

Key Points
Engineered human models of high-fatality pediatric leukemia are relevant to uncover disease biomarkers and therapeutic vulnerabilities.
NUP98-KDM5A–associated AMKL expresses SELP, MPIG6B, and NEO1 biomarkers and is sensitive to pharmacologic inhibition with ruxolitinib.

Abstract
Acute megakaryoblastic leukemia (AMKL) represents ∼10% of pediatric acute myeloid leukemia cases and typically affects young children (<3 years of age). It remains plagued with extremely poor treatment outcomes (<40% cure rates), mostly due to primary chemotherapy refractory disease and/or early relapse. Recurrent and mutually exclusive chimeric fusion oncogenes have been detected in 60% to 70% of cases and include nucleoporin 98 (NUP98) gene rearrangements, most commonly NUP98-KDM5A. Human models of NUP98-KDM5A–driven AMKL capable of faithfully recapitulating the disease have been lacking, and patient samples are rare, further limiting biomarkers and drug discovery. To overcome these impediments, we overexpressed NUP98-KDM5A in human cord blood hematopoietic stem and progenitor cells using a lentiviral-based approach to create physiopathologically relevant disease models. The NUP98-KDM5A fusion oncogene was a potent inducer of maturation arrest, sustaining long-term proliferative and progenitor capacities of engineered cells in optimized culture conditions. Adoptive transfer of NUP98-KDM5A–transformed cells into immunodeficient mice led to multiple subtypes of leukemia, including AMKL, that phenocopy human disease phenotypically and molecularly. The integrative molecular characterization of synthetic and patient NUP98-KDM5A AMKL samples revealed SELP, MPIG6B, and NEO1 as distinctive and novel disease biomarkers. Transcriptomic and proteomic analyses pointed to upregulation of the JAK-STAT signaling pathway in the model AMKL. Both synthetic models and patient-derived xenografts of NUP98-rearranged AMKL showed in vitro therapeutic vulnerability to ruxolitinib, a clinically approved JAK2 inhibitor. Overall, synthetic human AMKL models contribute to defining functional dependencies of rare genotypes of high-fatality pediatric leukemia, which lack effective and rationally designed treatments.
Introduction
Acute megakaryoblastic leukemia (AMKL) is a cytological subgroup of acute myeloid leukemia (AML) that harbors megakaryocytic lineage markers (eg, CD41, CD42, and CD61). AMKL occurrence is almost entirely restricted to children (usually <3 years of age), occurring in only 1% of adult AML1 cases, often after progression from myelodysplastic syndromes. AMKL represents ∼10% of pediatric AML cases and has been long recognized for its poor prognosis.2 Clinically, AMKL diagnosis remains challenging due to marked myelofibrosis and resulting hypocellular bone marrow (BM) aspirates, lack of disease-specific cell surface markers and, until recently, limited molecular markers. Interestingly, children with Down syndrome (DS; trisomy 21) are at increased risk of developing AMKL,3,4 but they have better survival than non-DS patients,5 suggesting 2 distinct types of disease.
Oncogenic gene fusions are recurrent, mutually exclusive mutations found in more than 70% of non-DS pediatric AMKL.6 These include CBFA2T3-GLIS2 (∼18%), MLL rearrangements (MLLr; ∼17%), HOX rearrangements (HOXr; ∼15%), NUP98-KDM5A (N5A; ∼11%), and RBM15-MKL1 (∼10%).6 Patients presenting with AMKL assigned to the CBFA2T3-GLIS2, MLLr, and N5A molecular subsets typically experience poor outcomes.6,7
Oncoproteins encoded by NUP98 fusion transcripts comprise the N-terminal glycine-leucine-phenylalanine-glycine repeat motifs of NUP98 and the C-terminal domains of the fusion partner (∼31 NUP98 fusion partners reported).8,9 N5A fusions typically comprise the C-terminal plant homeodomain of KDM5A (H3K4 methylation recognition) fused to NUP98, but not the Jumonji catalytic domain of the demethylase. Although N5A is most frequently identified in AMKL (French-American-British [FAB] M7 AML), it is occasionally associated with other FAB subtypes of childhood AML (all of them except FAB M3 were excluded from the analysis)9,10 and is now emerging as a recurrent fusion in pediatric acute erythroid leukemia (FAB M6 AML).11,12 In general, NUP98 oncogenic rearrangements (NUP98r) are associated with a variety of de novo and therapy-related acute myeloid and T-cell lymphoblastic leukemia subtypes, in addition to other hematopoietic malignancies.8,9 Evidence suggests that NUP98 fusions sustain the activation of key cell fate regulator genes, such as the HOXA (similar to MLLr) and HOXB cluster genes,13,14 in part by aberrantly recruiting epigenetic modifier complexes.15,16
N5A is sufficient to transform mouse cells, giving rise to CD34+CD117+ AML in vivo, characterized by transcriptional upregulation of Hoxa5, -7, -9, and -10; Gata3; Meis1; Eya; and Pbx1, with confirmed epigenetic activation of the Hoxa gene cluster.14 However, the CD34−CD41+CD61+ leukemic blasts typically seen in pediatric AMKL are not found in this animal model.14 This may be explained by physiological differences between mouse and human systems,17,18 differences in the developmental transcriptional programs of the initiating cells (fetal vs adult hemopoietic stem cells),19 and/or the diversity of cells susceptible to N5A-driven transformation. The paucity of N5A pediatric leukemia samples greatly limits molecular and functional studies of AMKL. In addition, human models of de novo N5A AMKL are currently lacking, hampering biomarker and potential drug target discovery.
Here, we present a validated protocol to generate renewable AMKL models in the physiological context of primitive human hematopoietic cells, driven by the overexpression of N5A in umbilical cord blood (CB) cells. In this model, the N5A fusion oncogene was a potent inducer of maturation arrest, sustaining long-term proliferative and progenitor capacities of engineered cells in our optimized culture conditions. Adoptive transfer of N5A-transformed cells led to de novo AMKL and other leukemia subtypes in xenograft models. N5A-driven human AMKL models faithfully mimicked the pediatric disease phenotypically and molecularly. The integrated transcriptomic and proteomic characterization of human models and primary samples of NUP98r AMKL revealed SELP, MPIG6B, and NEO1 to be distinctive disease biomarkers and pointed to JAK-STAT signaling pathway upregulation. Using an in vitro pharmacological approach, we show that primary xenografts of NUP98r AMKL are sensitive to JAK-STAT pathway inhibition with ruxolitinib and tofacitinib, as opposed to normal CD34+ CB cells or an MLLr cell line. Overall, the synthetic human models of NUP98r leukemias reported here will pave the way to defining subtype-specific functional dependencies of rare diseases plagued by dismal cure rates.
Methods
Patient samples
All pediatric AML samples (n = 6 patients) and associated clinical information described in supplemental Table 1 were collected after receiving informed consent from the patients and project approval from the Research Ethics Boards of Centre Hospitalier Universitaire (CHU) Sainte-Justine, in agreement with the Declaration of Helsinki. Biobanking was conducted by the Québec Leukemia Cell Bank (BCLQ, Montréal, QC, Canada).
Lentiviral vectors
FLAG-tagged NUP98-KDM5A coding sequence (kindly provided by David Allis, Rockefeller University, New York, NY)14 was subcloned using standard procedures into a MNDU lentiviral expression vector containing a GFP reporter gene (a gift from Keith Humphries, BC Cancer Agency, Vancouver, BC, Canada, and Donald B. Kohn, UCLA, Los Angeles, CA),20,21 as indicated in Figure 1A. VSV-G pseudotyped lentiviral vectors were produced and titered with HEK293T cells, according to standard protocols.
![Overexpression of NUP98-KDM5A efficiently induces maturation block and sustains the proliferative and progenitor capacities of CB-CD34+cells. (A) Experimental procedures used to establish in vitro models of N5A-driven leukemia. CD34+ cells isolated from single-donor CB were seeded in 96-well plates and infected with lentiviral particles carrying the chimeric NUP98-KDM5A oncogene. The lentiviral vector encodes FLAG-tagged NUP98-KDM5A and a GFP reporter gene, driven by MNDU3 and PGK promoters, respectively. Independent cell lines derived from each well were grown for 3 to 5 days in optimized culture conditions before GT evaluation and further in vitro expansion (20% of the cells from each well). (B) CD34+GFP+ enrichment in long-term cultures of CB-CD34+ cells transduced with a control (CTL, n = 4) or NUP98-KDM5A (N5A, n = 12) vector. (C) Short-term proliferation kinetic of transduced cells in independent cultures of CB-CD34+ cells transduced with N5A or control lentiviral vector. Cultures were initiated from 2 independent CBs (eg, CB1 and CB2) transduced with control (n = 6 per CB) or N5A (n = 14 per CB) lentiviral vector, as indicated. (D) Fluorescence-activated cell sorting profiles showing the time course of GFP and CD34 expression in 2 independent samples transduced with control (eg, CTL_C) or N5A lentiviral vector (eg, N5A_A). Transduced CB-CD34+ cells were derived from a single donor. (E) Giemsa-stained cytospins showing immature cellular morphology of an N5A-expressing cell line (N5A_C, bottom) at day 80 and differentiation of matched-CTL cells at day 59. Original magnification ×1000. (F) Acquisition by flow cytometry showing differentiation of control cells (GFP+CD34− C-KIThi) and a maturation arrest of N5A-transduced cells (GFP+CD34+ C-KITlow). (G) Graph showing the percentage of GFP+KITlow immature cells in each indicated culture, defined as median fluorescence intensity <1.5 × 104 for KITlow cells; n = 3 independent experiments, n = 4 CB units, n = 43 cultures of N5A cells, and n = 19 cultures of CTL-cells. (H) Clonogenic progenitor frequency for freshly isolated (day 0, n = 2) and CTL or N5A-transduced CB-CD34+ cells, plated at days 8 and 88 of culture (n = 2 for CTL; n = 4 for N5A; mean ± standard error of the mean [SEM]). Phenotypic classification of clonogenic progenitors is presented in supplemental Figure 1. (I) Representative image of a typical long-term colony generated from the N5A_C cell line at day 60 of culture. Top: bright field microscopy; bottom: epifluorescence microscopy. Original magnification ×10. **** P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/21/10.1182_bloodadvances.2019030981/3/m_advances030981f1.png?Expires=1764961219&Signature=j4xbgrXopEVyuXwXgPmYORNbuHft66B879o0-bSJNOYI91AMjr4n1W6sVFSLPQ~xEQ5t2b~wsm3H~aGKlKOq-EuLkMwdH3xPUmPf3CH7RnBuDR9LF~d3NGR55hnUtLcTS~YUu1TmbDZgFjLrzTstGqILhBIDOcj2wMjCgdL3a4nhyPpN5ZZkNmlleaufCS1Fdl4AQ4w6fJ3gwqhIXoH9XG20EnHFeaycK6XRSTqIqiiG9WRuw12p-2MqNa2iNNDdlMDNqPUAgxMFYHAmaaGyAFgq0X4OCtj5ehC9uk4YF-lcRLnhHMuN5zlGhpW7dOc3stpMKOsVWZDoysKCpFbatg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Overexpression of NUP98-KDM5A efficiently induces maturation block and sustains the proliferative and progenitor capacities of CB-CD34+cells. (A) Experimental procedures used to establish in vitro models of N5A-driven leukemia. CD34+ cells isolated from single-donor CB were seeded in 96-well plates and infected with lentiviral particles carrying the chimeric NUP98-KDM5A oncogene. The lentiviral vector encodes FLAG-tagged NUP98-KDM5A and a GFP reporter gene, driven by MNDU3 and PGK promoters, respectively. Independent cell lines derived from each well were grown for 3 to 5 days in optimized culture conditions before GT evaluation and further in vitro expansion (20% of the cells from each well). (B) CD34+GFP+ enrichment in long-term cultures of CB-CD34+ cells transduced with a control (CTL, n = 4) or NUP98-KDM5A (N5A, n = 12) vector. (C) Short-term proliferation kinetic of transduced cells in independent cultures of CB-CD34+ cells transduced with N5A or control lentiviral vector. Cultures were initiated from 2 independent CBs (eg, CB1 and CB2) transduced with control (n = 6 per CB) or N5A (n = 14 per CB) lentiviral vector, as indicated. (D) Fluorescence-activated cell sorting profiles showing the time course of GFP and CD34 expression in 2 independent samples transduced with control (eg, CTL_C) or N5A lentiviral vector (eg, N5A_A). Transduced CB-CD34+ cells were derived from a single donor. (E) Giemsa-stained cytospins showing immature cellular morphology of an N5A-expressing cell line (N5A_C, bottom) at day 80 and differentiation of matched-CTL cells at day 59. Original magnification ×1000. (F) Acquisition by flow cytometry showing differentiation of control cells (GFP+CD34− C-KIThi) and a maturation arrest of N5A-transduced cells (GFP+CD34+ C-KITlow). (G) Graph showing the percentage of GFP+KITlow immature cells in each indicated culture, defined as median fluorescence intensity <1.5 × 104 for KITlow cells; n = 3 independent experiments, n = 4 CB units, n = 43 cultures of N5A cells, and n = 19 cultures of CTL-cells. (H) Clonogenic progenitor frequency for freshly isolated (day 0, n = 2) and CTL or N5A-transduced CB-CD34+ cells, plated at days 8 and 88 of culture (n = 2 for CTL; n = 4 for N5A; mean ± standard error of the mean [SEM]). Phenotypic classification of clonogenic progenitors is presented in supplemental Figure 1. (I) Representative image of a typical long-term colony generated from the N5A_C cell line at day 60 of culture. Top: bright field microscopy; bottom: epifluorescence microscopy. Original magnification ×10. **** P < .0001.
Overexpression of NUP98-KDM5A efficiently induces maturation block and sustains the proliferative and progenitor capacities of CB-CD34+cells. (A) Experimental procedures used to establish in vitro models of N5A-driven leukemia. CD34+ cells isolated from single-donor CB were seeded in 96-well plates and infected with lentiviral particles carrying the chimeric NUP98-KDM5A oncogene. The lentiviral vector encodes FLAG-tagged NUP98-KDM5A and a GFP reporter gene, driven by MNDU3 and PGK promoters, respectively. Independent cell lines derived from each well were grown for 3 to 5 days in optimized culture conditions before GT evaluation and further in vitro expansion (20% of the cells from each well). (B) CD34+GFP+ enrichment in long-term cultures of CB-CD34+ cells transduced with a control (CTL, n = 4) or NUP98-KDM5A (N5A, n = 12) vector. (C) Short-term proliferation kinetic of transduced cells in independent cultures of CB-CD34+ cells transduced with N5A or control lentiviral vector. Cultures were initiated from 2 independent CBs (eg, CB1 and CB2) transduced with control (n = 6 per CB) or N5A (n = 14 per CB) lentiviral vector, as indicated. (D) Fluorescence-activated cell sorting profiles showing the time course of GFP and CD34 expression in 2 independent samples transduced with control (eg, CTL_C) or N5A lentiviral vector (eg, N5A_A). Transduced CB-CD34+ cells were derived from a single donor. (E) Giemsa-stained cytospins showing immature cellular morphology of an N5A-expressing cell line (N5A_C, bottom) at day 80 and differentiation of matched-CTL cells at day 59. Original magnification ×1000. (F) Acquisition by flow cytometry showing differentiation of control cells (GFP+CD34− C-KIThi) and a maturation arrest of N5A-transduced cells (GFP+CD34+ C-KITlow). (G) Graph showing the percentage of GFP+KITlow immature cells in each indicated culture, defined as median fluorescence intensity <1.5 × 104 for KITlow cells; n = 3 independent experiments, n = 4 CB units, n = 43 cultures of N5A cells, and n = 19 cultures of CTL-cells. (H) Clonogenic progenitor frequency for freshly isolated (day 0, n = 2) and CTL or N5A-transduced CB-CD34+ cells, plated at days 8 and 88 of culture (n = 2 for CTL; n = 4 for N5A; mean ± standard error of the mean [SEM]). Phenotypic classification of clonogenic progenitors is presented in supplemental Figure 1. (I) Representative image of a typical long-term colony generated from the N5A_C cell line at day 60 of culture. Top: bright field microscopy; bottom: epifluorescence microscopy. Original magnification ×10. **** P < .0001.
Human CD34+ hematopoietic stem/progenitor cell isolation and culture and lentiviral transduction
Umbilical CB units were collected with the consent of the mothers and distributed by Héma-Québec's Public Cord Blood Bank (Montréal, QC, Canada). The protocol was approved by the Research Ethics Board of CHU Sainte-Justine (approval number 3453). CD34+ human hematopoietic stem/progenitor cells (eg, CB-CD34+ cells) were isolated from fresh CB (≤30 hours) using the EasySep Human CD34 Positive Selection Kit (StemCell Technologies, Vancouver, BC, Canada). Preparations with ≥90% CB-CD34+ cells were used, as determined by flow cytometry. CB-CD34+ cells were maintained in optimized culture conditions22,23 at 37°C with 5% CO2 in humidified air. Lentiviral transduction and the clonogenic progenitor cell assay are described in the supplemental Methods.
Xenotransplantation
NOD-scid IL2Rgnull (NSG) and NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ (NSG-SGM3) mice from Jackson Laboratories (Bar Harbor, ME) were bred in pathogen-free conditions at the animal care facility of CHU Sainte-Justine. Mice were kept in ventilated microinsulator cages and provided with sterilized food and acidified water. Mouse husbandry, xenotransplantation, and follow-up procedures were approved by the CHU Sainte-Justine Institutional Animal Ethic Committee (supplemental Tables 2 and 3; supplemental Methods).
Flow cytometry
Cell surface marker staining was performed with antibodies listed in supplemental Table 4, as described in the supplemental Methods. Details related to automated cell counting and cytometers are summarized in the supplemental Methods. Data were analyzed with FACSDiva (BD Biosciences) and FlowJo (BD Biosciences) software.
Histopathology and microscopy
Peripheral blood smears, cytospin preparations of BM and spleen cells, and organ touch preparations were stained with Giemsa according to standard protocols. Sagittal sections of tibial BM were stained with hematoxylin-phloxine-saffron according to standard procedures. Detailed procedures are described in the supplemental Methods.
Molecular studies
RNA isolation.
Total RNA was extracted from patient BM stored in RNAlater with a QIAamp RNA Blood Mini or RNeasy Micro kit (Qiagen, Germantown, MD). Total RNA from CB-CD34+ cells, NUP98-KDM5A cell lines, and xenografts was extracted with TRIzol and Direct-zol RNA MiniPrep (Thermo Fisher Scientific, Waltham, MA, and Zymo Research, Irvine, CA).
RNA sequencing and bioinformatics analysis.
Samples used for RNA sequencing are listed in supplemental Table 5. Sequencing was performed using an Illumina HiSeq2000 or Nextseq500 with 200- and 150-bp cycle paired-end runs, respectively. Sequences were aligned to the reference human genome version GRCh38 (Gencode, version 26), using STAR version 2.5.1b.24 Gene expression was estimated directly from STAR as read count values and computed using RSEM, version 1.2.28.25 RNA sequencing was performed at the Institute for Research in Immunology and Cancer (IRIC) Genomics Platform (Montréal, QC, Canada). Additional details about library preparation, RNA sequencing, variant calling, gene ontology term enrichment, and principal component and differential expression analyses are presented in the supplemental Methods and supplemental Table 6.
RT-PCR.
Detection of N5A, NEO1, and KDM5B transcripts was performed by RT-PCR, using primers and conditions listed in supplemental Table 7 and supplemental Methods.
Statistical analysis
Statistical analyses were performed with GraphPad Prism v7.03. Bars and error bars represent means and SEM, respectively, unless indicated otherwise.
Chromatin immunoprecipitation sequencing, cell surface proteomics, and pharmacological inhibition analyses are described in the supplemental Methods.
Results
NUP98-KDM5A is a potent inducer of proliferation and maturation block ex vivo
An experimental protocol was established to overexpress NUP98-KDM5A (N5A) in CB stem/progenitor cells, a physiologically relevant context in terms of cell of origin and ontogeny with respect to pediatric AMKL (Figure 1A; supplemental Methods). Following initial gene transfer (GT) assessment, based on GFP fluorescence determined by flow cytometry, independent cultures were evaluated for total cell counts, cytology, immunophenotype, and progenitor cell frequencies at regular intervals. There was a progressive enrichment of CD34+GFP+ N5A cells over time, completely overriding the culture by day 80, even when initial GT rates were below 10% (>80% GFP+ cells by day 80; Figure 1B-C). Control cells were terminally differentiated into C-KIThi CD34− mastocytes and largely exhausted by days 40 to 60 of culture (Figure 1B,D-F). The N5A-transduced cells displayed maturation arrest on cytological examination (Figure 1E) and were CD34+ and C-KITlow (Figure 1F). This was observed in 57% to 79% of wells at day 42 of culture in all experiments (Figure 1G), pointing to a strong impact on cell fate from overexpression of the N5A fusion in human stem/progenitor cells. Although clonogenic progenitors were not detected at day 28 of culture in the control condition (frequency >1 in 200 seeded cells), they were maintained in vitro at high frequency for the N5A-transduced cells for at least 3 months (supplemental Figure 1; Figure 1H-I). Overall, these results suggest that the N5A fusion oncogene is a potent inducer of maturation arrest, sustaining long-term proliferative and progenitor capacities in vitro.
Generation of NUP98-KDM5A AMKL xenografts
A fraction of the fusion oncogene-carrying cells (N5A-cells) were used to induce de novo leukemias in mouse xenograft models (70% of each independent transduction; Figure 2A). Leukemia development showed a latency of 27 to 71 weeks at a frequency of 2 of 9 to 2 of 3 animals (n = 6 experiments; 3 to 28 mice/experiment; supplemental Tables 2 and 3). Mice presented clinically overt signs of disease (tachypnea, hunchback, weight loss, fatigue, and pallor). Grafts engineered to overexpress N5A gave rise to multiple leukemia subtypes in recipient mice, including AMKL, other acute myeloid leukemia subtypes/non-AMKL myeloid leukemia (AML-O), B- and T-cell acute lymphoblastic leukemia (B-ALL and T-ALL; Figure 2B; supplemental Tables 2 and 3). Expression of the N5A fusion transcript was confirmed in leukemic cells by RT-PCR (Figure 2C).

Overexpression of N5A fusion in CB-CD34+cells induces acute megakaryoblastic leukemia and multilineage leukemia subtypes in xenograft models. (A) Representation of the experimental procedures used to establish in vivo models of N5A-driven leukemia. Human CD34+ cells were isolated from CB and transduced as described in Figure 1A. After GT evaluation, 70% of the cells from each well were injected into a primary recipient mouse. Leukemia xenograft cells were collected and characterized phenotypically, molecularly, and functionally. (B) Distribution of generated xenograft models classified by leukemia subtypes based on molecular markers and cytology analyses (supplemental Table 3). Models originated from 6 experimental groups initiated from 7 independent CB samples (supplemental Table 2). (C) Detection of N5A fusion transcript expression by RT-PCR with RNA isolated from BM or spleen (Sp) cells of leukemic mice, as indicated. Normal nontransduced CB-CD34+ sample was used as negative control (CTL). (D-J) AMKL xenograft models. (D) Brittle white bones and mild splenomegaly were observed in AMKL xenograft models (xAMKL-3, N5A vector) compared with control xenograft-recipient mice (CTL, empty vector). (E) Representative hematoxylin-phloxine-saffron–stained longitudinal sections of tibia BM harvested from xCTL and xAMKL-3 recipient mice. (F) Blasts infiltration percentage (hCD45lowCD41+/CD61+ cells) in BM and spleen of primary AMKL (n = 6). (G) Giemsa-stained peripheral blood (PBL) smear and BM cytospin from an AMKL primary (1ary) recipient mouse (xAMKL-3) highlighting the presence of leukemic blasts (48 weeks after transplantation). (H) Fluorescence-activated cell sorting (FACS) profiles revealing typical hCD45lowCD34−CD41+ megakaryoblasts in the BM of a primary recipient mouse (xAMKL-3), along with CD45hiCD3+-activated T cells. The spleen is infiltrated by CD45hiCD3+ activated T cells with hCD45lowCD34−CD41+ barely detectable. Characterization of an additional mouse xenograft model (xAMKL-1) with detailed T-cell immunophenotype is shown in supplemental Figure 2. (I) Giemsa-stained BM cytospin and spleen touch preparations showing leukemic blasts derived from a secondary (2ary) recipient mouse with a 2.2 × 106 xAMKL-3 BM–cell transplant (33 weeks after transplantation). (J) FACS profiles revealing hCD45lowCD34−CD41+GFP+ megakaryoblasts in the BM and spleen of the secondary recipient mouse. *P < .05. HSC, hemopoietic stem cell; LSC, leukemic stem cell.
Overexpression of N5A fusion in CB-CD34+cells induces acute megakaryoblastic leukemia and multilineage leukemia subtypes in xenograft models. (A) Representation of the experimental procedures used to establish in vivo models of N5A-driven leukemia. Human CD34+ cells were isolated from CB and transduced as described in Figure 1A. After GT evaluation, 70% of the cells from each well were injected into a primary recipient mouse. Leukemia xenograft cells were collected and characterized phenotypically, molecularly, and functionally. (B) Distribution of generated xenograft models classified by leukemia subtypes based on molecular markers and cytology analyses (supplemental Table 3). Models originated from 6 experimental groups initiated from 7 independent CB samples (supplemental Table 2). (C) Detection of N5A fusion transcript expression by RT-PCR with RNA isolated from BM or spleen (Sp) cells of leukemic mice, as indicated. Normal nontransduced CB-CD34+ sample was used as negative control (CTL). (D-J) AMKL xenograft models. (D) Brittle white bones and mild splenomegaly were observed in AMKL xenograft models (xAMKL-3, N5A vector) compared with control xenograft-recipient mice (CTL, empty vector). (E) Representative hematoxylin-phloxine-saffron–stained longitudinal sections of tibia BM harvested from xCTL and xAMKL-3 recipient mice. (F) Blasts infiltration percentage (hCD45lowCD41+/CD61+ cells) in BM and spleen of primary AMKL (n = 6). (G) Giemsa-stained peripheral blood (PBL) smear and BM cytospin from an AMKL primary (1ary) recipient mouse (xAMKL-3) highlighting the presence of leukemic blasts (48 weeks after transplantation). (H) Fluorescence-activated cell sorting (FACS) profiles revealing typical hCD45lowCD34−CD41+ megakaryoblasts in the BM of a primary recipient mouse (xAMKL-3), along with CD45hiCD3+-activated T cells. The spleen is infiltrated by CD45hiCD3+ activated T cells with hCD45lowCD34−CD41+ barely detectable. Characterization of an additional mouse xenograft model (xAMKL-1) with detailed T-cell immunophenotype is shown in supplemental Figure 2. (I) Giemsa-stained BM cytospin and spleen touch preparations showing leukemic blasts derived from a secondary (2ary) recipient mouse with a 2.2 × 106 xAMKL-3 BM–cell transplant (33 weeks after transplantation). (J) FACS profiles revealing hCD45lowCD34−CD41+GFP+ megakaryoblasts in the BM and spleen of the secondary recipient mouse. *P < .05. HSC, hemopoietic stem cell; LSC, leukemic stem cell.
AMKL xenograft recipients were generated in 5 independent experiments (n = 6 cases; supplemental Table 2). Mice with AMKL had white bones and mild splenomegaly at necropsy (Figure 2D; supplemental Table 3). Infiltration by leukemic blasts was observed on blood smear and BM histocytological preparations and consisted of CD45loCD34−CD41+CD61+ cells with characteristic megakaryoblast features, including cytoplasmic blebs (Figure 2E,G-H; supplemental Figure 2A-B), faithfully phenocopying human AMKL. The extent of BM and spleen infiltration by GFP+CD41+CD61+ megakaryocytic leukemic blasts ranged between 11% and 55% and 0.1% and 4%, respectively (Figure 2F; supplemental Figure 2B,D; supplemental Table 3). A population of CD45hiCD3+ cells was observed in the blood, BM, and spleen of 3 of 6 AMKL-bearing mice (Figure 2H; supplemental Figure 2B-F), in the absence of clinical graft-versus-host disease. The CD3+ cells co-expressed HLA-DR and either CD4 or CD8, and on cytology appeared as activated lymphocytes (supplemental Figure 2C-D; n = 1 analyzed xenograft). Tumor-reactive T-lymphoid cell populations have been detected in association with human AML transplanted into NSG mice.26 We detected a T-cell population in 3 of 6 AMKL xenograft models, with or without expression of GFP (a marker expressed by the vector bearing the fusion oncogene; Figure 2H; supplemental Figure 2B). Transcriptomic and gene ontology term enrichment analyses of 2 independent CD3+ cell populations isolated along AMKL xenografts support a signature of activated T-cells (supplemental Figure 2G; supplemental Table 5).
To assess the self-renewal capacity of leukemic cells derived from 1 representative AMKL xenograft, freshly collected BM cells were transplanted into 2 secondary recipient mice. Both recipients developed AML with shorter latency than the disease in the primary host (33.3 and 32.9 vs 48 weeks). Compared with the primary host, secondary recipients presented similar clinical signs apart from severe splenomegaly. Only the leukemic cells from the myeloid compartment were detected in both secondary recipients, either leading to AMKL (CD41+CD71+; Figure 2I-J) or AML-O (CD41−CD71+, data not shown). These results suggest the presence of self-renewing primary megakaryoblast cells that can be serially transplanted in addition to other leukemic clones and/or primitive leukemia-initiating cells poised for alternative lineage decisions.
Overexpression of N5A in CB-CD34+ cells generates multiple subtypes of leukemia
In addition to AMKL, overexpression of N5A in CB-CD34+ cells led to the development of AML-O (CD34−CD41−CD33+/−CD36+), with histological features of myeloid blasts (supplemental Figure 3B-C; supplemental Table 3). Furthermore, in 8 of 25 synthetic primary leukemias generated, the blasts harbored molecular markers and cytological characteristics of either B-ALL (n = 7; CD34+CD38+/−CD19+CD10+) or T-ALL (n = 1; CD3+CD4+CD8−CD7+/−HLA-DR+; supplemental Figure 3D-G; supplemental Table 3). Splenomegaly and splenic infiltration by leukemic blasts in primary recipients were generally more profound for B- and T-ALL and AML-O, compared with AMKL (Figure 2F; supplemental Figure 3A; supplemental Table 3).
Individual leukemias presented a subtype-specific expression signature and clustered according to immunophenotypic subgroups (Figure 3). Principal component analysis of transcriptomic data sets from N5A synthetic models, a local pediatric AMKL cohort, and the TARGET AML cohort revealed that N5A AMKL or AML-O xenografts cluster with AMKL and acute monocytic leukemia (eg, FAB M5), respectively (Figure 3B; supplemental Figure 4A). The multilineage leukemogenic potential of N5A probably reflects distinct cells of origin for each disease entity or can be attributed to other parameters, such as fusion expression levels or cooperating mutations. Nonetheless, the generation of specific leukemia subtypes was highly constant and reproducible (Figure 3A; supplemental Figure 4B; supplemental Tables 2 and 3). Overall, the heterogeneity observed in the engineered N5A leukemia subtypes mirrors that of human disease driven by NUP98 fusions.
![Distinct expression profiles of NUP98-KDM5A leukemia subtypes compared with normal CB CD34+cells. (A) Hierarchical clustering heat map of the top 100 genes differentially expressed between the 3 leukemia subtypes observed in N5A xenograft models (AMKL xenograft [xAMKL], n = 4; B-ALL xenograft [xB-ALL], n = 4; AML-O xenograft [xAML-O], n = 3) and normal CB CD34+ cells (CB-CD34+ cells, n = 4). Read count data were converted into log2 values and represented according to a blue-yellow-red–colored gradient scale. The size factor values, reflecting the correction factor for normalization of the relative depth of sample libraries, are represented by a gradient of green tones according to the size factor scale bar. Sample conditions are described by a binary (pink, C/control; blue, E/experimental leukemia) color code. (B) Principal component analysis plots of the first and second principal components of NUP98-KDM5A models, patients, and controls, calculated using the 500 most variable genes between all conditions.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/21/10.1182_bloodadvances.2019030981/3/m_advances030981f3.png?Expires=1764961219&Signature=UqWfNhDScUIDy8Xgu0DfLh7C82BaaK1hnFRCdCkAl9ZMPrrxQfb6KNR3FtrzuoHICFnx3wy7VL8RbwuOTYRtIFBZcaLqrUf6MgbODoL6ZHsfg1HVUhDJt5gqjC2n1afVe-EjcRMhVwd~cAFkg1tT8DXDQjC2VzQWDOoYytx2-WKqNiaWOV9Q7wrI~iZS6KZ1tBlGaVliHjwshy2Qqa7Fqa~HM6tzQjpsTkSUxci95YY~45dyP2vQ-iquZHTxwyTyQ--gSouEb4nQh-7hFuHjny~uclo24EC4V9ZqUn2QDWHum4df9bAOu4vfj2n~q188zw1dAZKprQ5t7Ysw7eOdTw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Distinct expression profiles of NUP98-KDM5A leukemia subtypes compared with normal CB CD34+cells. (A) Hierarchical clustering heat map of the top 100 genes differentially expressed between the 3 leukemia subtypes observed in N5A xenograft models (AMKL xenograft [xAMKL], n = 4; B-ALL xenograft [xB-ALL], n = 4; AML-O xenograft [xAML-O], n = 3) and normal CB CD34+ cells (CB-CD34+ cells, n = 4). Read count data were converted into log2 values and represented according to a blue-yellow-red–colored gradient scale. The size factor values, reflecting the correction factor for normalization of the relative depth of sample libraries, are represented by a gradient of green tones according to the size factor scale bar. Sample conditions are described by a binary (pink, C/control; blue, E/experimental leukemia) color code. (B) Principal component analysis plots of the first and second principal components of NUP98-KDM5A models, patients, and controls, calculated using the 500 most variable genes between all conditions.
Distinct expression profiles of NUP98-KDM5A leukemia subtypes compared with normal CB CD34+cells. (A) Hierarchical clustering heat map of the top 100 genes differentially expressed between the 3 leukemia subtypes observed in N5A xenograft models (AMKL xenograft [xAMKL], n = 4; B-ALL xenograft [xB-ALL], n = 4; AML-O xenograft [xAML-O], n = 3) and normal CB CD34+ cells (CB-CD34+ cells, n = 4). Read count data were converted into log2 values and represented according to a blue-yellow-red–colored gradient scale. The size factor values, reflecting the correction factor for normalization of the relative depth of sample libraries, are represented by a gradient of green tones according to the size factor scale bar. Sample conditions are described by a binary (pink, C/control; blue, E/experimental leukemia) color code. (B) Principal component analysis plots of the first and second principal components of NUP98-KDM5A models, patients, and controls, calculated using the 500 most variable genes between all conditions.
Expression profiles of engineered AMKL xenografts correlate with human disease
Transcriptomic analyses of the generated N5A-AMKL xenografts closely matched those of N5A-patient AMKL samples overall, with correlation coefficients >0.90 (Figure 4A; see supplemental Tables 1, 3, and 5 for sample descriptions). A list of the most differentially expressed genes between N5A-AMKL patient samples and xenografts, as compared with CB-CD34+ cells is presented in Figure 4B (≥10-fold difference and ≥10 fragments per kilobase million [FPKM] in AMKL; a complete list is presented in supplemental Figure 7). There is a marked upregulation of the 5′HOXB cluster genes and MEIS2 in N5A-AMKL, with HOXB7, -8, and -9 ranking among the top 10 most differentially expressed genes (Figure 4B). As reported for most NUP98r and MLLr AML,8,27,28 there is concomitant upregulation of the 5′HOXA cluster genes (in particular HOXA9) and expression of MEIS1 in N5A-AMKL (Figure 4C). Epigenetic activation of HOXB8 was further confirmed by chromatin immunoprecipitation sequencing analyses (increased H3K4me3-active marks, decreased H3K27me3-repressive marks on promoters of N5A-transduced cell lines vs controls; supplemental Figure 5). Gene Set Enrichment Analyses (GSEAs) of transcriptomic data sets of N5A-AMKL identified several significant matches to signatures that correlate with pediatric AMKL samples,27 STAT5A signaling,29 and NPM1-mutated AML30 (HOXA and -B cluster genes upregulation; Figure 4D-E). A list of consistently upregulated genes matching each signature is provided in supplemental Figure 6.
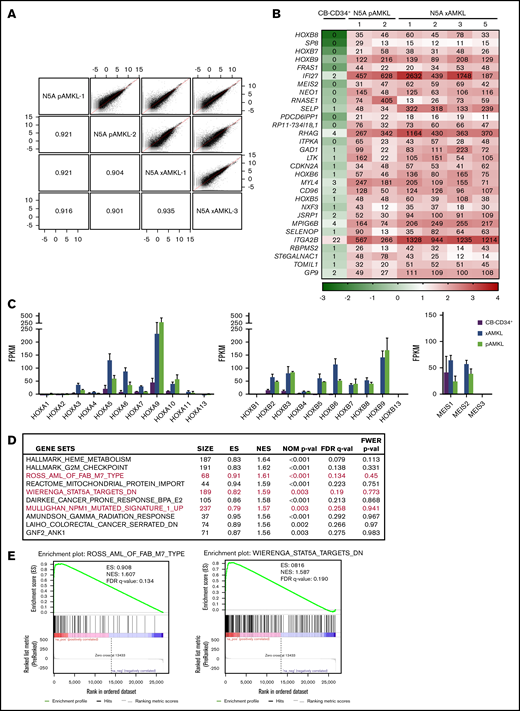
Molecular characterization of N5A AMKL. (A) Scatterplots showing pairwise correlation of gene expression values (log FPKM, RNAseq) between N5A AMKL derived from pediatric patients and xenograft models (N5A pAMKL and xAMKL, respectively). For each scatterplot the Pearson correlation coefficients (r) are indicated in mirroring cells. The red diagonal line represents a perfect correlation (r = 1.0). (B) Top 30 genes differentially expressed by at least 10-fold in a sampling of BM cells derived from 2 patients and 4 xenograft models presenting N5A AMKL, as compared with normal CB-CD34+ cells (n = 4). Genes with expression values of ≥10 FPKM for all N5A AMKL samples and fold changes ≥10 compared with CB-CD34+ samples are displayed (see supplemental Figure 7 for the complete list). The indicated FPKM values are represented by a logarithmic color scale (log10). (C) Expression of HOXA/HOXB cluster and MEIS genes in N5A AMKL samples derived from patients (n = 2) and xenograft models (n = 4) compared with control CB-CD34+ cells (n = 4). Values are presented as the mean ± SEM. (D) Top 10 ranked gene sets in the GSEA of genes upregulated in N5A AMKL patients and xenograft models compared with CB-CD34+ samples. Enrichment plots for selected gene sets (in red) are depicted in panel E and in supplemental Figure 6.
Molecular characterization of N5A AMKL. (A) Scatterplots showing pairwise correlation of gene expression values (log FPKM, RNAseq) between N5A AMKL derived from pediatric patients and xenograft models (N5A pAMKL and xAMKL, respectively). For each scatterplot the Pearson correlation coefficients (r) are indicated in mirroring cells. The red diagonal line represents a perfect correlation (r = 1.0). (B) Top 30 genes differentially expressed by at least 10-fold in a sampling of BM cells derived from 2 patients and 4 xenograft models presenting N5A AMKL, as compared with normal CB-CD34+ cells (n = 4). Genes with expression values of ≥10 FPKM for all N5A AMKL samples and fold changes ≥10 compared with CB-CD34+ samples are displayed (see supplemental Figure 7 for the complete list). The indicated FPKM values are represented by a logarithmic color scale (log10). (C) Expression of HOXA/HOXB cluster and MEIS genes in N5A AMKL samples derived from patients (n = 2) and xenograft models (n = 4) compared with control CB-CD34+ cells (n = 4). Values are presented as the mean ± SEM. (D) Top 10 ranked gene sets in the GSEA of genes upregulated in N5A AMKL patients and xenograft models compared with CB-CD34+ samples. Enrichment plots for selected gene sets (in red) are depicted in panel E and in supplemental Figure 6.
Recurrent cooperating mutations were not identified with RNA sequencing of the model leukemia, including variants in gene candidates associated with AMKL (eg, RB1, GATA1, and MPL; see supplemental Table 6 for the list of evaluated genes).6,31 Specifically, RB1 deletions that are associated with N5A AMKL6 were not detected; however, the number of samples remained too limited to draw definitive conclusions about their absence. In summary, using an optimized strategy, it is possible to generate synthetic human models of N5A-AMKL that closely match human disease in terms of expression profiles.
Synthetic human N5A-AMKL models unravel disease-specific biomarkers
To identify cell membrane markers that are specifically upregulated in N5A-AMKL, we mined transcriptomic data sets of synthetic and patient N5A-AMKL samples. Using a manually curated list of cell membrane–associated proteins, we generated a set of gene candidates encoding potential cell surface markers. Candidate genes that were expressed on N5-AMKL (FPKM >5 in all samples) at levels at least 10-fold higher than on CB-CD34+ cells (upper limit of CB-CD34+ expression of FPKM <5; leukemia-specific expression) are listed in Figure 5A and supplemental Table 8. Most putative AMKL biomarkers are essentially not expressed in N5A B-ALL or in AML-O xenografts (supplemental Table 8). External validation, using data from the largest cohort of rare pediatric AMKL (n = 73 RNAseq data sets)6 and the TARGET pediatric AML project32 (RNA-seq from 284 pediatric AML cases sequenced at the British Columbia Cancer Research Centre-Genome Sciences Centre; Figure 5B), showed that expression of megakaryocyte and platelet inhibitory receptor G6b (MPIG6B) and neogenin (NEO1) was highly specific to AMKL (FAB M7) cases, irrespective of genetic subtype (P ≤ .05 for all pairwise comparisons between FAB M7 and other FAB; Figure 5A-B). P-selectin (SELP) was expressed only in a subset of AMKL genetic subgroups and, notably, was not expressed in CBFA2T3-GLIS2 (INV16) AMKL characterized by high NCAM1 (CD56) expression33 (Figure 5A). High expression of Rh-associated glycoprotein (RHAG) was associated with M6 and M7 subtypes (Figure 5B). In sharp contrast, markers such as CD96 or LTK were expressed in most AML cases, irrespective of cytological subgroups. High expression level of RHAG or NEO1 or GP9/CD42a combined to ITGA2B/CD41 was highly predictive of an AML subtype M7 (Figure 5C-D). Expression of SELP was confirmed at the protein level by flow cytometry on hCD45+CD41+GFP+ blasts derived from the BM of an N5A AMKL primary xenograft recipient (Figure 5E). The expression of NEO1 was validated by RT-PCR in 2 AMKL xenograft samples and in primary specimens of NUP98-BPTF AMKL34 (diagnosis and measurable residual disease [MRD] monitoring), given the absence of commercially available antibodies robustly validated for flow cytometry (Figure 5F-G). Using proteomic analysis, we validated the expression of 411 proteins at the surface of NUP98-BPTF AMKL patient–derived xenograft (PDX) cells, including NEO1, MPIG6B, and SELP (Figure 6A; supplemental Table 9). Overall, our human models of N5A-driven leukemia revealed SELP, MPIG6B, and NEO1 to be specific biomarkers that define AMKL.

Specific biomarkers of AMKL. (A) Heat map of expression values in FPKM (RNAseq) of 7 top-ranked genes encoding cell surface proteins that are differentially expressed by a least 10-fold in leukemic BM cells derived from patients (pAMKL) or mouse xenograft models (xAMKL) in the middle panel, as compared with normal CB-CD34+ cells in the left panel, and expressed at low levels (≤5 FPKM) in CB-CD34+ cells. Genes with expression values of ≥5 FPKM in all N5A AMKL samples, fold change ≥10 compared with CB-CD34+ samples, and low expression levels (≤5 FPKM) in CB-CD34+ cells are listed in supplemental Table 8. Expression of the selected genes in leukemic BM cells derived from patients presenting other genetic subtypes of AMKL (non-N5A pAMKL) or non-AMKL leukemia subtype involving NUP98 rearrangement (NUPr pAML) are also indicated. Expression of ITGA2B/CD41, ITGB3/CD61, and NCAM1 is indicated in red for comparison. Expression of the selected genes in the validation cohort6 are shown in the right panel, represented as mean expression per indicated genetic group. (B) Distribution of selected gene expression values (FPKM) in BM-derived pediatric AML cells classified according to the FAB nomenclature (M0-M7); n = 284 pediatric AML cases from the National Cancer Institute (NCI) TARGET database. Horizontal lines represent median values. Pairwise gene expression comparisons between M7 and other FAB categories were performed with a Mann-Whitney rank sum test with the Benjamini-Hochberg correction (shown below graphs). M7 leukemia (n = 11) involved the following exclusive genetic lesions: NUP98-KDM5A (n = 1), CBFA2T3-GLIS2 (n = 4), KMT2A-MLLT10 (n = 1), and RBM15-MKL1 (n = 1). (C) Pairwise scatterplot representations showing correlative expression of the indicated genes in a pediatric AML (NCI, TARGET database). Representations were created with the bioinformatic tool MiSTIC.51 AML classified as FAB M7 or M6 are indicated in red and blue, respectively. (D) Selection of specimens expressing the highest levels of RHAG, NEO1, GP9, or ITGB3/CD61 combined with ITGA2B/CD41 significantly enriches for FAB M7 AML (eg, AMKL). M7: 8 of 9 selected P = 6.1e-11. See data set and bioinformatic tool in panel C. (E) SELP expression, as assessed by flow cytometry in freshly isolated CB-CD34+ cells and in AMKL BM cells from an N5A mouse xenograft model. (F) Expression of NEO1 detected by RT-PCR using RNA derived from the BM of leukemic xenograft models or from CB-CD34+ cells. KDM5B expression was used as the endogenous control. (G) Expression of NEO1 detected by RT-PCR using RNA derived from the BM of an infant with NUP98-BPTF AMKL. RNA was isolated at diagnosis (NUP98r pAMKL-3D) and after 2 cycles of chemotherapy when disease burden was ∼2% by cytology (NUP98r pAMKL-3 MRD). Human placental RNA was used as the nontumor control. ns/not significant P > .05; *P < .05; **P < .01; ***P < .001; ****P < .0001.
Specific biomarkers of AMKL. (A) Heat map of expression values in FPKM (RNAseq) of 7 top-ranked genes encoding cell surface proteins that are differentially expressed by a least 10-fold in leukemic BM cells derived from patients (pAMKL) or mouse xenograft models (xAMKL) in the middle panel, as compared with normal CB-CD34+ cells in the left panel, and expressed at low levels (≤5 FPKM) in CB-CD34+ cells. Genes with expression values of ≥5 FPKM in all N5A AMKL samples, fold change ≥10 compared with CB-CD34+ samples, and low expression levels (≤5 FPKM) in CB-CD34+ cells are listed in supplemental Table 8. Expression of the selected genes in leukemic BM cells derived from patients presenting other genetic subtypes of AMKL (non-N5A pAMKL) or non-AMKL leukemia subtype involving NUP98 rearrangement (NUPr pAML) are also indicated. Expression of ITGA2B/CD41, ITGB3/CD61, and NCAM1 is indicated in red for comparison. Expression of the selected genes in the validation cohort6 are shown in the right panel, represented as mean expression per indicated genetic group. (B) Distribution of selected gene expression values (FPKM) in BM-derived pediatric AML cells classified according to the FAB nomenclature (M0-M7); n = 284 pediatric AML cases from the National Cancer Institute (NCI) TARGET database. Horizontal lines represent median values. Pairwise gene expression comparisons between M7 and other FAB categories were performed with a Mann-Whitney rank sum test with the Benjamini-Hochberg correction (shown below graphs). M7 leukemia (n = 11) involved the following exclusive genetic lesions: NUP98-KDM5A (n = 1), CBFA2T3-GLIS2 (n = 4), KMT2A-MLLT10 (n = 1), and RBM15-MKL1 (n = 1). (C) Pairwise scatterplot representations showing correlative expression of the indicated genes in a pediatric AML (NCI, TARGET database). Representations were created with the bioinformatic tool MiSTIC.51 AML classified as FAB M7 or M6 are indicated in red and blue, respectively. (D) Selection of specimens expressing the highest levels of RHAG, NEO1, GP9, or ITGB3/CD61 combined with ITGA2B/CD41 significantly enriches for FAB M7 AML (eg, AMKL). M7: 8 of 9 selected P = 6.1e-11. See data set and bioinformatic tool in panel C. (E) SELP expression, as assessed by flow cytometry in freshly isolated CB-CD34+ cells and in AMKL BM cells from an N5A mouse xenograft model. (F) Expression of NEO1 detected by RT-PCR using RNA derived from the BM of leukemic xenograft models or from CB-CD34+ cells. KDM5B expression was used as the endogenous control. (G) Expression of NEO1 detected by RT-PCR using RNA derived from the BM of an infant with NUP98-BPTF AMKL. RNA was isolated at diagnosis (NUP98r pAMKL-3D) and after 2 cycles of chemotherapy when disease burden was ∼2% by cytology (NUP98r pAMKL-3 MRD). Human placental RNA was used as the nontumor control. ns/not significant P > .05; *P < .05; **P < .01; ***P < .001; ****P < .0001.

Primary NUP98r xenograft cells are vulnerable to JAK-STAT signaling inhibitors. (A) Schematic overview of the procedures used to perform the cell surface proteomic analysis of CD41+ AMKL cells isolated from the spleen of NUP98r (NUP98-BPTF) PDX mice. Cell surface proteins were biotinylated and isolated using streptavidin pulldown and stringent washes. Analysis by liquid chromatography-mass spectrometry allowed for identification of 411 cell surface proteins, including AMKL-specific biomarkers (supplemental Table 9). (B) Top 10 hallmark gene sets enriched in the analysis of the cell surface proteins detected on NUP98r AMKL primary xenograft using Metascape (see supplemental Methods for details). The complete analysis is provided in supplemental Table 10. (C) Dose-response curves and half maximal inhibitory concentrations (IC50) determined for each indicated cell type submitted to a viability assay in presence of an inhibitor or DMSO vehicle (Cell-Titer Glo, 6-day incubation). Experiments were conducted with 4 replicates. CMK, N5A xAMKL (E771, G662), and CB-CD34+ (no. 5/6 and 7); n = 1 experiment. M07e, ML-2, 2ary NTF AMKL PDX (I603, I604); n = 2 experiments.
Primary NUP98r xenograft cells are vulnerable to JAK-STAT signaling inhibitors. (A) Schematic overview of the procedures used to perform the cell surface proteomic analysis of CD41+ AMKL cells isolated from the spleen of NUP98r (NUP98-BPTF) PDX mice. Cell surface proteins were biotinylated and isolated using streptavidin pulldown and stringent washes. Analysis by liquid chromatography-mass spectrometry allowed for identification of 411 cell surface proteins, including AMKL-specific biomarkers (supplemental Table 9). (B) Top 10 hallmark gene sets enriched in the analysis of the cell surface proteins detected on NUP98r AMKL primary xenograft using Metascape (see supplemental Methods for details). The complete analysis is provided in supplemental Table 10. (C) Dose-response curves and half maximal inhibitory concentrations (IC50) determined for each indicated cell type submitted to a viability assay in presence of an inhibitor or DMSO vehicle (Cell-Titer Glo, 6-day incubation). Experiments were conducted with 4 replicates. CMK, N5A xAMKL (E771, G662), and CB-CD34+ (no. 5/6 and 7); n = 1 experiment. M07e, ML-2, 2ary NTF AMKL PDX (I603, I604); n = 2 experiments.
NUP98r xenograft models are vulnerable to in vitro pharmacologic inhibition
JAK-STAT pathway activation, initially suggested by GSEAs of model and NUP98 rearranged patient sample expression profiles (Figure 4D-E), is further supported by hallmark signatures of cell membrane proteomic experiments, using a patient-derived xenograft model (Figure 6A-B). These results corroborated recent studies in the field of pediatric AMKL, suggesting the importance of the JAK-STAT signaling pathway.35,36
As proof of principle, we have thus tested in vitro sensitivity of AMKL cells to clinically approved JAK inhibitors (ruxolitinib, a JAK2/1 inhibitor, and tofacitinib, a JAK3/1 inhibitor), using our model NUP98-KDM5A AMKL along with a NUP98r AMKL PDX,34 and gauged cross toxicity on normal CB cells. We chose the JAK inhibitors ruxolitinib and tofacitinib, as they are currently used in pediatrics in the context of cancer, graft‐versus‐host disease and rheumatoid arthritis. NUP98r AMKL show greater sensitivity to ruxolitinib (IC50, 0.217-0.662 µM) and tofacitinib (IC50, 0.663-3.008 µM) compared with normal CB cells and KMT2A rearranged cell line (IC50 >10 µM for both), showing that synthetic AMKL models can be exploited to test therapeutic vulnerabilities (Figure 6C). This selective therapeutic window was not observed with highly potent chemotherapeutic agents cytarabine and clofarabine inhibiting both normal and leukemic hematopoietic cell types in vitro (Figure 6C). Overall, our work highlights the JAK-STAT pathway along with other candidate mechanisms putatively sustaining NUP98r AMKL (Figures 4D and 6B) and provide the biological substrate to conduct further validation and discovery studies (Figure 6C).
Discussion
Diagnosis and treatment development of rare subtypes of highly lethal pediatric AML have been hampered, at least in part, by the restricted number of patient samples, the hypocellularity of BM specimens (related to fibrosis and dry taps in AMKL), and the lack of molecularly relevant cell lines or animal models reflecting the human disease. We propose a newly optimized pipeline to generate de novo NUP98-KDM5A cell lines and AMKL xenografts from diverse donor genetic backgrounds that phenocopy the human disease. This renewable resource opens the door to in vitro and in vivo studies to define AMKL-specific biomarkers and functional dependencies, something which has not been achievable with rare primary samples.
In vitro, overexpression of NUP98-KDM5A in CB-CD34+ cells induces maturation arrest and sustained proliferation with short latency and high frequency. In vivo, the engineered N5A xenografts give rise to multiple subtypes of leukemia, as seen in humans with hematological malignancies involving NUP98 fusions8 and as reported in NUP98-PHF23 mouse leukemia models yielding de novo acute myeloid/erythroid leukemia and T-ALL, B-ALL, and biclonal leukemia in transgenic mice with latencies of 5 to 18 months.37,38 Importantly, this system enabled the generation of synthetic human AMKL models that closely matched expression profiling of AMKL patient samples,27 providing valuable models to study the disease. The engineered models also demonstrated a strong propensity for N5A to promote the development of non-AMKL AML. This corroborates the fact that N5A is associated with various AML FAB subtypes.10 Thus, for rare pediatric leukemia subtypes, synthetic human leukemia models can contribute to elucidate the full spectrum of disease associated with a given oncogene. To date, N5A or NUP98 fusions are not recurrently associated with B-ALL in humans.8 Whether cytogenetically cryptic NUP98 fusions will be detected at low frequency in larger patient B-ALL cohorts remains a question to be addressed in future clinical studies. The N5A B-ALL generated can also reflect a potential bias in the model system, in particular the use of CB stem-progenitor cells that are equally poised for myeloid or lymphoid lineage commitment.39,40
Overall, expression profiling of the synthetic AMKL xenografts shows significant similarity to patterns reported for AMKL patients,27 with minimal genetic noise. As reported for AML with MLL and NUP98 rearrangements,27 the generated AMKL xenografts are HOXA9 and MEIS1 expressing leukemia, but they also markedly upregulate HOXB9 and MEIS2. A leukemogenic role for MEIS2 in the context of MN1 and AML1-ETO AML is emerging.41,42 However, the contribution of MEIS2 in the development of AMKL remains to be determined. HOXB9 was the gene that correlated most with the expression signature of NPM1-mutated AML30 (supplemental Figure 5). GSEA of transcriptomic data and cartography of proteins expressed at the cell surface of NUP98r leukemia point to activation of JAK-STAT signaling, as observed by other groups.35,36 As proof of principle, we demonstrated selective vulnerability of NUP98r AMKL xenograft cells to clinically approved JAK inhibitors (ruxolitinib and tofacitinib) and lack of cross-toxicity on normal CB cells in vitro.
AMKL xenograft models contribute to identify and validate disease-specific biomarkers, such as NEO1, MPIG6B, and SELP, which are minimally expressed in parental CB-CD34+ cells. NEO1 gene expression is the most highly correlated with the AMKL patient samples signature (supplemental Figure 6A). NEO1 encodes a cell membrane receptor of the immunoglobulin superfamily involved in multiple cellular processes including axonal guidance.43 It may potentially be a regulator of hemopoietic stem cell function in mice.44 MPIG6B encodes a platelet- and megakaryocyte-specific immunoreceptor tyrosine-based inhibitory motif receptor. MPIG6B loss-of-function mutations in humans and mice lead to thrombocytopenia, anemia, and BM myelofibrosis.45-47 NEO1 and MPIG6B are expressed across all AMKL genotypes comprising the largest pediatric AMKL cohort6 and are highly distinctive of a FAB M7 subtype (Figure 5A-B). SELP, expressed in a subset of AMKL genetic subtypes, encodes the receptor P-selectin translocated at the cell surface of stimulated platelets and endothelial cells, and also expressed on megakaryocytes and aged murine hematopoietic stem cells.48,49 Overall, the surface biomarkers upregulated in AMKL, as compared with parental stem/progenitor cells, can be clinically validated for MRD tracking, as observed in this study for NEO1 transcript expression in serial BM aspiration in a case of NUP98-BPTF AMKL34 (Figure 5G), which seemed to correlate with disease burden. At diagnosis, flow cytometry and/or RT-PCR–based AMKL subtype-specific cell marker panels can accelerate and tailor treatment decisions for high-risk leukemia in the challenging context of hypocellular BM specimen.
N5A synthetic leukemia models constitute valuable tools to identify novel and disease-specific biomarkers and will serve to dissect the role of potent oncogenes in leukemogenesis. As the heterogeneous genomic landscape of high-fatality leukemia is being unraveled, synthetic human models will be seminal in identifying physiologically relevant and disease subtype-specific biomarkers and functional dependencies to tailor therapies, such as the identification of RET in the context of MLL-AF9 AML.50
Acknowledgments
The authors thank the patients and their families and members of the pediatric Hematology-Oncology Team of CHU Sainte-Justine for their outstanding support; the members of the BCLQ and the Hematology-Oncology Division of CHU Sainte-Justine for their contribution to the biobanking initiative; Virginie Saillour, Charles Privé, and René Allard (Integrated Centre for Pediatric Clinical Genomics, CHU Sainte-Justine) and Jennifer Huber (IRIC) for assistance with sequencing and data analysis; Benoit Fiset and Julie Cardin (CHU Sainte-Justine) for contributing to bioinformatics analyses; Ines Boufaied of the flow cytometry facility, Elke Küster-Schöck of the microscopy facility, and all the personnel of the humanized mouse core facility (CHU Sainte-Justine); Isabel Boivin (IRIC) and Jean-François Delisle (CHU Sainte-Justine) for help with pharmacological inhibition experiments; Danièle Gagné and Gaёl Dulude of the flow cytometry facility, Julie Hinsinger of the histology division, and Christian Charbonneau of the microscopy facility (IRIC); David Allis for generously providing the NUP98-KDM5A retroviral construct; and Victor Kokta (CHU Sainte-Justine) for help with interpretation of the immunohistology slides.
This study was supported by seminal Cole Foundation Transition Awards, Terry Fox Research Institute (TFRI) New Investigator grants, and a Canadian Cancer Society Research Institute (CCSRI)/Cole Foundation Impact grant (S. Cellot and B.T.W.); an operating grant from the Leukemia and Lymphoma Society of Canada; and funding from the CHU Sainte-Justine Foundation (Fonds d’Innovation Thérapeutique), the Charles Bruneau Foundation (operating grants), and the Canada Foundation for Innovation (John R. Evans Leaders Fund) (S. Cellot). S. Cardin and M.R. are recipients of Cole Foundation postdoctoral and doctoral fellowships, respectively. The Quebec Leukemia Cell Bank is supported by the Cancer Research Network of the Fonds de Recherche du Québec-Santé.
Authorship
Contribution: S. Cardin and M.B. conceived of and performed the experiments, analyzed the data, generated the figures, and wrote the manuscript; M.R., L.L., J.D., and F.C. performed the experiments and reviewed the manuscript; T.M. performed ChIP-Seq experiments, analyzed the data, generated a figure, and reviewed the manuscript; A.R., L.J., J.-R.L., P.G., J.-F.S., J.M., D.S., and B.T.W. performed bioinformatics analyses and reviewed the manuscript; L.A and P.P.R. performed the cell surface proteomic analysis and reviewed the manuscript; S.M. generated a table with clinical annotations and reviewed the manuscript. É.H. shared humanized mouse recipients and reviewed the manuscript; R.K.H. provided a lentiviral construct and guidance in designing the experiments and reviewed the manuscript; H.D. provided guidance in designing the experiments, and reviewed the manuscript; J.H. supervised the biobanking of patient specimens at the BCLQ, and reviewed the manuscript; T.A.G. shared an essential pediatric AMKL RNAseq data set; and S. Cellot conceived the experiments, analyzed the data, supervised the biobanking of patient specimens at the BCLQ, and wrote the manuscript.
Conflict-of-interest disclosure: J.-R.L is a founder of and has an ownership stake in Streamline Genomics. The remaining authors declare no competing financial interests.
Correspondence: Sonia Cellot, CHU Sainte-Justine, 3175 Côte-Sainte-Catherine, Room 6-17-007, Montréal, QC H3T 1C5, Canada; e-mail: sonia.cellot@umontreal.ca.

