participants are grouped by prior HCT status. Forest plots showing linear mixed-effects model estimates for the mean change in the total number of viruses (C) and epitopes (D) recognized by IgG. Exploratory subgroup estimates were computed from separate models, including the subgroup variable, the time period (post– vs pre–CD19-CARTx), and their interaction. Participants without a prior HCT had a greater decrement in the total virus and epitope IgG (C-D) compared with participants with a prior HCT, although the differences did not reach statistical significance (interaction P = .07 and .12, respectively). Squares represent the change estimates, and bars indicate the 95% CIs. Dashed vertical reference lines are shown at our prespecified noninferiority margins and at no change.
The mean number of detected viruses and epitopes were similar post–CD19-CARTx compared with pre–CD19-CARTx in linear mixed-effects models (mean differences, −0.3 viruses and −6 epitopes; 95% CIs, −1 to 0.5 and −17 to 5.5, respectively) and met the noninferiority criteria (Figure 5C-D). These findings were similar in models stratified by age and underlying disease. Participants who received an HCT prior to CD19-CARTx had a lower mean number of viruses and epitopes detected pre–CD19-CARTx compared with participants who had not received a prior HCT (9.3 vs 11.5 viruses and 142 vs 155 epitopes, respectively; P < .01), and the change in the mean number of detected viruses and epitopes after CD19-CARTx was smaller in participants with a history of HCT (Figure 5C-D). When stratified by early and late time periods post–CD19-CARTx, no significant change in the mean number of viruses or epitopes remained for either time period (supplemental Table 4). The number of viruses with low or high epitope losses or gains post–CD19-CARTx did not vary substantially overall, when stratified by pre–CD19-CARTx HCT status or post–CD19-CARTx periods of CD19+ B-cell aplasia (supplemental Table 5), or in individuals with hypogammaglobulinemia (supplemental Figure 8).
Together, these findings demonstrate substantial variability in the breadth of the antivirome per participant, with a lower mean number of detected viruses and epitopes in participants who underwent HCT prior to CD19-CARTx. However, there did not appear to be an effect of CD19-CARTx or CD19+ B-cell detection on these outcomes.
New antiviral IgG was identified after CD19-CARTx
Given that we did not find substantial changes in measles IgG levels or the net change in the antivirome after CD19-CARTx, we wanted to determine whether CD19-CARTx recipients recovered virus-specific humoral immunity. Among the 8 participants lacking seroprotective measles IgG levels pre–CD19-CARTx, 1 participant recovered a seroprotective measles IgG level 114 days post–CD19-CARTx without vaccination but in conjunction with blood CD19+ B-cell detection on day 90 (4.5% of blood leukocytes, 414 cells per microliter). Among the 28 subjects with VirScan results pre- and post–CD19-CARTx, the virus and epitope-specific profiles remained similar on a per-participant basis in 23 (82%) and 16 (57%) participants, respectively (McNemar’s test of homogeneity, P > .05). We next evaluated the number of gains and losses of IgG to specific viruses and epitopes. New antibody responses to previously unrecognized viruses occurred in 19/28 patients (median, 1 virus/patient), and most participants gained IgG to ≥2 epitopes for ≥2 viruses (Figures 5 and 6). These data suggest that CD19-CARTx recipients may be able to recover or generate antibody responses to viral infections.

The net change in viral epitopes to which serum IgG antibodies were directed. For each participant depicted in a column, the panels are colored according to the change in the number of epitopes (range indicated in the legend) recognized by IgG for a given virus (indicated in rows) between the pre– and last post–CD19-CARTx samples. The participants are grouped by prior HCT status. Empty cells indicate that the participant did not have any IgG directed against an epitope for a given virus in the pre– and post–CD19-CARTx samples or that there was only a change of 1 epitope recognition. This figure includes data for 28 participants with serum sample results from both the pre– and post–CD19-CARTx time periods; 2 participants who did not have data for both periods were excluded. Participant 8 had the greatest losses in IgG responses to specific viruses and viral epitopes. There were no distinct clinical features of this individual who had NHL, did not receive a prior HCT, received dose level 2 of CAR T cells with cyclophosphamide and fludarabine conditioning, had grade 1 CRS, never received IVIG, never had a total IgG concentration <400 mg/dL, and had 1 mild upper respiratory tract infection after CARTx.
The net change in viral epitopes to which serum IgG antibodies were directed. For each participant depicted in a column, the panels are colored according to the change in the number of epitopes (range indicated in the legend) recognized by IgG for a given virus (indicated in rows) between the pre– and last post–CD19-CARTx samples. The participants are grouped by prior HCT status. Empty cells indicate that the participant did not have any IgG directed against an epitope for a given virus in the pre– and post–CD19-CARTx samples or that there was only a change of 1 epitope recognition. This figure includes data for 28 participants with serum sample results from both the pre– and post–CD19-CARTx time periods; 2 participants who did not have data for both periods were excluded. Participant 8 had the greatest losses in IgG responses to specific viruses and viral epitopes. There were no distinct clinical features of this individual who had NHL, did not receive a prior HCT, received dose level 2 of CAR T cells with cyclophosphamide and fludarabine conditioning, had grade 1 CRS, never received IVIG, never had a total IgG concentration <400 mg/dL, and had 1 mild upper respiratory tract infection after CARTx.
Discussion
The development of hypogammaglobulinemia and potential for infections associated with humoral immunodeficiency are emphasized prior to CD19-CARTx. However, the direct impact of CD19-CARTx on these outcomes is poorly described. In this cohort of 39 adults with relapsed or refractory ALL, CLL, or NHL with complete responses for >6 months after CD19-CARTx, we demonstrated a modest decrement in serum total IgG concentrations but preservation of virus-specific antibody concentrations. These findings support the hypothesis that CD19-CARTx may have limited impact on preexisting humoral immunity in adults for up to 1 year after CD19-CARTx and may challenge current treatment paradigms of prophylactic IVIG. However, these findings may differ in children because of fewer established plasma cell clones.
Despite CD19+ B-cell depletion, viral infections occurred with an incidence rate of 0.91 per person-year, which is similar to that observed earlier after CARTx and in healthy adults43,44 but may be underestimated in this retrospective study. We observed only 2 serious viral infections involving the lower respiratory tract in a single patient with cytopenias and a concurrent bacterial pneumonia. The majority of viral infections involved the upper respiratory tract where the role of prophylactic IVIG is unclear,45 and exploratory analyses suggested that infection was not more frequent in the subgroup of patients not receiving IVIG.
We analyzed longitudinal changes in serum total IgG after CD19-CARTx. Hypogammaglobulinemia was common pre–CD19-CARTx. After CD19-CARTx, there was an estimated 17.5% mean decrement in IgG (corresponding to −100 mg/dL) that occurred in the first 100 days and remained stable, although the clinical significance of this decrease is unclear.
Despite the decrease in total IgG, there was no decrease in virus-specific IgG based on stable mean quantitative measles IgG levels, as well as stable numbers of viruses and viral epitopes recognized by IgG using the VirScan assay. Importantly, 27% of tested participants were not seroprotected for measles pre–CD19-CARTx, underscoring the extent of preexisting humoral immune deficits. Only 1 participant lost a seroprotective measles IgG level post–CD19-CARTx, although this individual received autologous and allogeneic HCTs prior to CD19-CARTx without revaccination. Interestingly, this participant had a concomitant increase in serum total IgG after CD19-CARTx (open green squares, Figures 2A and 4A), supporting a lack of correlation between total and pathogen-specific IgG.13,46 VirScan findings after kidney transplantation were similar to our data.47
We analyzed the associations of age, underlying malignancy, HCT pre–CD19-CARTx, blood CD19+ B detection, and peak CD19-directed CAR T-cell concentrations with total and virus-specific IgG concentrations. The receipt of an HCT prior to CD19-CARTx was the only factor associated with differences in virus-specific IgG. Participants who had an HCT pre–CD19-CARTx had lower virus-specific immunity but a smaller decrease post–CD19-CARTx, which may be due to the lower starting point. Together, our data support the hypothesis that a CD19− population of LLPCs may persist after CD19-CARTx and produce IgG.
Whether patients can generate immune responses to infections after CD19-CARTx has not been described. We found that 45% of participants had new detection of ≥0.01% normal CD19+ B cells in blood after CD19-CARTx, demonstrating evidence of at least partial CD19+ B-cell reconstitution. Other groups have shown recovery of polyclonal B cells without relapse after CD19-CARTx,48 which likely occurs earlier in marrow and lymphoid tissues. VirScan results demonstrated that patients may generate new antibodies after CD19-CARTx, which is supported by recovery of a seroprotective measles IgG level, without revaccination, concurrent with blood detection of CD19+ B cells in 1 individual. It remains uncertain whether our findings represent de novo responses or expansion of existing B-cell clones, and the findings are hypothesis generating. Although prophylactic IVIG is approved to reduce serious bacterial infections in patients with primary immunodeficiencies,49 routine use after other cancer therapies, including HCT, is being reconsidered based on limited benefit, the possibility of harm, and high costs.30 Vaccination may be a more cost-effective and durable approach to protect patients from infections after CD19-CARTx. Studies of vaccination strategies are critical in this population, particularly among children and patients who received a prior HCT,46 and this is underscored by the current measles outbreak.50 As CARTx products evolve, it will be important to define effects on immunity. For example, CARTx targeting B-cell maturation antigen51 received US Food and Drug Administration breakthrough designation in 2017 for adults with multiple myeloma. In contrast to CD19, B-cell maturation antigen is selectively expressed by LLPCs52 and may have different effects on humoral immunity.
Strengths of this study include longitudinal data and sample collection over a median of 508 days in a unique cohort of CD19-CARTx recipients with sustained CR. This allowed time to detect an effect of CD19-CARTx on serum IgG concentrations, given an estimated IgG half-life of 20 to 30 days.41,42 IgG levels may decline with longer follow-up, but LLPCs can live for decades, even in the absence of regeneration by B cells.38 We excluded results obtained within 16 weeks of IVIG administration (>4 half-lives) to mitigate the influence of exogenous IgG. We recognize that exclusion of participants receiving frequent IVIG may bias our analysis to less immunocompromised participants, but this affected a minority of the cohort, and the timing of included vs excluded samples was similar. Additionally, the linear mixed-effects models allowed us to maximize patient inclusion by incorporating data from subjects with single time points. We leveraged the VirScan assay to obtain a comprehensive understanding of the impact of CD19-CARTx on the antivirome. VirScan has high sensitivity and specificity for a range of viruses,39,40,47 but its performance for all viruses and correlation with clinical outcomes are unknown. Additionally, VirScan does not detect IgG to certain viruses and is nonquantitative except in the breadth of detected epitopes. Conclusions from subgroup analyses were limited by sample size, and this study was not powered to compare viral infection rates or severity with other populations. Studies in pediatric patients will be important, because they may have fewer plasma cell clones with limited preestablished immunity.53
In conclusion, we demonstrated that, in patients who achieved durable remission not requiring additional antitumor therapy, CD19-CARTx did not profoundly impact preexisting antiviral IgG concentrations, and serious viral infections were infrequent >90 days after treatment. The utility and cost-effectiveness of prophylactic IVIG after CD19-CARTx are unclear. Randomized trials of the utility of prophylactic IVIG and vaccination will be important to establish cost-effective approaches to protect patients after B-cell–targeted CAR T-cell therapies. These and other data provide clinical equipoise to perform such studies.
Acknowledgments
The authors thank the staff of the Fred Hutchinson Cancer Research Center Program in Immunology and Therapeutic Products Program and the Bezos Family Immunotherapy Clinic. They also acknowledge the Elledge Laboratory (Harvard Medical School, Boston, MA) for providing the VirScan library.
This study was supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases grant K23 AI119133, National Institutes of Health, National Cancer Institute grants R01 CA136551 and R35 CA197734, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant P30 DK56465, National Institutes of Health, National Cancer Institute grant P30 CA15704, the Life Science Discovery Fund, the Bezos family, the University of British Columbia Clinical Investigator Program, the National Institutes of Health, National Heart, Lung, and Blood Institute–funded National Gene Vector Biorepository at Indiana University (contract 75N92019D00018), and Juno Therapeutics, a Celgene company.
Authorship
Contribution: J.A.H., E.M.K., K.A.H., S.D., and C.J.T. designed the study; J.A.H., E.M.K., K.A.H., S.D., R.A.B.I., M.J.B., and C.J.T. interpreted the data; J.A.H., E.M.K., S.D., T.S.-A., and G.P. analyzed the data and created the figures; J.A.H., K.A.H., S.C., X.C., S.R.R., J.M., M.B., and D.G.M. collected data; J.A.H. drafted the initial manuscript with help from E.M.K; and all authors contributed to the writing and revision of the manuscript and approved the final version.
Conflict-of-interest disclosure: J.A.H. has served as a consultant for Nohla Therapeutics and Amplyx and has received research support from Nohla Therapeutics, Karius, and Takeda (formerly Shire), all unrelated to this research. K.A.H. has served as a consultant for Celgene. M.J.B. has served as a consultant and received research support from Merck Research Laboratories, Chimerix, Glaxo Smith Kline, Gilead Sciences, Vir Biotechnology, Ansun BioPharma, and Takeda (formerly Shire), all unrelated to this research. D.G.M. and S.R.R. received research funding from Juno Therapeutics and hold patents. C.J.T. receives research funding from and has patents licensed or pending with Juno Therapeutics, a Celgene company, and Nektar Therapeutics; has served on advisory boards and has equity in Caribou Biosciences, Eureka Therapeutics, and Precision Biosciences; and has served on advisory boards for Aptevo Therapeutics, bluebird bio, Adaptive Biotechnologies, Juno Therapeutics, a Celgene company, Kite, a Gilead Company, Humanigen, Nektar Therapeutics, Novartis, T-CURX, and Allogene Therapeutics. S.R.R. is a cofounder of Juno Therapeutics. E.M.K. has received research funding from Global Life Technologies. The remaining authors declare no competing financial interests.
Correspondence: Joshua A. Hill, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Mail Stop E-400, Seattle, WA 98109; e-mail: jahill3@fredhutch.org.