

Key Points
Preinfusion dim CD19 expression and rare CD19– events in B-ALL do not affect relapses or responses to CD19-directed CAR T-cells.
Prior blinatumomab treatment increases the rate of failure to achieve MRD– remission and CD19– MRD and relapse.

Abstract
Tisagenlecleucel, a chimeric antigen receptor (CAR) T-cell product targeting CD19 is approved for relapsed/refractory B-cell acute lymphoblastic leukemia (B-ALL). However, the impact of pretreatment variables, such as CD19 expression level, on leukemic blasts, the presence of CD19– subpopulations, and especially prior CD19-targeted therapy, on the response to CAR T-cell therapy has not been determined. We analyzed 166 patients treated with CAR T-cell therapy at our institution. Eleven patients did not achieve a minimal residual disease (MRD)– deep remission, whereas 67 patients had a recurrence after achieving a MRD– deep remission: 28 patients with CD19+ leukemia and 39 patients with CD19– leukemia. Return of CD19+ leukemia was associated with loss of CAR T-cell function, whereas CD19– leukemia was associated with continued CAR T-cell function. There were no significant differences in efficacy of CAR T cells in CD19-dim B-ALL, compared with CD19-normal or -bright B-ALL. Consistent with this, CAR T cells recognized and lysed cells with very low levels of CD19 expression in vitro. The presence of dim CD19 or rare CD19– events by flow cytometry did not predict nonresponse or recurrence after CAR T-cell therapy. However, prior therapy with the CD19-directed, bispecific T-cell engager blinatumomab was associated with a significantly higher rate of failure to achieve MRD– remission or subsequent loss of remission with antigen escape. Finally, immunophenotypic heterogeneity and lineage plasticity were independent of underlying clonotype and cytogenetic abnormalities.
Introduction
CD19 is a key B-cell lineage marker that is expressed almost universally on newly diagnosed B-cell acute lymphoblastic leukemia (B-ALL). CD19-targeted immunotherapies induce high response rates (complete remission: 34%-92%) in relapsed/refractory B-ALL, when compared with salvage chemotherapy.1-3 Tisagenlecleucel and blinatumomab are both CD19-targeting immunotherapies that are commercially available in the United States and other countries.4 Tisagenlecleucel is a chimeric antigen receptor (CAR)–modified autologous T-cell product that targets CD19, whereas blinatumomab is a bispecific, T-cell–engaging protein that binds both CD3 and CD19. Although the initial response rate for CAR T-cell therapy is 82% to 94%, long-term responses are impacted by relapses.5 CD19+ relapses are thought to be related to poor persistence and/or function of CAR T cells. CD19– relapses are associated with abnormalities in CD19 gene function and expression.6,7 However, it is not clear whether CD19– relapses arise from preexisting CD19– blasts present at the time of infusion or they occur de novo under treatment pressure.
Our prior work revealed the heterogeneity of CD19 expression in both de novo and relapsed B-ALL.8 Although most B-ALL showed normal to bright expression of CD19, a subset of cases had dim CD19 expression without exposure to any CD19-targeted therapy.8 It is unknown whether B-ALL with dim CD19 expression will respond as well to CAR T-cell therapy as does B-ALL with bright CD19 expression. Although no cases of de novo and/or relapsed B-ALL were completely negative for CD19 in our prior study,8 abnormalities have been found in CD19 after blinatumomab therapy.9-12 Therefore, it is also not clear whether prior blinatumomab therapy affects responses to subsequent CD19-directed CAR T-cell therapy.13
We addressed these questions in a large single-institution cohort of B-ALL patients treated with CD19-directed CAR T-cell therapy. We analyzed the impact of CD19 expression, the presence of CD19– blasts, and prior exposure to blinatumomab on response to CAR T-cell therapy.
Methods
Immunophenotypic analysis of patients infused with CAR T cells
Consecutive cases of B-ALL treated with CD19-directed CAR T-cell therapy and evaluable for response from April 2012 through December 2017 at the Children’s Hospital of Philadelphia (CHOP) were identified from the pathology archives in a retrospective study approved by the CHOP institutional review board. All the patients received a CAR T-cell product with a single-chain variable fragment directed against CD19, CD8a hinge, 4-1BB costimulatory domain, and CD3-ζ signaling domain. Outcomes in a subset (n = 34) of these patients have been reported as part of prior studies.1,5 Patients who previously received CAR T-cell therapy were excluded from the analysis. Flow cytometric data from diagnosis, relapse, and postlymphodepletion pre-CAR and post-CAR time points (1, 3, 6, 9, and 12 months and any relapses) were analyzed and correlations sought with laboratory, radiological, and follow-up data from the electronic medical record. For the purposes of this analysis, deep response was defined as minimal residual disease (MRD) >0.01% of white blood cells (WBCs), in addition to National Comprehensive Cancer Network standard response criteria, which define complete remission (CR) as <5% bone marrow blasts by morphologic determination, with no evidence of extramedullary disease or refractory disease; no response (NR) as failure to achieve CR; and relapse as loss of CR. CD19 expression by flow cytometry and/or immunohistochemistry was used to classify leukemia as CD19– or CD19+. Aberrant expression of multiple antigens was used to define and follow the immunophenotype of lymphoblasts in multiparameter flow cytometry.14 B-cell aplasia was determined by the absence of CD19+ B cells in the peripheral blood and/or CD19+ B-cell precursors (hematogones) in the bone marrow.
Classification of CD19 expression and calculation of CD19– blasts in B-ALL
Surface CD19 expression by flow cytometry was classified as dim, normal, or bright, based on comparison with normal B cells and isotype controls, as described previously.8 MRD analysis for low-level blasts (>0.01% of WBCs) was performed at the University of Washington flow cytometry laboratory.14,15 Gating strategy was modified to detect and quantify CD19– events (supplemental Figure 2A-C). Quantification of low-level CD19– events in MRD flow cytometric data was performed retrospectively in a subset of patients (n = 36).
Human IgH sequencing and immune repertoire analysis
Genomic DNA was extracted from archived bone marrow aspirates with the Qiagen Gentra DNA purification kit. IgH rearrangements were amplified in duplicate (2 biological replicates per sample) using BIOMED2 primers adapted for next-generation sequencing (NGS); first-round polymerase chain reaction amplicons were purified and subjected to second-round amplification to generate sequencing libraries using the Illumina Nextera XT paired-end kit, as previously described.16,17 Paired-end sequencing (2 × 300 bp) was performed on an Illumina MiSeq instrument in the Human Immunology Core Facility at the University of Pennsylvania. Forward and reverse reads were paired, and consensus sequences were filtered for quality score, deletions, and length.18 Related sequences were collapsed into clones using distance- and lineage-tree–based methods.19,20 MiXCR-2.0 with default settings was used at a copy number cutoff of 2 for B-cell receptor sequencing, unless otherwise specified. VDJtools-1.1.1 and ImmuneDB21,22 were used for clonal lineage analysis. IgH sequences are provided in the supplemental Data.
CAR T-cell production for in vitro studies
T cells were isolated, activated, and transduced with a lentiviral vector encoding the FMC63-based CD19-targeted CAR with 4-1BB-ζ signaling domains, as previously described.23 Cultures were expanded every 2 days with the addition of rhIL-2 (Proleukin) at 100 IU/mL. Nontransduced (NTD) T cells and T cells transduced with the lentivirus containing the CAR construct (19-BBz) were used in functional assays at between 10 and 14 days of expansion.
CD19 RNA synthesis
RNA encoding truncated human CD19 comprising the extracellular and transmembrane domains (amino acids 1-315 of NP_001171569) was produced by in vitro transcription from a pGEM plasmid.23 In vitro RNA transcription was performed according to the manufacturer’s instructions (T7 mScript Standard mRNA Production System; CellScript, Madison, WI).
RNA electroporation
K562 cells were electroporated, as described previously.23 In brief, increasing amounts of CD19 RNA were electroporated into 400 000 cells with a BTX 620 instrument, according to the manufacturer’s recommendations (ECM 830; Holliston, MA). Electroporated cells were then analyzed by flow cytometry and used in coculture assays. One day after electroporation, CD19 expression was quantitatively assessed by flow cytometry separately, using 2 clones of anti-CD19: FMC63 (Millipore-Sigma, St. Louis, MO) and J3-119 (Beckman Coulter, Indianapolis, IN). CD19 expression on K562 was compared with Nalm6, which was processed in parallel. Antibodies bound per cell were estimated by using anti-mouse IgG quantitative beads (Bangs Laboratories, Fishers, IN) and Quantibrite PE beads (BD Biosciences, San Jose, CA).
Cytotoxicity and T-cell activation assays
K562 cells were plated in 96-well plates at 10 000 cells per well, along with various numbers of T cells at the indicated effector-to-target ratios, in medium alone, to measure spontaneous lysis, or in 5% sodium dodecyl sulfate, to measure maximum lysis. After 18 hours, luciferin was added to each well, and luminescence was measured with a Synergy HTX Multi-Mode Microplate Reader (BioTek Instruments, Winooski, VT). Percentage-specific lysis was calculated by using the following formula: 100 × (experimental − spontaneous)/(spontaneous − maximum).23 Cytotoxicity against electroporated K562 cells, measured using 3 replicates of a luciferase killing assay, was normalized by comparing killing by T cells at each dose of CD19 RNA with the killing level displayed by the same type of T cell (NTD or CAR) against mock-electroporated K562 cells.
The proportion of T cells expressing CD69 and CD137 (BioLegend, San Diego, CA) after coculture with each group of CD19 expressing K562 cells was compared with the proportion in their respective negative control conditions (T cells cocultured with mock-electroporated K562 cells). Anti-CD3/anti-CD28–coated beads were used as stimulation for positive controls, and 10 000 events were acquired on an LSR Fortessa (BD Biosciences) and analyzed with FlowJo software (BD Biosciences).
Statistics
Statistical analyses were performed with GraphPad Prism software (San Diego, CA). The χ2 test was used to compare ≥2 unmatched groups (with binomial outcomes). One-way analysis of variance (ANOVA) was used to compare ≥3 matched groups (with Gaussian distribution). The Student t test was used for experiments with CAR T cells.
Results
Patient characteristics
Consecutive patients (n = 166) with relapsed/refractory B-ALL who received initial CD19-redirected CAR T-cell therapy from 2012 through 2017 were analyzed.1,5 These included 91 males and 75 females, with a median age of 12 years (range, 1-29). Sixteen patients received 1 to 4 cycles (median, 1 cycle) of blinatumomab 1 to 15 months (median, 4 months) before CAR T-cell therapy. As the most proximate measurement of disease burden, bone marrow involvement was assessed just before CAR infusion and after lymphodepleting chemotherapy was complete. The median bone marrow involvement was 2.75% (range, 0%-95%). Pre-CAR CD19 disease burden was not significantly different between patients with early loss of B-cell aplasia (1, 3, 6, and 12 months) compared with patients who never lost B-cell aplasia. The median follow-up period at data cutoff, 1 February 2019, was 29 months after infusion. Eleven patients did not achieve MRD– deep remission at assessment on day 28 (Figure 1): 7 with NR and 4 with morphologic CR with persistent MRD. Twenty-eight patients had recurrence with CD19+ MRD (n = 6) or overt relapse (n = 22), and 39 patients with CD19– MRD (n = 5) or overt relapse (n = 34) after achieving an MRD− remission (Figure 1). Eighty-eight patients were in continued remission at the cutoff date. A low-level, aberrant B-cell population was initially reported as MRD+ on 2 patients (HP-4 and HP-53). However, the aberrant populations cleared without further intervention. Retrospective analysis of MRD flow cytometric data revealed that the phenotype of the reportedly aberrant B cells was markedly different from the pre-CAR aberrant B-lymphoblast phenotype in both patients. Those unusual B-cell populations are now considered to be very early normal B-cell precursors and were first described to be expanded in this setting.15 Hence, they were considered false-positive MRD and categorized as deep CR for this study.
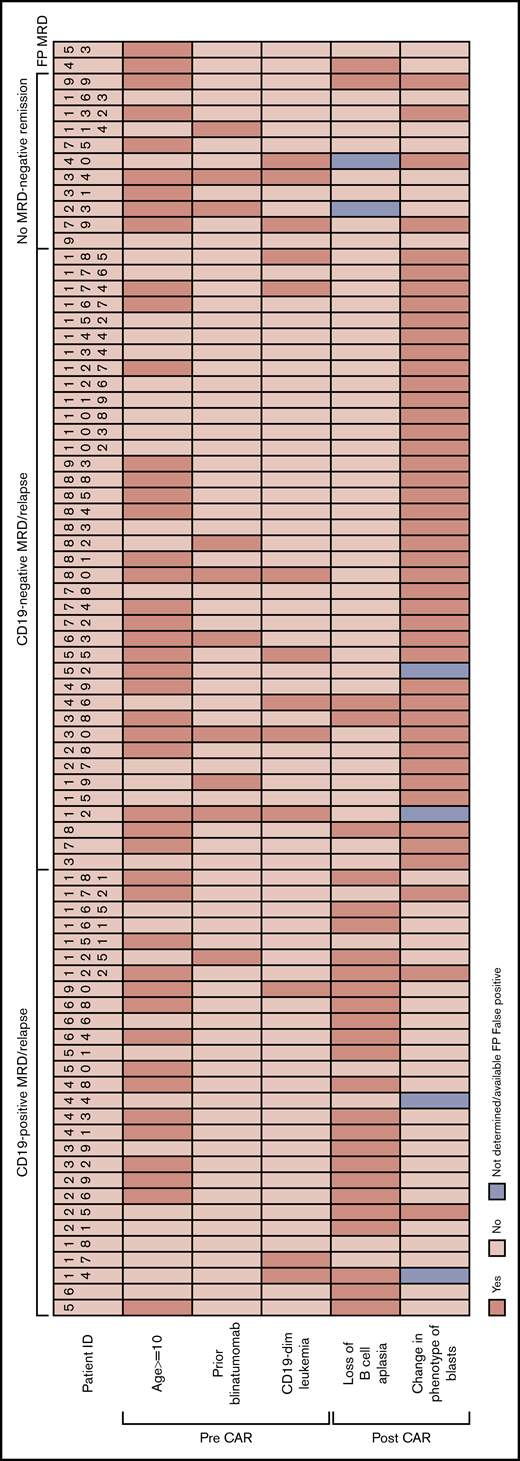
Clinicopathological characteristics of the CD19+MRD/relapse, CD19–MRD/relapse, and no-MRD–-remission categories after CAR T-cell therapy. Age, prior blinatumomab treatment, and CD19 expression levels on blasts, before CAR T-cell infusion are shown. HP-4 and H-53 had false-positive MRD at day 28 assessment and remained in remission without further therapy at data cutoff.
Clinicopathological characteristics of the CD19+MRD/relapse, CD19–MRD/relapse, and no-MRD–-remission categories after CAR T-cell therapy. Age, prior blinatumomab treatment, and CD19 expression levels on blasts, before CAR T-cell infusion are shown. HP-4 and H-53 had false-positive MRD at day 28 assessment and remained in remission without further therapy at data cutoff.
Characterization of nonresponse, CD19+ MRD/relapse, and CD19– MRD/relapse after CAR T-cell therapy
Eleven patients never achieved MRD– remission. Of those, 3 were CD19 dim or negative before CAR T-cell therapy, and 3 received prior blinatumomab with overlap of both in 1 patient (HP-34). Three CD19 dim/negative patients of the 11 nonresponders was a higher rate than 6 CD19 dim patients of 59 achieving remission, but the difference was not significant (Fisher’s exact test; P = .14). Although most nonresponders showed the same immunophenotype as the pre-CAR disease, patients HP-40, HP-79, HP-99, and HP-132 showed CD19– blasts at day 28.
All 28 patients who developed CD19+ MRD/relapse initially showed complete aplasia of CD19+ B cells in the peripheral blood and marrow, consistent with functional CAR T cells. However, B-cell aplasia and functional persistence was lost at a median of 3 months (range, 1-9 months) after infusion in 78% of those patients. CD19+ normal B cells were noted in peripheral blood or bone marrow (Figure 2A), at or before CD19+ MRD/relapse that occurred at a median of 11 months (range, 3-29 months) after infusion. The proportion of patients with CD19+ relapse was significantly higher (χ2 test; P = .0002) in those with early loss of B-cell aplasia (<12 months), compared with patients who never lost B-cell aplasia. The immunophenotype of post-CAR blasts, as assessed by expression levels of multiple unique aberrant markers, was exactly the same as preinfusion disease in 88% of the relapses (Figure 2B), consistent with return of the pre-CAR immunophenotypic clone. Pre-CAR and post-CAR blasts showed the same CD19 expression level in CHOP and MRD flow cytometry. Recurrences in 2 patients (HP-25 and HP-172) were predominantly CD19+ but showed a minor CD19– subset. NGS analysis of antibody VH gene rearrangements in 5 patients (HP-6, HP-25, HP-51, HP-58, and HP-161) showed that the dominant clones were identical in both the pre-CAR and recurrence samples (Figure 2D-E; sequences provided in supplemental Data). The cytogenetic characteristics of CD19+ recurrences were similar to those in the overall treatment population. No abnormalities of the CD19 gene locus were detected by karyotype or genome-wide single-nucleotide polymorphism (SNP) array analysis in most of the recurrences (8 of 9). One patient (HP-25) showed heterozygous deletion of chromosome 16. Interestingly, 4 of 6 patients who relapsed when B-cell aplasia was intact, relapsed with isolated extramedullary and/or central nervous system disease (HP-17, HP-18, HP-44, and HP-151).
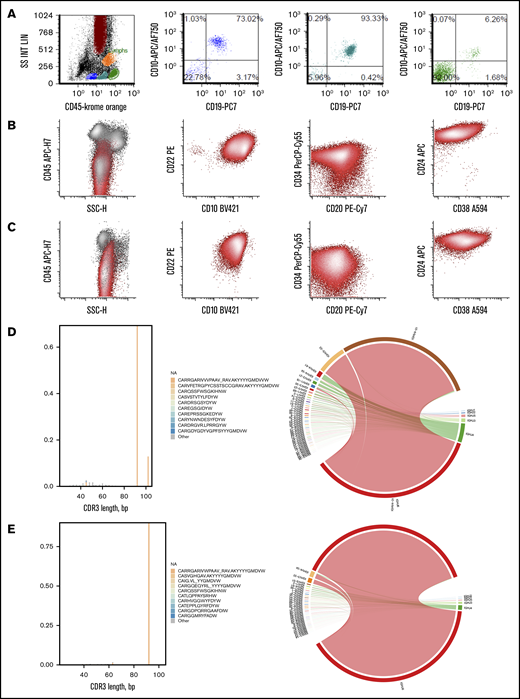
Immunophenotypic and clonotypic comparison of representative CD19+MRD/relapse. (A) Early reappearance of normal hematogones in the bone marrow at 3 months after infusion in HP-181 who subsequently developed CD19+ MRD. Normal maturation pattern of B cells with progressive acquisition of CD45 and CD19 and decrease in CD10 as they mature from stage 1 (blue) to stage 2 (teal) to stage 3 (green) hematogones.31 Other markers such as CD34, CD24, CD22, and CD20 (not shown) were also consistent with normal maturation pattern. Events in green also include mature T cells (CD19– events). Pre-CAR blasts (B) and 12-month post-CAR blasts (C) from HP-6 with identical CD45 dim variable; CD22+,CD10+,CD34 dim variable; CD20 variable; CD24 bright; and CD38 dim phenotype. Immune repertoire analysis showed identical CDR3 sequences (CARRGARIVVVPAAV_RAVAKYYYYGMDVW) and identical variable region (V) joining region (J) family usage (V4-31 J6) in pre-CAR blasts (D) and 12-month post-CAR blasts (E) from HP-51.
Immunophenotypic and clonotypic comparison of representative CD19+MRD/relapse. (A) Early reappearance of normal hematogones in the bone marrow at 3 months after infusion in HP-181 who subsequently developed CD19+ MRD. Normal maturation pattern of B cells with progressive acquisition of CD45 and CD19 and decrease in CD10 as they mature from stage 1 (blue) to stage 2 (teal) to stage 3 (green) hematogones.31 Other markers such as CD34, CD24, CD22, and CD20 (not shown) were also consistent with normal maturation pattern. Events in green also include mature T cells (CD19– events). Pre-CAR blasts (B) and 12-month post-CAR blasts (C) from HP-6 with identical CD45 dim variable; CD22+,CD10+,CD34 dim variable; CD20 variable; CD24 bright; and CD38 dim phenotype. Immune repertoire analysis showed identical CDR3 sequences (CARRGARIVVVPAAV_RAVAKYYYYGMDVW) and identical variable region (V) joining region (J) family usage (V4-31 J6) in pre-CAR blasts (D) and 12-month post-CAR blasts (E) from HP-51.
Thirty-nine patients developed CD19– MRD/relapse, at a median of 5 months (range, 2-33 months) after infusion, despite ongoing B-cell aplasia, indicating functional CAR T-cell persistence at the time of recurrence, in 94% of cases. Six of these patients received prior blinatumomab and 7 patients were CD19 dim before treatment (Figure 1). Three patients were both pretreated with blinatumomab and CD19 dim (HP-12, HP-30, and HP-80). The cytogenetic characteristics of CD19– recurrences were similar to the overall treatment population.
Correlation of pretreatment CD19 expression and blinatumomab with remission and MRD/relapse
Dim CD19 expression before CAR T-cell infusion (Figure 3A) was noted in 12% of patients. Among the CD19-dim patients, 4 of 19 (21%) had been pretreated with blinatumomab, compared with 8 of 111 (7.2%) among the CD19-normal/bright patients (P = .068). We assessed whether the level of CD19 on blasts before CAR therapy correlated with failure to achieve MRD– remission and CD19– MRD/relapse, compared with the remission group. There was a trend toward a higher rate of failure to achieve deep remission and CD19– MRD/relapse in CD19-dim B-ALL (Figure 3B; χ2 test; P = .14). Four of the 16 (25%) blinatumomab-treated patients were CD19 dim, compared with 16 among the 150 (10.67%) non–blinatumomab-treated patients (Figure 3C; P = .067). Stratification of patients by prior blinatumomab status revealed a higher rate of remission and nonresponse/CD19– MRD/relapses between the blinatumomab-pretreated group compared with the no-prior-blinatumomab group (Figure 3D; χ2 test; P = .043). In 1 patient with CD19– recurrence (HP12), a decrease in CD19 level happened after blinatumomab but before CAR T-cell therapy (Figure 3E). CD19 RNA sequencing of the recurrence in this patient revealed aberrant splicing that resulted in loss of exon 2, as previously reported.24 The aberrantly spliced isoform was also detected at low levels before CAR T-cell therapy.
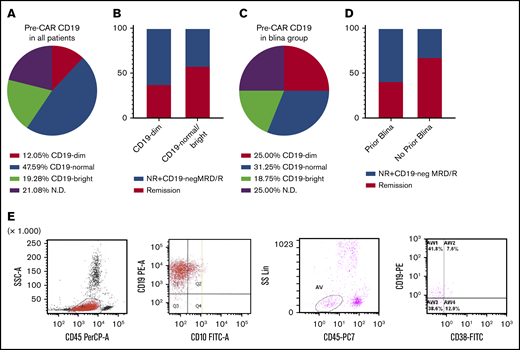
CAR T cells are effective for CD19-dim B-ALL, but there is a higher rate of failure to achieve MRD–remission and CD19–MRD/relapse in blinatumomab-pretreated patients. (A) Distribution of CD19 expression before CAR T-cell therapy in all patients (n = 166). (B) Proportion of remission and NR/CD19-negMRD/R among the CD19-normal/bright and CD19-dim B-ALL cases. (C) Distribution of CD19 expression among the patients previously treated with blinatumomab (blina group; n = 16). (D) Proportion of remission and NR/CD19− MRD/R among patients stratified by prior blinatumomab therapy. (E) Flow cytometric data from HP-12 at relapse (2 plots on the left) and postblinatumomab pre-CAR (2 plots on the right) time points show loss of CD19 expression after blinatumomab.
CAR T cells are effective for CD19-dim B-ALL, but there is a higher rate of failure to achieve MRD–remission and CD19–MRD/relapse in blinatumomab-pretreated patients. (A) Distribution of CD19 expression before CAR T-cell therapy in all patients (n = 166). (B) Proportion of remission and NR/CD19-negMRD/R among the CD19-normal/bright and CD19-dim B-ALL cases. (C) Distribution of CD19 expression among the patients previously treated with blinatumomab (blina group; n = 16). (D) Proportion of remission and NR/CD19− MRD/R among patients stratified by prior blinatumomab therapy. (E) Flow cytometric data from HP-12 at relapse (2 plots on the left) and postblinatumomab pre-CAR (2 plots on the right) time points show loss of CD19 expression after blinatumomab.
The bone marrow CD19 disease burden was significantly higher (Mann-Whitney U test; P < .00001) in patients with CD19− MRD/relapse and nonresponders (mean, 51.88%), compared with patients who achieved remission (13.72%).
Threshold of CD19 recognition by CAR T cells in vivo and in vitro
The earliest CD19-expressing B-cell precursors (stage 1 hematogones) in the bone marrow were conspicuous by their complete absence after CAR T-cell infusion. The only normal B-cell populations seen were minor populations of normal very early CD19– B-cell precursors. These are characteristically positive for CD22, variably positive for CD10 and CD34, negative for CD24 (Figure 4A), and distinct from aberrant leukemic blasts.15
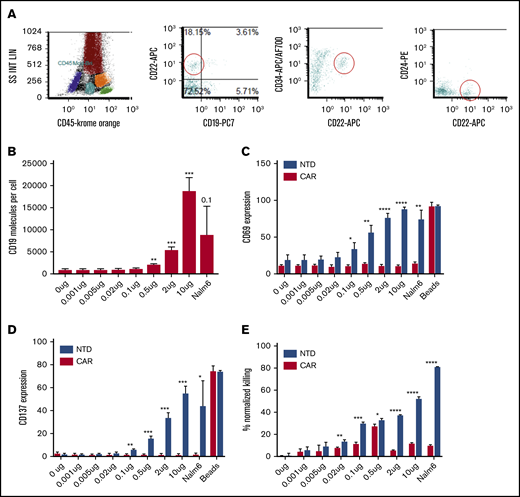
CAR T cells recognize very low levels of CD19. (A) Expanded populations of CD19– precursors (also CD22+CD24–CD10 variable CD34 variable) that are seen after CAR T-cell therapy. (B) Flow cytometric estimation of CD19 expression (antigen-binding capacity) in CD19– K562 cells transduced with increasing doses of CD19 mRNA (n = 3). (C) Significant CD69 expression in CAR T cells at 0.005 μg CD19 mRNA (n = 3). (D) Significant CD137 expression at 0.1 μg CD19 mRNA (n = 3). (E) Significant killing by CAR (19-BBz CAR construct transduced) at 0.02 μg CD19 mRNA at a 10:1 effector-to-target ratio (n = 3). Student t test; *P < .05, **P < .01, ***P < .001, and ****P < .0001.
CAR T cells recognize very low levels of CD19. (A) Expanded populations of CD19– precursors (also CD22+CD24–CD10 variable CD34 variable) that are seen after CAR T-cell therapy. (B) Flow cytometric estimation of CD19 expression (antigen-binding capacity) in CD19– K562 cells transduced with increasing doses of CD19 mRNA (n = 3). (C) Significant CD69 expression in CAR T cells at 0.005 μg CD19 mRNA (n = 3). (D) Significant CD137 expression at 0.1 μg CD19 mRNA (n = 3). (E) Significant killing by CAR (19-BBz CAR construct transduced) at 0.02 μg CD19 mRNA at a 10:1 effector-to-target ratio (n = 3). Student t test; *P < .05, **P < .01, ***P < .001, and ****P < .0001.
We assessed the level of CD19 surface expression that is necessary to initiate killing by CAR T cells in vitro. K562 cells, which lack endogenous CD19, were electroporated with increasing concentrations of CD19 mRNA. Significant surface CD19 expression above the background was first detected at 0.5 µg CD19 mRNA dose (Figure 4B). Quantitative flow cytometry estimated an average of 2086 surface molecules of CD19 per cell at that dose. In comparison, the earliest discrete population of B-cell precursors (CD19+CD10+CD34+ stage 1 hematogones) in normal bone marrows showed an average of 2352 molecules per cell (n = 3; range, 1979-2682). CD19 expression increases with B-cell maturation and is highest in activated mature B cells in lymph nodes.8 CD19 expression in 93% of B-ALL is brighter than that of stage 1 hematogones.8 We assessed upregulation of activation markers CD69 and CD137 (4-1BB) on CAR T cells after overnight coculture with CD19-expressing K562 cells. CD69 upregulation on total CAR T cells was detected, even at the low level of 0.005 µg of CD19 mRNA (Figure 4C). CD137 upregulation was detected starting at 0.1 µg of CD19 RNA (Figure 4D). The pattern of activation was similar in the CD4 and CD8 T-cell compartments. Killing by CAR T cells increased in proportion to target cell CD19, starting from the 0.02 µg CD19 mRNA dose (mean, 956 molecules per cell) for a 10:1 CAR T cell: target ratio (Figure 4C). Killing was noted at 2 µg (mean, 5402 molecules per cell), 0.02 µg, and 0.1 µg (mean, 1123 molecules per cell) CD19 mRNA for the 30:1, 3:1, and 1:1 ratios, respectively (supplemental Figure 1). Hence, activation and killing by CAR T cells started even before detection of surface CD19 by flow cytometry.
Correlation of rare CD19– events before CAR, with remission and MRD/relapse
We assessed whether presence of CD19– events before CAR T-cell therapy was associated with CD19– MRD/relapse. Our prior work revealed low level but discrete populations of CD19– blasts in 17% of de novo and relapsed B-ALL.8 We also detected low levels of blasts (>1%) As MRD-level analyses were not performed on those cases, we determined whether MRD levels (>0.01% of WBCs) of CD19– blasts could be detected in our cohort before CAR T-cell therapy (supplemental Figure 2). Surprisingly, low levels of CD19– events (median, 0.8%; range, 0%-41.1%) could be detected in 94% of cases analyzed (36 cases analyzed). There were no significant differences in CD19– events between the various groups (P = .56).
Immunophenotype and immune repertoire in CD19– MRD/relapse
Although the mechanism of loss of CD19 in CD19– relapses has been elucidated in limited cases,7 it is unclear whether CD19– blasts arise from preexisting events or occur de novo. Comparison of pre-CAR and post-CAR blasts in our cohort revealed that, of the multiple antigens that were monitored, only CD19 was lost in the 71% of patients with CD19– MRD/relapse (Figures 1 and 5A-B). The non-CD19 antigens were identical to the pretreatment disease, which suggests the occurrence of a CD19-specific event. NGS analysis of antibody VH gene rearrangements in 6 patients (HP-15, HP-19, HP-74, HP-78, HP-84, and HP-152) revealed that the identical dominant clone was present in the pre-CAR and CD19– recurrence samples (Figure 5C-D; supplemental Data).7 The CD19– MRD/relapse group also includes 3 B-ALL (HP-78, HP-144, and HP-152) that switched to acute myelogenous leukemia (AML) after CAR T-cell therapy. Whereas 2 of these had KMT2A rearrangements, 1 had a TCF3-ZN384 rearrangement that is known to be associated with mixed-phenotype leukemia.25 Immune repertoire analysis of KMT2A-rearranged CAR T-cell–treated cases showed the identical IgH clone in both the B-ALL and AML samples, despite the marked difference in their morphology, immunophenotype, and lineage (supplemental Figure 3; supplemental Data). Although the overall cytogenetic category was the same as that in preinfusion disease, additional genetic alterations were noted in many cases consistent with clonal evolution. Genome-wide SNP array analyses of 5 CD19– relapses (HP-3, HP-15, HP-19, and HP-83) showed deletions and loss of heterozygosity (LOH) in the CD19 gene locus at 16p11.2. Notably, SNP array data for HP-83 were consistent with 16p copy-neutral LOH in ∼20% to 25% of cancer cells, with tandem Sanger sequencing revealing separate frameshift mutations in exons 3 and 4 (both with wild-type greater than mutant chromatogram signal), suggesting a multiclonal pattern in this relapse. Aberrant splicing leading to overexpression of isoforms lacking exon 2 (and the FMC63 epitope) were also noted in 2 samples (HP-72 and HP-79).24 Masking of the CD19 epitope by transduction of CAR construct in leukemic cells was reported in a single case (HP-72), but this mechanism of CD19 loss has not been detected in any other samples.7 Interestingly, CD19 gene alterations were not found in either of the KMT2A-rearranged leukemias (HP-78 and H-152).
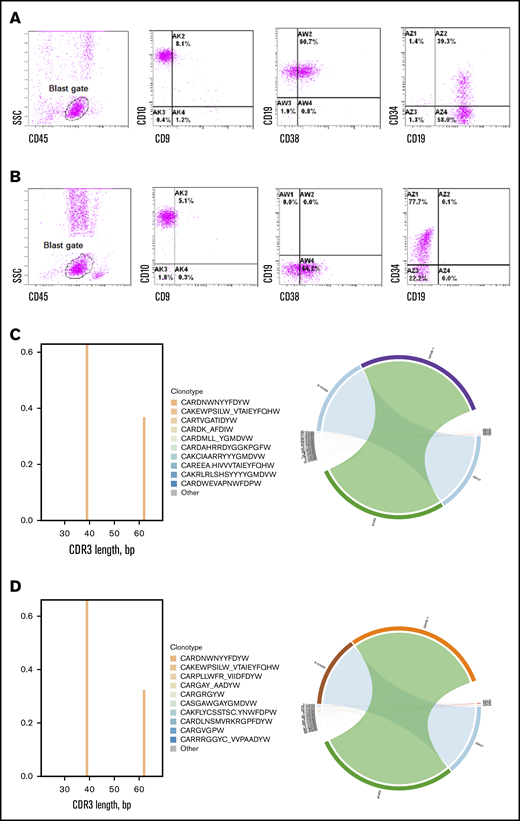
Immunophenotypic and clonotypic comparison of representative CD19–recurrence. Immunophenotype of blasts pre-CAR (A) and 3 months after CAR MRD (B) from HP-15 showed complete loss of CD19. Immune repertoire analysis of IgH variable (V) region revealed identical CDR3 sequences (CARNWNYYFDYW) and identical VJ family usage (V6-1 J-4) in pre-CAR B-ALL (C) and post-CAR CD19– leukemia (D) from HP-78.
Immunophenotypic and clonotypic comparison of representative CD19–recurrence. Immunophenotype of blasts pre-CAR (A) and 3 months after CAR MRD (B) from HP-15 showed complete loss of CD19. Immune repertoire analysis of IgH variable (V) region revealed identical CDR3 sequences (CARNWNYYFDYW) and identical VJ family usage (V6-1 J-4) in pre-CAR B-ALL (C) and post-CAR CD19– leukemia (D) from HP-78.
Discussion
CAR T-cell products are US Food and Drug Administration approved for use for the treatment of relapsed/refractory B-ALL and diffuse large B-cell lymphoma, and they are under active investigation for the treatment of other hematological malignancies and solid tumors. Important questions, such as the correlation of antigen expression with response, and the factors underlying nonresponse or relapse remain to be elucidated. CAR T-cell therapy of B-ALL is ideally suited to answer these questions, given that correlates of CAR T-cell function and disease status can be easily and routinely assessed from peripheral blood and bone marrow examination through quantification of normal and aberrant CD19-expressing B cells and blasts. Aberrancies of antigen expression in B-ALL are well described and form the basis of MRD detection by multiparameter flow cytometry.14 Hence, unique antigenic aberrancies can be used to follow specific clones from diagnosis to relapse. We performed immunophenotypic analyses along with genetic and immune repertoire characterization and correlated with B-cell aplasia.
CD19+ recurrences have been associated with loss of CAR T cells.5,26 We found that CD19+ MRD/relapses had the identical immunophenotype and IgH gene rearrangement as the pre-CAR leukemia and were preceded by loss of CAR T-cell function in most cases. In our experience, a pharmacodynamic assessment of CAR T-cell function by persistent B-cell aplasia is superior to direct assessment of CAR T cells by flow cytometry or polymerase chain reaction. It is significant that the CD19+ recurrences with ongoing B-cell aplasia had an isolated extramedullary relapse, suggesting site-specific dysregulation. Although some patients had early recurrence, there were many who experienced relapse >3 months after infusion. The occurrence of late CD19+ MRD or relapse in our cohort highlights the need for long-term persistence and surveillance by CAR T cells. However, the minimum duration of CAR T-cell persistence for optimum disease control is unknown and may differ between CAR designs. CAR T-cell therapies vary widely in the persistence of engineered CAR T cells and duration of B-cell aplasia.27,28 CAR T cells with a 4-1BB costimulatory domain are observed to persist longer than CAR T cells with a CD28 costimulatory domain.29 Among the patients treated with a 4-1BB CAR, it is not clear why some patients have functional persistence with long-term B-cell aplasia, whereas others do not. Until we understand those factors, consolidation of CAR T-cell–induced remissions with a hematopoietic stem cell transplant may be considered in a subset of patients with short CAR T-cell persistence.
In contrast to CD19+ recurrences, CD19– recurrences occurred despite functional persistence and ongoing B-cell aplasia. Hence, they are likely to be independent of CAR T-cell parameters per se and to be related to other disease factors, such as genetics or CD19 expression level. Relapses after CD22-targeted CAR T cells have been associated with lower CD22 site density.3 However, in our cohort, CAR T cells targeting CD19 were equally effective for CD19-dim B-ALL compared with CD19 normal or bright B-ALL. Importantly, CD19 antibody binding capacity assessment, reported previously on a small subset of the cases, also did not reveal differences between nonresponders and clinical responders.1 The differences between CD19- and CD22-CAR likely reflects the differences in baseline antigen expression. CD19 is more widely expressed at a higher level than CD22 in B-ALL.8
CD19 antigen burden has been associated with early loss of B-cell aplasia.30 However, in our large cohort, we did not find an association between low disease burden and CD19+ MRD/relapses. Paradoxically, the disease burden was higher in patients with CD19+ MRD/relapses. The differences could be attributed to the CAR T products used.
B-cell precursors increase their CD19 expression as they mature.31 Our in vivo data showed that CAR T cells lysed even the earliest B-cell precursors as soon as they began to express CD19. In vitro studies confirmed that CAR T cells were able to recognize and lyse target cells expressing very low levels of CD19. Crucially, this activity was noted even before flow cytometric detection of CD19. We did not find a correlation between rare (<1%) CD19– blasts and response to CAR T-cell therapy. Hence, the presence of dim CD19 or rare CD19– events by flow cytometry does not predict nonresponse or recurrence after CAR T-cell therapy and is not a reason to consider excluding such patients from receiving this therapy. The only cases of leukemia that may not respond are those that are predominantly or completely negative for CD19, and it is important to acknowledge that patients with this expression pattern were not treated in our studies. CD19– de novo B-ALL is very rare but reported.32,33 A more common scenario is the loss of CD19 because of prior use of CD19-targeted immunotherapy.11 We found that patients who were previously treated with blinatumomab had a significantly higher rate of failure to achieve MRD– remission, and CD19– MRD/relapse. Interestingly, only a subset of the prior blinatumomab-treated patients showed dim CD19 before CAR T-cell infusion. It is possible that CD19 was reexpressed after cessation of blinatumomab,34,35 or there were other abnormalities in CD19 that were not apparent by routine immunophenotyping. Pediatric trials of blinatumomab are limited, but show lower rates (31%) of complete remission compared with CAR T-cell therapy.13 Advantages of CAR T-cell therapy over blinatumomab include rapid expansion in vivo, long-term persistence, and excellent penetration of sanctuary sites, such as the central nervous system. A limitation of our study is the low number of blinatumomab-treated patients. A large multicenter study of blinatumomab- and CAR-treated patients must be performed to definitively answer this question. Until then, we must be cognizant of the potential impact of blinatumomab on subsequent CAR T-cell therapy.
CD19– recurrences form the largest category of recurrences in our cohort, and these patients have limited treatment options.11,24 Multiple mechanisms of CD19 loss have been described including mutations and deletions, with or without LOH of CD19 gene,6 retention of misfolded protein in the endoplasmic reticulum and lineage switch.7,24,34,36 We found that several such alterations could be detected by clinical SNP array testing. An unresolved question is whether blasts with aberrations in CD19 were present before immunotherapy.37 BCR-ABL1 fusion transcripts were noted in CD19– hematopoietic stem cell precursors before and after blinatumomab in 2 Philadelphia chromosome+ B-ALL patients who relapsed with CD19– myeloid leukemia.38 However, genetic alterations in CD19 gene were not detected before CAR infusion in an analysis of CD19– relapses, with the caveat that the sensitivity of the molecular method to detect very low level events was not established.7 We used flow cytometric MRD analysis to detect very low-level CD19– events (<0.01%). Interestingly, rare CD19– blasts could be detected before infusion in most patients and did not correlate with responses. The loss of only CD19 in the majority of the CD19– recurrences with identical IgH gene rearrangement raises the possibility of active loss of CD19. In addition to the previously characterized genetic mechanisms, trogocytosis of CD19 antigen from targets to CAR T cells provides an explanation for isolated loss of CD19.39 KMT2A and TCF3-ZN384 rearranged B-ALL that switched lineage to AML possessed identical dominant IgH gene rearrangements, despite the dramatic difference in their immunophenotype. This pattern suggests plasticity of the leukemic cell and support emerging data that immunophenotypic plasticity in mixed-phenotype leukemia is independent of the underlying immune repertoire or genetic heterogeneity.25 To summarize, we addressed several important questions pertaining to immunotherapy of B-ALL that have therapeutic implications for the use, and possibly sequence, of CD19-directed immunotherapies.
Acknowledgments
The authors thank the Children's Hospital of Philadelphia (CHOP) flow cytometry laboratory, the CHOP hematology laboratory, the Human Immunology Core facility, the CAR T-cell therapy team, and the Division of Genomic Diagnostics for contributions to the study; Robert Doms and Stephen Hunger for critical reading of the manuscript; and Marybeth Helfrich and Joseph McMann for help with the study.
This work was supported by a Cancer Research Institute Clinic and Laboratory Integration Program Grant (V.P.); a research grant from Alex’s Lemonade Stand Foundation and Love Your Melon (J.R. and K.M.); National Institutes of Health (NIH), National Cancer Institute Grant P30CA016520 (W.M.); a Young Investigator Award from Alex’s Lemonade Stand Foundation (A.B.); NIH, National Cancer Institute Grant U01CA232563, Stand Up To Cancer-St. Baldrick’s Pediatric Dream Team Grant SU2C-AACR-DT1113, a 2016 research grant from William Lawrence and Blanche Hughes Foundation, and The V Foundation for Cancer Research Grant T2018-014 (A.T.-T.); and the St. Baldrick’s Foundation, the V Foundation, Stand Up To Cancer-St. Baldrick’s Pediatric Dream Team Grant SU2C-AACR-DT1113, and NIH, National Cancer Institute Grant 1P01CA214278-01 (S.L.M.).
Authorship
Contribution: V.P. conceived of and designed the study; J.R., K.M., A.B., W.M., J.V.A., A.M.D., J.J.M., and V.P. acquired the data; K.M., S.C., J.R.F., D.A.O., W.M., D.M., E.T.L.P., G.W., M.P., M.L., C.H.J., V.G.B., S.L.M., S.A.G., S.R.R., and V.P. analyzed and interpreted the data; K.M., W.M., E.T.L.P., D.M., V.G.B., A.T.-T., S.A.G., S.L.M., S.R.R., and V.P. wrote, reviewed, and/or revised the manuscript; and V.P. supervised the study.
Conflict-of-interest disclosure: S.L.M. is a consultant/advisory board member for Novartis Pharmaceutical Corporation (NPC) and Kite Pharma. S.A.G. has received research and/or clinical trial support from NPC, Servier, and Kite and has served as consultant for and on study steering committees or scientific/clinical advisory boards of NPC, Cellectis, Adaptimmune, Eureka, TCR2, Juno, GlaxoSmithKline, Vertex, Cure Genetics, Humanigen, and Roche. C.H.J. reports receiving research funding from NPC and Immune Design and is a consultant/advisory board member for NPC, Tmunity Therapeutics, and Immune Design. The remaining authors declare no competing financial interests.
Correspondence: Vinodh Pillai, Department of Pathology, Children's Hospital of Philadelphia, 3400 Civic Center Blvd, Philadelphia, PA 19104; e-mail: pillaiv1@email.chop.edu.

