Key Points
Blast phenotype is an independent predictor of outcome in NPM1-mutated AML.
Patients with a DN blast phenotype (lacking CD34 and HLA-DR expression) harbor TET2/IDH mutations and show superior outcomes.

Abstract
Recent work has identified distinct molecular subgroups of acute myeloid leukemia (AML) with implications for disease classification and prognosis. AML with mutated NPM1 (AML-NPM1) represents a distinct entity in the revised 2017 World Health Organization classification, but relatively little work has examined the clinical significance of phenotypic and genetic heterogeneity within this group. A multi-institutional cohort of 239 AML-NPM1 cases included 3 phenotypic groups: cases with blasts showing monocytic differentiation (n = 93; monocytic AML-NPM1), cases lacking monocytic differentiation (n = 72; myeloid AML-NPM1), and cases where blasts were negative for both CD34 and HLA-DR (n = 74; double-negative [DN] AML-NPM1). Genotypic diversity typical of AML-NPM1 was seen, with comutations occurring most commonly in DNA methylation genes (81% of cases), FLT3 (48%; including internal tandem duplication and tyrosine kinase domain mutations), and RAS pathway genes (30%). However, the comutation pattern differed by blast phenotype. TET2 and IDH1/2 mutations were significantly more common in DN AML-NPM1 (96% of cases) than in myeloid (39%) or monocytic AML-NPM1 (48%; P < .0001). Conversely, DNMT3A mutations were significantly less common in DN AML-NPM1 (27%) than in myeloid (44%) or monocytic cases (54%; P = .002). Furthermore, the 3 phenotypic groups showed significant differences in outcome, with DN AML-NPM1 showing significantly longer relapse-free (RFS) and overall survival (OS) (64.7 and 66.5 months, respectively) than monocytic AML-NPM1 (RFS, 20.6 months; OS, 44.3 months) or myeloid AML-NPM1 (RFS, 8.4 months; OS, 20.2 months; P < .0001 and P = .01 for RFS and OS, respectively). Our findings highlight biologic differences within immunophenotypically defined subgroups of NPM1-mutated AML that may impart prognostic significance.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease caused by the acquisition of genetic alterations within hematopoietic precursor cells. Recent work supports a genetic classification of AML based on the presence of a recurrent cytogenetic abnormality, such as a PML-RARA or RUNX1-RUNX1T1 translocation, or the presence of recurrent mutations in genes such as TP53, CEBPA, or NPM1.1,2 AML with mutated NPM1 (AML-NPM1) represents the largest of these genetically defined groups, comprising ∼30% of adult AMLs,1,3,4 and is now recognized as a distinct entity in the 2017 World Health Organization (WHO) classification.5 NPM1 mutations typically occur in de novo AML and are associated with a normal karyotype and relatively favorable prognosis.6
Although all cases of AML-NPM1, by definition, demonstrate an NPM1 mutation, NPM1 mutations typically do not occur in isolation.1,4 Most commonly, comutations occur in the FLT3 gene (including both FLT3–internal tandem duplication [ITD] and FLT3–tyrosine kinase domain [TKD] mutations) and in genes regulating DNA methylation, such as DNMT3A, TET2, IDH1, and IDH2.7 DNA methylation mutations have been described in 70% of AML-NPM1s, with DNMT3A mutations seen in ∼50% of cases and TET2, IDH1, and IDH2 mutations each occurring in ∼15% of cases.1,7-13 TET2 and IDH1/2 mutations are thought to act through a common mechanism leading to inhibition of TET2 function, resulting in loss of TET2-mediated hydroxymethylcytosine production and subsequent DNA demethylation.14 As a result, cells with TET2 and IDH1/2 mutations show aberrant DNA hypermethylation, which is thought to impair myeloid differentiation.14,15 The poor prognostic impact of FLT3-ITD mutations in AML-NPM1 (particularly when occurring at a high allelic burden) has been well described.6 However, the prognostic impact of other comutations in AML-NPM1 has remained unclear, with recent studies showing conflicting results.7
In addition to genetic diversity, AML-NPM1 is known to show phenotypic heterogeneity, with a subset of cases showing evidence of monocytic differentiation, as assessed by blast morphology, positivity for nonspecific esterase (NSE) by cytochemical staining, and positivity for monocytic surface markers, such as CD11b, CD14, and CD64, by flow cytometry.16 Additionally, studies have shown that many cases of AML-NPM1 are negative for CD34, including cases both with and without monocytic differentiation, whereas a smaller subset has been shown to be negative for HLA-DR.3,16-18 Furthermore, we and others have demonstrated that a significant subset of cases of AML-NPM1 lacking monocytic differentiation is negative for both CD34 and HLA-DR, potentially mimicking acute promyelocytic leukemia in immunophenotype.19,20
The other genetically defined AMLs specified by the WHO classification predominantly include AMLs with recurrent cytogenetic abnormalities, such as those involving core-binding factor genes t(8;21);RUNX1-RUNX1T1 and inv(16)/t(16;16);CBFB-MYH11 or PML-RARA. Although these AML categories have traditionally been considered to be relatively homogeneous, with generally favorable prognoses, the prognostic effect of certain comutations (eg, KIT mutations in core-binding factor AML) is becoming increasingly appreciated.6 Accordingly, KIT mutation testing is recommended for core-binding factor AMLs in the recent American Society of Hematology/College of American Pathologists guidelines on the initial diagnostic workup of acute leukemias.21 AML-NPM1 represents 1 of 3 new genetic categories of AML in the 2017 WHO classification defined by the presence of a recurrent gene mutation, but it is currently unknown whether the phenotypic and genetic (aside from FLT3-ITD) variations within this disease category may hold prognostic significance. Furthermore, it remains unclear whether genotype-phenotype correlations might exist within this heterogeneous disease category, as is seen in some other AML subtypes, such as the association of t(8;21) with CD19 positivity on myeloblasts.22 In the current study, we characterize the phenotypic and genetic diversity within a large multi-institutional cohort of AML-NPM1s and show that distinct blast phenotypes are associated with significant differences in comutation pattern and patient outcome.
Methods
Patient cohort
The study cohort included 239 cases of NPM1-mutated, de novo AML with flow cytometric, cytogenetic, and next-generation sequencing data identified from the pathology archives at Brigham and Women’s Hospital (BWH; n = 108 cases), Vanderbilt University Medical Center (VUMC; n = 62 cases), University of Pittsburgh Medical Center (UPMC; n = 41 cases), and Massachusetts General Hospital (MGH; n = 29 cases). Patients with a history of myelodysplastic syndrome or myeloproliferative neoplasm, exposure to cytotoxic chemotherapy, or cytogenetic findings defining AML with myelodysplasia-related changes were excluded. A subset of patients in this cohort has been described previously.19,23 Institutional review boards at all institutions approved this research.
Immunophenotyping
Flow cytometric analysis was performed at BWH, MGH, and UPMC on a FACSCanto II flow cytometer (BD Biosciences, San Jose, CA), with data analyzed using FACSDiva software (BD Biosciences), and at VUMC on a FACSCanto II flow cytometer, with data analyzed using Winlist software (Verity Software House, Topsham, MA). Immunophenotypic data were collected on expression of the following markers for all cases: CD11b, CD13, CD14, CD33, CD34, CD56, CD64, CD117, and HLA-DR. Myeloperoxidase assessment was performed by either flow cytometry or cytochemistry for a majority of cases. Cases were categorized as showing monocytic differentiation based on blast morphology (presence of monoblasts, with round nuclei, lacy chromatin, and abundant basophilic, often vacuolated cytoplasm, and promonocytes, with folded, irregular, or convoluted nuclei), cytochemical studies (positivity for NSE), and/or surface marker expression by flow cytometry (positivity for ≥2 of the following: CD11b, CD14, and CD64). Cases with myelomonocytic features were included in the monocytic phenotypic group. Cases were characterized as myeloid based on the absence of expression of ≥2 monocytic markers and absence of monocytic blast morphology. In reporting the peripheral blood blast percentage, monoblasts and promonocytes were counted as blasts.
Molecular genetic analysis
For patients seen at BWH, VUMC, and UPMC, next-generation sequencing was performed on an Illumina MiSec (Illumina, Inc., San Diego, CA) using targeted amplicon sequencing targeting genes recurrently mutated in hematologic malignancies. The BWH panel covered 95 genes.24 VUMC and UPMC sequencing was performed at GenPath Diagnostics (Elmwood Park, NJ) and targeted 37 genes. MGH sequencing used anchored multiplex polymerase chain reaction and targeted 39 genes. Variant allele frequencies (VAFs) >60% were considered to represent copy-neutral loss of heterozygosity. When multiple variants were present within the same gene, the highest VAF was used in the analysis.
Statistical analysis
Fisher’s exact and χ2 tests were used to calculate statistical significance for categorical variables, and Mann-Whitney U tests were used for continuous variables. Log-rank tests were used to analyze Kaplan-Meier curves, which were generated using Graphpad Prism 5.02 (www.graphpad.com). P ≤ .05 was considered statistically significant. The Benjamini-Hochberg method was used to correct for multiple comparisons where indicated. Univariate (log-rank test) and multivariable analyses (Cox proportional hazards model) were performed using XLSTAT (version 2017.5) to evaluate for variables independently associated with relapse-free (RFS) and overall survival (OS). Variables with P < .20 in univariate analysis were included in the multivariable analysis, with stepwise elimination of nonsignificant variables to arrive at the final model.
Results
Clinical characteristics
The cohort included 239 patients with AML-NPM1, with a slight female predominance (female/male ratio, 1.17) and a median age at diagnosis of 64.8 years (range, 14-89 years; Table 1). Cytogenetic data were available for 219 patients, of whom 189 (86%) had a normal karyotype. For patients with an abnormal karyotype, none of the cytogenetic abnormalities defined AML with a recurrent cytogenetic abnormality or AML with myelodysplasia-related changes. Clinical follow-up was available for 231 patients (97%), with a median length of follow-up of 14.2 months (range, 0.1-88.4 months). A majority of patients received standard induction chemotherapy (n = 172 patients), and 92 patients underwent SCT, which was performed in first complete remission for 71 patients. A subset of patients (19% overall) received FLT3 inhibitor therapy.
NPM1-mutated AML shows immunophenotypic diversity
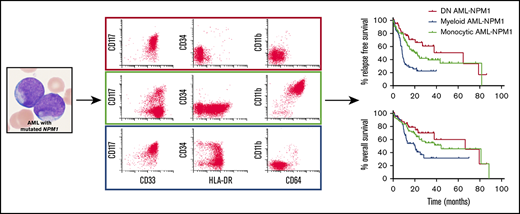
A total of 98 cases (41%) showed morphologic, cytochemical, and/or immunophenotypic evidence of monocytic differentiation (monocytic AML), whereas a majority of cases (n = 141; 59%) lacked evidence of monocytic differentiation (myeloid AML). Consistent with previous reports,3,16-18 flow cytometric analysis demonstrated that 77% of all AML-NPM1 cases (n = 184) were negative for CD34, and 33% (n = 79) were negative for HLA-DR. However, there was variation in blast phenotype between monocytic and myeloid cases (Figure 1). As expected, a majority of monocytic cases (80 [82%] of 98) were negative for CD34 and positive for HLA-DR. In contrast, negativity for both CD34 and HLA-DR was the most common blast phenotype in myeloid AML-NPM1 (69 [49%] of 141). This double-negative (DN) phenotype was uncommon in monocytic cases, seen in only 5 of 98 monocytic cases. We previously reported a subset of this cohort and showed that this DN group, which resembles acute promyelocytic leukemia immunophenotypically, demonstrates distinct phenotypic and molecular genetic characteristics as compared with other cases of myeloid AML-NPM1.19 Therefore, we divided the present cohort of AML-NPM1s into 3 immunophenotypic subgroups: myeloid AML-NPM1 (n = 72), monocytic AML-NPM1 (n = 93), and DN AML-NPM1 (n = 74; supplemental Figure 1). The DN AML-NPM1 cohort included 69 cases that would have otherwise been classified as myeloid AML-NPM1 and 5 cases that would have otherwise been classified as monocytic AML-NPM1. Of the 5 cases of DN AML-NPM1 that showed monocytic differentiation, 4 were positive for CD11b, CD14, and CD64 (2 of 2 tested cases were positive for NSE), and 1 was positive for CD11b, CD64, and NSE but negative for CD14.
![Immunophenotypic variation within AML with mutated NPM1. Myeloid and monocytic AML-NPM1 show distinct patterns of CD34 and HLA-DR expression. The most common blast phenotype in myeloid AML-NPM1 was a CD34−/HLA-DR− phenotype (69 [49%] of 141). In monocytic AML-NPM1, a majority of cases were negative for CD34 and positive for HLA-DR (80 [82%] of 98).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/21/10.1182_bloodadvances.2019000328/3/m_advancesadv2019000328f1.png?Expires=1767887123&Signature=gCIdeK5mKg7GuIernaDqPMyuZQvYEpvRxHeHd3X16HEGlf~Rd1Xm8hg6c~u3AX93~46SYyqYHlW~gGlS68i6xDbhdAFwuSUYJvm3ynCCNUduCG8G~x6XuN1KUuvgVi4Je2zbngz0wN0AumDgvpr9D91LTv2-oNLgYMC-XfQNjMJxx4jrm-eY2kT5v4M2YlAwAhL0hoWTxoHr7MDUTTiFAPIYj2ebjB~ezH94HFL9NipQX94kpBbK3qdU0YhZ0nxcsgKEfdKNXilB5VR9-sR8KglkQGJest9~Ju-CUlM-hkMckISIx8uoiR-8QshZWJdStPYtizeTS67QnTyoIUiwIw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Immunophenotypic variation within AML with mutated NPM1. Myeloid and monocytic AML-NPM1 show distinct patterns of CD34 and HLA-DR expression. The most common blast phenotype in myeloid AML-NPM1 was a CD34−/HLA-DR− phenotype (69 [49%] of 141). In monocytic AML-NPM1, a majority of cases were negative for CD34 and positive for HLA-DR (80 [82%] of 98).
Immunophenotypic variation within AML with mutated NPM1. Myeloid and monocytic AML-NPM1 show distinct patterns of CD34 and HLA-DR expression. The most common blast phenotype in myeloid AML-NPM1 was a CD34−/HLA-DR− phenotype (69 [49%] of 141). In monocytic AML-NPM1, a majority of cases were negative for CD34 and positive for HLA-DR (80 [82%] of 98).
The DN AML-NPM1 group included a higher proportion of female patients (female/male ratio, 1.96:1), and patients in this group were older at diagnosis (median age, 67.7 years) than patients in the myeloid (median age, 64.3 years) or monocytic group (median age, 63.4 years), although these differences did not reach statistical significance (Table 1). Peripheral blood blast percentage was significantly lower for patients in the monocytic group (33.0%) than for patients in the DN (64.8%; P = .005 vs monocytic group) or myeloid group (40.3%; P = .046 vs monocytic group), although recognition of promonocytes as blast equivalents could potentially have contributed to this result.
Blast phenotype correlates with genetic findings in NPM1-mutated AML
NPM1 mutation as a sole aberration was seen in only 2% of cases, whereas additional comutations were present in 98% (234 of 239) of cases (Figure 2). The median number of total mutations per case (including NPM1 mutation) was 3 (range, 1-7 mutations), and there was no difference in the median number of mutations present in the 3 phenotypic groups. The most commonly comutated genes were FLT3 (48% of cases overall, including ITD [41%] and TKD mutations [10%]), genes regulating DNA methylation (81% of cases overall, including DNMT3A [43%], TET2 [23%], IDH1 [18%], and IDH2 [21%]), and components of the RAS pathway (30% of cases overall, including NRAS, KRAS, CBL, and PTPN11; Figure 3; Table 2). Splicing factor mutations were seen in 12% of patients (n = 29) and were predominantly SRSF2 mutations (n = 25; 10%), which occurred almost exclusively in combination with TET2 or IDH1/2 mutations (Figure 2). TET2, IDH1, and IDH2 mutations were generally mutually exclusive, but 5 cases harbored 2 of these mutations (2 with TET2 + IDH1, 2 with TET2 + IDH2, and 1 with IDH1 + IDH2). Patients with TET2 and IDH1/2 mutations were significantly older at diagnosis (median age, 65.7 years; range, 14-89 years) than patients without these comutations (median age, 61.9 years; range, 21-81 years; P = .002).
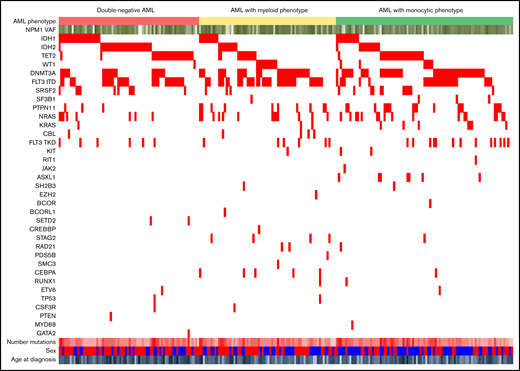
Comutations in AML with mutated NPM1. Each column represents a single patient. All identified comutations are shown. For AML phenotype, red = DN, yellow = myeloid, and green = monocytic. For age at diagnosis, NPM1 VAF, and number of mutations, darker colors correspond to higher values. For sex, blue = male and red = female.
Comutations in AML with mutated NPM1. Each column represents a single patient. All identified comutations are shown. For AML phenotype, red = DN, yellow = myeloid, and green = monocytic. For age at diagnosis, NPM1 VAF, and number of mutations, darker colors correspond to higher values. For sex, blue = male and red = female.
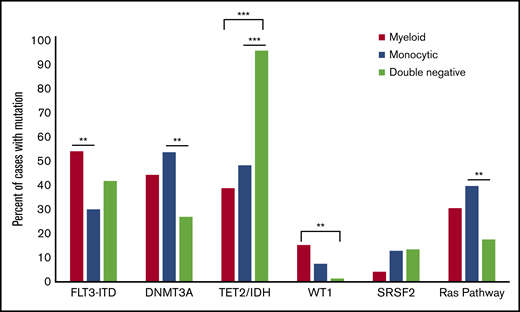
Comutations vary by blast phenotype in AML with mutated NPM1.TET2 and IDH1/2 comutations are significantly more frequent in cases with a DN blast phenotype (P < .0001 comparing all 3 groups), whereas DNMT3A mutations are significantly less common in this group (P = .002 comparing all 3 groups). RAS pathway mutations included NRAS, KRAS, CBL, and PTPN11 mutations. **P < .01, ***P < .0001.
Comutations vary by blast phenotype in AML with mutated NPM1.TET2 and IDH1/2 comutations are significantly more frequent in cases with a DN blast phenotype (P < .0001 comparing all 3 groups), whereas DNMT3A mutations are significantly less common in this group (P = .002 comparing all 3 groups). RAS pathway mutations included NRAS, KRAS, CBL, and PTPN11 mutations. **P < .01, ***P < .0001.
There were significant differences in the most common comutations, depending on blast phenotype (Figure 3; Table 2). FLT3-ITD mutations were significantly less common in monocytic AML-NPM1 (30%) compared with myeloid AML-NPM1 (54%; P = .008) but not compared with DN AML-NPM1 (42%). Seventy-one (96%) of 74 DN AML-NPM1 cases harbored TET2 and/or IDH1/2 mutations, which was significantly more frequent than in myeloid (39%) or monocytic cases (48%; P < .00001 for both). In contrast, DNMT3A mutations were significantly less common in DN AML-NPM1 (27%) as compared with monocytic cases (54%; P = .003) and were also less common compared with myeloid cases (44%), although this did not reach statistical significance after correction for multiple comparisons (P = .08). WT1 mutations were significantly less common in DN cases (1%) when compared with myeloid (15%; P = .008) but not monocytic cases (8%), whereas RAS pathway mutations were significantly less common in DN cases (18%) when compared with monocytic (40%; P = .008) but not myeloid cases (31%).
Figure 4A shows the distribution of VAFs at diagnosis for NPM1 and the most common comutations. The median VAFs for NPM1, DNMT3A, TET2, and IDH1/2 were all relatively high, ranging from 39.6% (NPM1) to 45.6% (IDH2), consistent with NPM1 and other comutations occurring together within the dominant clone in a majority of cases. VAFs for NRAS, PTPN11, and FLT3-TKD mutations were generally lower, suggesting these comutations were often present within subclones. The relatively high VAFs for NPM1, TET2, and IDH1/2 hindered determinations of mutational hierarchy with respect to clonal architecture. However, there was variation in the relationship between NPM1 and TET2 and IDH1/2 VAFs, depending on blast phenotype (Figure 4B). In patients with DN AML-NPM1, there was a strong correlation between NPM1 VAF and TET2 or IDH1/2 VAF (r = 0.8), such that patients with higher NPM1 VAFs showed higher comutation VAFs. In contrast, there was no significant relationship between NPM1 VAF and TET2 or IDH1/2 VAF in patients with monocytic AML-NPM1 (r = 0.05). The myeloid AML-NPM1 group contained fewer patients and showed an intermediate pattern (r = 0.39).
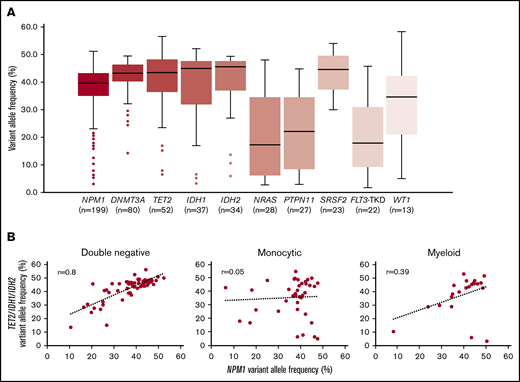
Correlation between NPM1 and comutation VAFs. (A) Box and whisker plots show VAFs at diagnosis for NPM1 and the most common comutations (n = 199). (B) Scatter plots showing the correlation between NPM1 VAF and TET2 and IDH1/2 VAFs in cases with DN (n = 61), monocytic (n = 40), and myeloid (n = 22) immunophenotypes.
Correlation between NPM1 and comutation VAFs. (A) Box and whisker plots show VAFs at diagnosis for NPM1 and the most common comutations (n = 199). (B) Scatter plots showing the correlation between NPM1 VAF and TET2 and IDH1/2 VAFs in cases with DN (n = 61), monocytic (n = 40), and myeloid (n = 22) immunophenotypes.
NPM1 VAF at diagnosis was recently shown to correlate with outcome.23 Patients with a myeloid phenotype showed a median NPM1 VAF of 42.1%, which was significantly higher than that in patients with monocytic (P = .002) or DN phenotypes (P = .02). There was no significant difference in median NPM1 VAF at diagnosis between patients with DN (38.4%) and monocytic blast phenotypes (38.3%).
Patient outcome varies by blast phenotype in AML-NPM1
We found significant differences in patient outcome associated with the different blast phenotypes. RFS and OS were evaluated in patients who received standard induction chemotherapy (n = 172). Comparing the 3 immunophenotypic groups, patients with a DN blast phenotype had the longest median RFS (64.7 months; Figure 5A) and OS (66.5 months; Figure 5B), whereas patients with a myeloid phenotype had the shortest RFS (8.4 months) and OS (20.2 months), with patients with a monocytic phenotype having intermediate RFS (20.6 months; P < .0001 comparing all 3 groups) and OS (44.3 months; P = .01 comparing all 3 groups). Importantly, there was no difference in the rate of SCT overall or the rate of SCT in first complete remission between the 3 groups (Table 1). The effects of SCT in first complete remission and age at diagnosis on OS are shown in supplemental Figure 2A-B, respectively.

Kaplan-Meier analysis of patients with NPM1-mutated AML with different blast phenotypes who received standard induction chemotherapy. All analyses included patients who received standard induction chemotherapy (n = 172 in total). (A-B) Patients with a DN blast phenotype (n = 44) showed significantly prolonged RFS (64.7 months) (A) and OS (66.5 months) (B). Patients in the myeloid group (n = 55) showed significantly shortened RFS (8.4 months) and OS (20.2 months), with patients with a monocytic phenotype (n = 77) showing intermediate RFS (20.6 months; P < .0001 comparing all 3 groups) and OS (44.3 months; P = .01 comparing all 3 groups). (C-D) Looking specifically at patients harboring TET2 or IDH1/2 comutations, patients with a DN blast phenotype (n = 42) showed significantly prolonged RFS (64.7 vs 11.0 months; P = .0004) (C) and OS (66.5 vs 21.3 months; P = .01) (D), compared with patients lacking a DN phenotype (n = 54).
Kaplan-Meier analysis of patients with NPM1-mutated AML with different blast phenotypes who received standard induction chemotherapy. All analyses included patients who received standard induction chemotherapy (n = 172 in total). (A-B) Patients with a DN blast phenotype (n = 44) showed significantly prolonged RFS (64.7 months) (A) and OS (66.5 months) (B). Patients in the myeloid group (n = 55) showed significantly shortened RFS (8.4 months) and OS (20.2 months), with patients with a monocytic phenotype (n = 77) showing intermediate RFS (20.6 months; P < .0001 comparing all 3 groups) and OS (44.3 months; P = .01 comparing all 3 groups). (C-D) Looking specifically at patients harboring TET2 or IDH1/2 comutations, patients with a DN blast phenotype (n = 42) showed significantly prolonged RFS (64.7 vs 11.0 months; P = .0004) (C) and OS (66.5 vs 21.3 months; P = .01) (D), compared with patients lacking a DN phenotype (n = 54).
DN AML-NPM1 showed a very high frequency of TET2 and/or IDH1/2 mutations (96%), raising the possibility that the outcomes in this group were related to the presence of these mutations. However, when compared specifically with the subset of non-DN cases with TET2/IDH1/2 comutations (n = 73), patients with a DN blast phenotype still showed significantly prolonged median RFS (64.7 vs 11.0 months; P = .0004; Figure 5C) and median OS (66.5 vs 21.3 months; P = .01; Figure 5D). This was despite a higher rate of FLT3-ITD positivity in DN cases (44%) as compared with non-DN cases (27%; P = .003) when looking specifically at patients with TET2/IDH1/2 mutations. There was notably no significant difference in outcome in the DN blast group between patients who harbored TET2 comutations (undefined median RFS and OS) and those who had IDH1/2 comutations (RFS, 37.8 months; OS, 66.5 months).
We next examined the impact of TET2 and IDH1/2 mutations on survival and found that there was no significant difference in RFS or OS in patients with and without these mutations in the cohort as a whole (Figure 6A-B). However, when present in monocytic AML-NPM1, these mutations were associated with shortened RFS and OS (Figure 6C-D), although this difference reached significance only for RFS. Conversely, despite the nearly ubiquitous presence of TET2 or IDH1/2 mutations in the DN group, these patients had the longest survival of all phenotypic groups. Of the 3 patients with DN AML-NPM1 lacking TET2/IDH1/2 comutations, only 1 had follow-up data available and received standard induction chemotherapy; this patient had an OS of 14.4 months. The presence or absence of TET/IDH1/2 comutations had no impact on survival within the non-DN myeloid AML-NPM1 group (data not shown). DNMT3A mutations had no effect on outcome in the cohort overall (RFS, 15.2 months; OS, 26.5 months for patients with DNMT3A mutations vs RFS, 17.4 months; OS, 37.8 months for patients without DNMT3A mutations) or when looking specifically within any of the 3 phenotypic groups (data not shown). Previous work has suggested that the negative effect of FLT3-ITD mutations in the context of AML-NPM1 is most pronounced in the presence of a DNMT3A comutation.1 Similarly, we found that FLT3-ITD mutations showed a more adverse effect on outcome in the presence of DNMT3A mutations, a difference which trended toward significance (supplemental Figure 3A-B). However, there was no significant difference in the frequency of NPM1/DNMT3A/FLT3-ITD cooccurrence between DN, myeloid, and monocytic AML-NPM1 groups (supplemental Figure 3C).
![Kaplan-Meier analysis examining the effect of TET2 or IDH1/2 mutations in patients with NPM1-mutated AML who received standard induction chemotherapy. All analyses included patients who received standard induction chemotherapy (n = 172 in total). (A-B) The presence of TET2 or IDH1/2 mutations had no effect on RFS (A) or OS (B) when examining the entire cohort of AML-NPM1s (RFS, 17.4 months; OS, 37.8 months for patients with TET2/IDH mutations [n = 96] vs RFS, 15.2 months; OS, 44.3 months for patients without TET2/IDH mutations [n = 80]). (C-D) In patients with monocytic AML-NPM1, TET2 and IDH1/2 comutations (n = 35) were associated with shortened RFS (13.8 vs 32.3 months in patients without TET2/IDH comutations [n = 42]; P = .026) (C) and OS (38.1 vs 88.4 months in patients without TET2/IDH comutations) (D).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/21/10.1182_bloodadvances.2019000328/3/m_advancesadv2019000328f6.png?Expires=1767887123&Signature=jgeJMTpWwWQExQE7D58YtT7rEeTwXZkUZjtCQdVUx2Thpq6zTUBBMzEnDgOIewZfLO1rud~i6EEEHzeeDqs32~qwmabltBNT0DYWJxXZOhfFxwkapdEH4MZfWXvGD30KqbDlHXwlu9ooZdWZlTq63opTQY-aRR7y1tkrt71gnXMqbH9MaAUWEu92KHjmU27QdCBsK0-N~vqA0AlU5hBcENhWVjDihBeWoncemvkYA6c70lTdl0~~yODRztcFklEuiJpIUdAwa1V4MnapGlxVUKfVCVlbkEzCcJzS5xPKfBL-fZFSrqp-MQ5h~kA4Bpqi5qfqsVD89Dk7xaT6A~F3bA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Kaplan-Meier analysis examining the effect of TET2 or IDH1/2 mutations in patients with NPM1-mutated AML who received standard induction chemotherapy. All analyses included patients who received standard induction chemotherapy (n = 172 in total). (A-B) The presence of TET2 or IDH1/2 mutations had no effect on RFS (A) or OS (B) when examining the entire cohort of AML-NPM1s (RFS, 17.4 months; OS, 37.8 months for patients with TET2/IDH mutations [n = 96] vs RFS, 15.2 months; OS, 44.3 months for patients without TET2/IDH mutations [n = 80]). (C-D) In patients with monocytic AML-NPM1, TET2 and IDH1/2 comutations (n = 35) were associated with shortened RFS (13.8 vs 32.3 months in patients without TET2/IDH comutations [n = 42]; P = .026) (C) and OS (38.1 vs 88.4 months in patients without TET2/IDH comutations) (D).
Kaplan-Meier analysis examining the effect of TET2 or IDH1/2 mutations in patients with NPM1-mutated AML who received standard induction chemotherapy. All analyses included patients who received standard induction chemotherapy (n = 172 in total). (A-B) The presence of TET2 or IDH1/2 mutations had no effect on RFS (A) or OS (B) when examining the entire cohort of AML-NPM1s (RFS, 17.4 months; OS, 37.8 months for patients with TET2/IDH mutations [n = 96] vs RFS, 15.2 months; OS, 44.3 months for patients without TET2/IDH mutations [n = 80]). (C-D) In patients with monocytic AML-NPM1, TET2 and IDH1/2 comutations (n = 35) were associated with shortened RFS (13.8 vs 32.3 months in patients without TET2/IDH comutations [n = 42]; P = .026) (C) and OS (38.1 vs 88.4 months in patients without TET2/IDH comutations) (D).
Multivariable analysis was performed to evaluate for factors independently associated with RFS and OS. Patients with a monocytic blast phenotype showed differences in outcome (particularly RFS) depending on the presence or absence of TET2/IDH comutations in univariate analysis of the subgroups; therefore, the monocytic group was further divided into 2 groups of patients, those with and without TET2/IDH comutations, for the purposes of the multivariable analysis. Variables in the univariate analysis were: age (continuous), total number of mutations (continuous), NPM1 VAF at diagnosis (continuous), presence of FLT3-ITD, DNMT3A, and TET2/IDH mutations, phenotypic group (myeloid, monocytic ± TET2/IDH comutation, and DN), SCT in first complete remission, white blood cell count at diagnosis (continuous), platelet count at diagnosis (continuous), and peripheral blood blast percentage at diagnosis (continuous). In the final multivariable model (Table 3), FLT3-ITD mutations were independently associated with both RFS and OS. Age at diagnosis was independently associated with OS, and SCT in first complete remission was independently associated with RFS. Additionally, blast phenotype independently influenced both RFS and OS; as compared with the myeloid group, both the presence of a DN blast phenotype and the presence of a monocytic blast phenotype in the absence of TET2/IDH comutations were associated with significantly decreased risk of relapse or death. In contrast, the risk of relapse or death associated with a monocytic blast phenotype in combination with TET2/IDH comutations was not significantly different from that in the myeloid phenotypic group.
Discussion
Here we describe, to our knowledge, the largest cohort of AML with mutated NPM1 with complete immunophenotypic and genetic data. We have identified phenotypic and genetic heterogeneity within AML-NPM1, including subgroups with significantly different outcomes after induction chemotherapy. A large subset of AML-NPM1 (31%) lacks both CD34 and HLA-DR expression. Patients with this blast phenotype (DN AML-NPM1) almost universally harbor TET2 or IDH1/2 comutations and show significantly prolonged RFS and OS compared with other patients with AML-NPM1. In contrast, patients with non-DN AML-NPM1 who also lack evidence of monocytic differentiation (myeloid AML-NPM1) showed significantly shortened RFS and OS. Our results suggest that, in addition to clinical and genetic variables, blast phenotype should be considered when assessing prognosis in AML-NPM1.
The genetic heterogeneity within our data set is similar to that reported in recently published large AML studies.1,8-13 The frequency of DNMT3A mutations in our cohort was slightly lower than the frequencies published in these studies (43% in our cohort vs 45%-54% in cited studies), whereas TET2, IDH1, and IDH2 comutation frequencies were slightly higher (TET2: 23% in our cohort vs 8%-49%; IDH1: 18% in our cohort vs 7%-18%; IDH2: 21% in our cohort vs 14%-17%). Interestingly, these previously published studies included a predominance of younger patients. In our cohort of unselected cases of AML-NPM1, patients with TET2 or IDH1/2 comutations were significantly older at diagnosis (median age, 65.7 years) than patients without these comutations (median age, 61.9 years; P = .002). Therefore, the slightly higher frequencies of these comutations in our cohort may reflect the inclusion of older patients as well as normal population variation.
Although the presence of TET2 or IDH1/2 mutations was a feature common to nearly all patients in the DN cohort, the improved outcomes seen in patients with a DN blast phenotype did not seem to be related solely to the presence these comutations, because patients in the DN group continued to show significantly improved outcomes when compared specifically with non-DN patients with TET or IDH1/2 comutations. Moreover, these mutations conferred an adverse prognosis to the monocytic group, where they were associated with shortened RFS. We observed a strong positive relationship between NPM1 VAFs and TET2 and IDH1/2 VAFs in the DN group but not in the monocytic group; this difference may suggest that DN AML-NPM1 is a more homogeneous disease, with a distinct mechanism of pathogenesis from monocytic AML-NPM1, which may represent a more heterogeneous disease with more varied clonal architecture. Taken together, these findings highlight the importance of interpreting genetic findings in the context of blast phenotype within AML-NPM1. Past studies have shown conflicting results regarding the prognostic impact of IDH and TET2 mutations in AML-NPM1.10,25-28 Our results may help to explain this uncertainty, because prior studies did not account for blast phenotype when assessing the prognostic significance of these comutations.
The improved survival in the DN group cannot be explained merely by genetic factors known to affect prognosis in AML-NPM1; the rate of FLT3-ITD mutations in the DN group was not significantly different from that seen in the myeloid group and was in fact higher than the rate of FLT3-ITD mutations seen in the monocytic group. There have been conflicting recent reports on whether NPM1 VAF is independently associated with prognosis.23,29 Although the median NPM1 VAF in the myeloid group (the group with the poorest outcome) was the highest of the 3 groups, we did not find a significant effect of NPM1 VAF on RFS or OS in the entire cohort. There have also been conflicting data as to whether DNMT3A mutations affect outcome in AML-NPM1.30 In our cohort, the presence of a DNMT3A mutation was not associated with worse outcome, although we did find a borderline stronger negative effect of FLT3-ITD mutations when present in the context of a DNMT3A comutation, consistent with previous reports.1 We found that DNMT3A mutations were significantly less common in patients with a DN blast phenotype. Studies in mice have suggested that, in the context of NPM1 and IDH2 mutations, DNMT3A mutations confer stem/progenitor cell properties and promote maintenance of an undifferentiated state31 ; this may explain why, in our cohort, cases with a more mature, DN blast phenotype showed a lower frequency of DNMT3A mutations. Interestingly, patients in the DN cohort showed superior outcomes despite older age, which might have negatively affected prognosis. Patients in the monocytic group also showed longer survival compared with patients in the non-DN myeloid group, which could be related to the higher rate of FLT3-ITD positivity and the higher median NPM1 VAF at diagnosis in the myeloid group as compared with the monocytic group. Overall, our data suggest that the stage of blast differentiation, as reflected by blast immunophenotype, is an important contributor to patient outcome in AML-NPM1, as evidenced by its retention as a significant predictor of outcome in the multivariable model.
A recent study by van Galen et al32 demonstrated heterogeneity in cell-type composition in AML, with different clusters of cases showing varying abundances of cells at distinct stages of differentiation, and showed prognostic associations with cell-type composition. Cases with a high abundance of HSC- or progenitor-like cells did poorly compared with cases with a predominance of more differentiated granulocyte-monocyte precursor–like cells, all of which represented acute promyelocytic leukemia based on genetic findings. This is in line with prior studies demonstrating that a higher proportion of primitive leukemia stem cells correlates with poor outcome, possibly because of chemotherapy resistance within this cell population.33 Although van Galen et al32 found that AML with mutated NPM1 fell predominantly into the progenitor-like or monocyte-like clusters, our data suggest that DN AML-NPM1 may be characterized by more differentiated blasts, similar to the granulocyte-monocyte precursor–like group, which may explain the superior outcomes in DN AML-NPM1 patients. Additional studies are needed to understand the differential effects of TET or IDH mutations based on blast phenotype in AML-NPM1.
Although TET2/IDH mutations were present in nearly all cases of AML-NPM1 with a DN blast phenotype, these mutations were not exclusive to this phenotypic group, suggesting that these comutations are necessary but not sufficient to promote the DN phenotype. Both TET2 and IDH mutations are thought to lead to loss of TET2 function,14,34 and recent work has suggested that WT1 mutations also affect TET2 function, leading to loss of TET2-mediated hydroxymethylcytosine production.35,36 However, we found that WT1 mutations were extremely uncommon in cases with a DN blast phenotype, present in only 1 (1%) of 74 cases. This suggests that WT1 mutations are not conducive to a DN phenotype and that, at least in this subgroup of patients, the overall functional impact of WT1 mutations may be distinct from that of TET2 and IDH mutations.
With the recent approval of targeted IDH1 and IDH2 inhibitors to treat IDH-mutated AML, rapid identification of patients with IDH mutations will become increasingly important. We identified a DN blast phenotype in >50% (49 of 92) of patients with AML-NPM1 harboring IDH1/2 comutations. Although patients with a DN blast phenotype showed superior outcomes when treated with standard induction chemotherapy, patients with this phenotype were generally older in age, and a subset did not undergo induction therapy. Therefore, our findings suggest that identification of a DN blast phenotype in the context of AML-NPM1 may help to quickly identify patients who might benefit from targeted IDH inhibition.
The accumulated evidence to date suggests that AML with mutated NPM1 is not a uniform group, but rather encompasses biologically diverse diseases. Our data identify 3 distinct phenotypes in AML with mutated NPM1: a DN phenotype, a monocytic phenotype, and a non-DN myeloid phenotype. The former represents the cohort with the best outcome and is almost universally associated with TET2/IDH comutations. In contrast, the latter group represents the cohort with the worst outcome; the median OS of 20.2 months and RFS of 8.4 months were substantially shorter than those in the other groups. Although validation in other patient cohorts is warranted, these results emphasize the utility of immunophenotyping newly diagnosed AMLs with mutated NPM1 in identifying patients at higher risk. Future studies in a larger cohort of patients could potentially address the question of whether patients with a non-DN myeloid phenotype may benefit from alternative treatment strategies.
Acknowledgment
The authors acknowledge Frank C. Kuo for his invaluable contributions to this and many other projects. He was brilliant and kind and will be greatly missed.
Authorship
Contribution: E.F.M. and O.P. designed the study, collected data, performed data analysis, and prepared the manuscript; and R.P.H., N.A., and A.C.S. contributed to data collection, data analysis, and manuscript review.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Emily F. Mason, Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, 1301 Medical Center Dr, 4603A TVC, Nashville, TN 37232; e-mail: emily.f.mason@vumc.org.