Key Points
CMML monocytes exhibit a proinflammatory transcriptional signature, contributing to malignant expansion and increased cardiovascular risk.

Abstract
Chronic myelomonocytic leukemia (CMML) is an aggressive myeloid neoplasm of older individuals characterized by persistent monocytosis. Somatic mutations in CMML are heterogeneous and only partially explain the variability in clinical outcomes. Recent data suggest that cardiovascular morbidity is increased in CMML and contributes to reduced survival. Clonal hematopoiesis of indeterminate potential (CHIP), the presence of mutated blood cells in hematologically normal individuals, is a precursor of age-related myeloid neoplasms and associated with increased cardiovascular risk. To isolate CMML-specific alterations from those related to aging, we performed RNA sequencing and DNA methylation profiling on purified monocytes from CMML patients and from age-matched (old) and young healthy controls. We found that the transcriptional signature of CMML monocytes is highly proinflammatory, with upregulation of multiple inflammatory pathways, including tumor necrosis factor and interleukin (IL)-6 and -17 signaling, whereas age per se does not significantly contribute to this pattern. We observed no consistent correlations between aberrant gene expression and CpG island methylation, suggesting that proinflammatory signaling in CMML monocytes is governed by multiple and complex regulatory mechanisms. We propose that proinflammatory monocytes contribute to cardiovascular morbidity in CMML patients and promote progression by selection of mutated cell clones. Our data raise questions of whether asymptomatic patients with CMML benefit from monocyte-depleting or anti-inflammatory therapies.
Introduction
Chronic myelomonocytic leukemia (CMML) is a genetically heterogeneous hematopoietic stem cell disorder that combines features of a myelodysplastic syndrome (MDS) and a myeloproliferative neoplasm (MPN) and occurs almost exclusively in older individuals.1,-3 The epigenetic regulators TET2 (40%-60% of patients) and ASXL1 (30%-60% of patients)4 and the splicing factor SRSF2 (30%-50%)5,6 are the most frequently mutated genes in CMML. Mutational activation of the RAS signaling pathway is also common,7 and ∼25% of CMML patients have structural or numerical chromosomal aberrations.8 Multivariate analyses suggest that somatic mutations explain only one quarter of the heterogeneity of CMML outcomes, inferring that leukemia-specific factors other than somatic mutations and/or host factors have a major impact on prognosis.9 For instance, aberrant DNA methylation correlates with outcome and response to hypomethylating agents, irrespective of somatic mutations.10,11
Aging causes profound changes in the hematopoietic system. Immune cell subsets and cytokine profiles exhibit patterns of enhanced inflammation, and there is a bias toward polyclonal myelomonocytic differentiation.12,,,-16 At the same time, detection of somatic mutations in blood cells of hematologically normal individuals, variably referred to as clonal hematopoiesis of indeterminate potential (CHIP) or age-related clonal hematopoiesis (ARCH), increases steeply with age.17,,,,,,-24 An increasing body of evidence supports an intimate interplay between a systemic inflammatory state and myeloid neoplasms. The risk of myeloid malignancies is significantly elevated in patients with a history of infection or autoimmune disease,25 and a recent report demonstrated that bacterial infection can promote the expansion of Tet2-deficient hematopoietic cells in mice.25,26 The relationship between inflammation and somatic mutations may be particularly important in the context of monocyte-rich myeloid neoplasms, as a recent study found that CMML patients have a more than twofold increase in the rate of cardiovascular events compared with patients with chronic lymphocytic leukemia.27 Given the extreme skewing of the CMML age distribution (median age of onset, 70 years), we hypothesized that separating age-related from CMML-specific alterations will be critical to mechanistically understand CMML oncogenesis and associated CMML-specific risks.28 As a first step in this direction, we performed comparative transcriptional and DNA methylation analyses of CD14+ monocytes from CMML patients, age-matched (old) healthy controls, and young healthy controls.
Methods
Study population
CD14+ monocytes were isolated from the blood of untreated CMML patients (n = 12; median age 72 years; range, 57-87), age-matched healthy controls (old controls, n = 12; median age, 68 years; range, 62-74) and young healthy controls (young controls, n = 16; median age, 29 years; range, 24-44) and subjected to RNA sequencing. In addition, we performed DNA methylation profiling for the CMML patients and old controls. The University of Utah Institutional Review Board approved the study protocols, and all participants provided written informed consent before sample collection.
Sample processing, monocyte isolation, and DNA and RNA extraction
Standard methods were used. For details see the supplemental Information.
RNA sequencing
The Illumina TruSeq Stranded Total RNA Sample Prep with Ribo-Zero Gold rRNA removal kit (Illumina, San Diego, CA) was used for RNA library preparation. Sequencing was performed at the High Throughput Genomics and Bioinformatics Analysis Shared Resource of the Huntsman Cancer Institute on an Illumina HiSeq 2500 (Illumina) system. The MultiQC report is included in supplemental Table 1.
Genome-wide DNA methylation profiling
DNA methylation was assessed at whole-genome level in a cohort of 12 CMML patients and 12 age-matched controls with Illumina Infinium Human Methylation 450K BeadChip arrays, which interrogate >485 000 methylation sites per sample at single-nucleotide resolution, as previously described.29
Analysis pipeline
Human GRCh38 FASTA and GTF files were downloaded from Ensembl,30 release 87, and the reference database was created using STAR,31 version 2.5.2b, with splice junctions optimized for 50-bp reads. Reads were aligned to the reference database by using STAR in 2-pass mode to output a BAM file sorted by coordinates. Mapped reads were assigned to annotated genes in the GTF, using featureCounts,32 version 1.5.1. The output files from FastQC, STAR, and featureCounts were summarized with MultiQC,33 to check for any sample outliers. Samples with unacceptably low mapping rates to coding regions in the MultiQC report were excluded from the analysis. The Huntsman Cancer Institute Bioinformatics Analysis Shared Resource assisted with data analysis.
Identification of differentially expressed genes
Differentially expressed genes were identified by using a 1% false discovery rate (FDR) with DESeq2,34 version 1.16.0. The analysis included 12 CMML patients, 12 age-matched (old) healthy controls, and 16 young controls. Pairwise comparisons were carried out between transcriptomes of the 3 defined groups. The resulting sets of differentially expressed genes were overlapped in a Venn diagram, and changes were identified that were specific among the comparisons. CMML-specific genes were identified by subtracting all genes with significant differences in the young controls vs old controls comparison from the CMML vs old controls comparison.
Identification of DMRs
The DNA methylation arrays from 12 CMML patients and 12 age-matched controls were analyzed with the minfi package.35 The arrays were normalized using the functional normalization algorithm and quality control checked using interactive plots from the shinyMethyl package.36 Probes containing a single-nucleotide polymorphism at the CpG interrogation or at a single-nucleotide extension were removed. The bumphunter package37 was then used to find and annotate differentially methylated regions (DMRs). The DMRs near promoters and significant CMML genes in RNA-Seq data were selected in a subsequent step for analysis. The β-value represents a quantitative measure of the DNA methylation level of specific CpG-sites and ranges from 0 (completely unmethylated) to 1 (completely methylated).
Gene Set Enrichment Analysis
Broad Institute Gene Set Enrichment Analysis (GSEA),38 version 3.0 (http://www.broadinstitute.org/gsea/), was used to determine whether defined sets of genes exhibit a statistically significant enrichment in their distribution within our ranked gene list. The rank lists were compared with gene sets in MSigDB 6.1 including pathways from the Kyoto Encyclopedia of Genes and Genomes database.
Ingenuity Pathway Analysis
Pathway-associated gene analyses were generated using commercially available software (Qiagen Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis).
Results
Clinical characteristics of the CMML patients
Consecutive newly diagnosed patients seen at the Hematologic Malignancies Clinic at the Huntsman Cancer Institute were included in our study (8 samples). The remaining 4 samples were obtained from consecutive newly diagnosed CMML patients treated at Oregon Health ’ Science University and were enrolled on a prospective clinical trial of 5-azacitidine (www.clinicaltrials.gov #NCT01350947). Based on the percentage of blast cells in the blood and bone marrow, CMML can be graded from 0-2, with 2 being the most severe. Of the 12 CMML patients studied, 8 were CMML-0, and 3 CMML-2.39 One patient was unclassifiable because no bone marrow biopsy was available. Seven cases were classified as proliferative and 4 as dysplastic CMML. The karyotype was low risk for 9 patients, high risk for 2 patients and unknown for 1 patient. TET2 mutations were detected in 75% of patients by whole exome sequencing. For clinical features see Table 1.
Identification of gene expression differences between CMML patients and healthy controls
We performed RNAseq on CD14+ monocytes from treatment-naive CMML patients and healthy controls (old and young) and compared gene expression between CMML vs old controls (Figure 1A,C) and young controls vs old controls (Figure 1B,D), using stringent statistical criteria (FDR < 0.01) as cutoff. Considering only protein-coding transcripts, 1565 genes were upregulated and 915 downregulated in monocytes from CMML patients vs old controls. The 24 genes with the most significant differential expression (sorted by P value: 12 upregulated and 12 downregulated) are summarized in Table 2. The comparison between young and old controls, using the same criteria, identified 1043 upregulated and 308 downregulated transcripts. The 24 genes with the most significant differential expression (sorted by P value: 12 upregulated and 12 downregulated) in the young controls vs old controls comparison are summarized in Table 2. Reynolds et al40 recently reported on age-related transcriptome-wide changes in monocytes collected from 1264 participants (aged 55-94 years) in the Multi-Ethnic Study of Atherosclerosis (MESA) cohort. A total of 2704 genes were differentially expressed with chronological age (FDR ≤ 0.001). Despite the higher age bracket of the MESA cohort, there was statistically significant overlap with the genes differentially expressed in young controls vs old controls in our study (627 of 1351 in common, Fisher’s exact test, P = 1.028 × 10−7), providing independent validation of our control dataset (supplemental Table 2A). In contrast, the overlap with genes differentially expressed between CMML and old controls failed to reach statistical significance (657 of 2840 in common; Fisher’s exact test P = .0614; supplemental Table 2B).

Identification of differentially expressed genes in CD14+cells from CMML patients and old and young controls. Highly purified CD14+ monocytes were subjected to RNA sequencing. (A) Differentially expressed genes in CMML CD14+ monocytes compared with old healthy controls were graphed according to fold change and FDR-adjusted P value (volcano plot). (B) Differentially expressed genes in CD14+ monocytes of healthy young controls compared with healthy old controls were graphed according to fold change and FDR-adjusted P value (volcano plot). (C) Heat map depicting the top 12 most significantly upregulated genes and the top 12 most significantly downregulated genes in the CMML vs old controls comparison clustered by rlog values. (D) Heat map depicting the top 12 most significantly upregulated genes and the top 12 most significantly downregulated genes in the young controls vs old controls comparison clustered by rlog values.
Identification of differentially expressed genes in CD14+cells from CMML patients and old and young controls. Highly purified CD14+ monocytes were subjected to RNA sequencing. (A) Differentially expressed genes in CMML CD14+ monocytes compared with old healthy controls were graphed according to fold change and FDR-adjusted P value (volcano plot). (B) Differentially expressed genes in CD14+ monocytes of healthy young controls compared with healthy old controls were graphed according to fold change and FDR-adjusted P value (volcano plot). (C) Heat map depicting the top 12 most significantly upregulated genes and the top 12 most significantly downregulated genes in the CMML vs old controls comparison clustered by rlog values. (D) Heat map depicting the top 12 most significantly upregulated genes and the top 12 most significantly downregulated genes in the young controls vs old controls comparison clustered by rlog values.
To identify CMML-specific changes, we cross-referenced the resulting sets of differentially expressed genes in the young controls vs old controls and CMML vs old controls comparisons (Figure 2A). Removing the 360 genes differentially expressed between young and old controls from the list of genes with differential expression between CMML and old controls resulted in a set of 2480 deregulated genes specific to CMML. We next performed unsupervised hierarchical clustering, using the CMML gene signature as input (Figure 2B). CMML patients clearly separated from controls, whereas young and old controls clustered together, as expected after removal of the genes with differential expression between them. The top 25 differentially regulated genes are listed in Figure 2C. For validation, we measured the expression of 4 transcripts by quantitative PCR; all were confirmed (Figure 2D).

Identification of a CMML transcriptional signature. (A) Venn diagram illustrating the number of differentially expressed genes in CMML vs old controls (blue) compared with young controls vs old controls (red) using an FDR < 0.01. (B) Unsupervised clustering heat map of the 2480 genes that are unique to the CMML vs old controls set, shown in CMML and normal old and young control monocytes. (C) Heat map of the top 25 most significant genes of the CMML signature. (D) RNAseq and qPCR gene expression normalized values for TEX14, PLK2, CAMKK2, RPA1, and CXCL8 transcripts. P values were calculated using the nonparametric Mann-Whitney U test. **P < 0.01; ****P < 0.0001.
Identification of a CMML transcriptional signature. (A) Venn diagram illustrating the number of differentially expressed genes in CMML vs old controls (blue) compared with young controls vs old controls (red) using an FDR < 0.01. (B) Unsupervised clustering heat map of the 2480 genes that are unique to the CMML vs old controls set, shown in CMML and normal old and young control monocytes. (C) Heat map of the top 25 most significant genes of the CMML signature. (D) RNAseq and qPCR gene expression normalized values for TEX14, PLK2, CAMKK2, RPA1, and CXCL8 transcripts. P values were calculated using the nonparametric Mann-Whitney U test. **P < 0.01; ****P < 0.0001.
Functional annotation of the top 25 gene expression signature of CMML monocytes revealed upregulation of several genes involved in inflammation. For instance, activating transcription factor 3 (ATF3) expression is induced by various stress signals and plays an important role in modulating inflammatory response.41 CXCL2 encodes the CXCR2 ligand, macrophage inflammatory protein 2 (MIP2) and is regulated by the granulocyte colony stimulating factor (G-CSF STAT3 signaling cascade.42 Oxidized low-density lipoprotein receptor-1 (OLR-1), is a type II transmembrane receptor of the C-type lectin family that induces oxidative stress, activates tumor necrosis factor α TNFα/nuclear factor-κB (NFκB), upregulates the expression of monocyte chemoattractant protein-1 (MCP1), a key mediator of inflammatory processes, and has a proinflammatory role in cardiovascular disease.43,-45 calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2) transcripts were downregulated in CMML CD14+ monocytes. As previous studies have demonstrated, a crucial role of the CAMKK2-5′-AMP–activated protein kinase catalytic subunit-1 (PRKAA1) axis in the differentiation of monocytes into macrophages, our data are consistent with the observation that the differentiation from monocytes to macrophages is compromised in CMML.46,47 Interestingly, we also found upregulation of testis-expressed sequence 14 (TEX14) in CMML monocytes. TEX14 is a tumor-testis antigen whose expression is restricted to malignancy and normal germ cells and may represent a target for immunotherapy.48
For an unbiased functional pathway annotation, we used the 2480 CMML-specific differentially expressed genes as input into the Ingenuity Pathway Analysis (IPA) application (Figure 3). In addition to enrichment for pathways involved in the regulation of proliferation and cell cycle, inflammatory pathways were highly represented, including lymphotoxin-β receptor signaling, interleukin (IL)-6 signaling, NFκB activation, CD40 signaling, IL-17A signaling and lipopolysaccharide-simulated MAPK signaling. These data suggest that, compared with normal aged-matched controls, CMML monocytes exhibit strongly increased inflammatory signaling. JAK2/STAT signaling was also enriched, in accordance with the granulocyte-macrophage colony-stimulating factor (GM-CSF) hypersensitivity of many CMML cases.49

IPA. The 2480-gene, CMML-specific signature was used as input into IPA. Pathways were ranked according to significance.
IPA. The 2480-gene, CMML-specific signature was used as input into IPA. Pathways were ranked according to significance.
A focused comparison of 378 cytokine/chemokine-encoding genes (defined using Gene Ontology Annotations, GO:0005125) identified 46 genes with significantly different expression in the comparison between CMML and old controls or between old and young controls (supplemental Figure 2A-B). Hierarchical clustering, using only this set of cytokine/chemokine genes, clearly distinguished among the 3 groups (supplemental Figure 2C; supplemental Table 3). Interestingly, a group of cytokine/chemokine-encoding genes was upregulated in the young controls compared with both old controls and CMML, including positive (eg, IL-16 and IL-32) and negative (eg, IL-1RN) regulators of inflammation (supplemental Figure 2C). Cytokines/chemokine transcripts with increased expression specific to CMML included IL-1A, IL-1B, IL-10, CSF110, and IL-6, among others (supplemental Figure 2D). CXCL8, IL-6, and VEGFA transcripts overlap with the high levels of cytokines present in CMML patients’ plasma compared with that of healthy controls identified by Niyongere et al.50 Altogether, these data show that CMML monocytes exhibit a unique cytokine/chemokine expression profile that is distinct from both young and old controls.
TET2 is the most commonly mutated CHIP gene and Tet2-deficient monocytes promote atherosclerosis in low-density lipoprotein receptor–deficient mice.51,52 Liu and colleagues53 used the Multiethnic Study of Atherosclerosis (MESA) cohort to establish correlations between the transcriptional signature of monocytes and the risk of human atherosclerosis, as defined by carotid plaque burden and coronary artery calcification. They identified 21 genes with expression associated with carotid plaque score and 104 genes associated with coronary artery calcification (FDR < 0.2). We tested whether the gene expression signature of CMML monocytes may overlap with the atherosclerosis signature of the MESA cohort. Twenty-four differentially expressed genes, detailed in supplemental Table 4, were shared, including ARID5B (expression increased in CMML monocytes and in atherosclerosis). ARID5B is a transcriptional coactivator implicated as a mediator of cardiovascular risk.53 However, as a whole, this overlap did not reach statistical significance (P = .1676).
Methylation differences between CMML and healthy monocytes and integrated analysis
Several studies have shown that CMML mononuclear cells exhibit profound changes in DNA methylation compared with normal controls.11,54,55 However, data specific to monocytes are unavailable. To understand to what extent differences in methylation correlate with differentially expressed genes identified by RNAseq, we compared DNA methylation between CMML and aged-matched normal CD14+ monocytes using high-density methylation arrays. DNA methylation profiling revealed 909 DMRs between CMML and age-matched controls (FDR < 0.05 and methylation difference ≥20%), with most regions being hypermethylated in CMML monocytes (Figure 4A). Of the hypermethylated DMRs, 37% were intronic, 22% were exonic, 14% were in promoter regions, 10% were downstream, 10% were upstream and the remaining 7% were in 5′-untranslated regions.
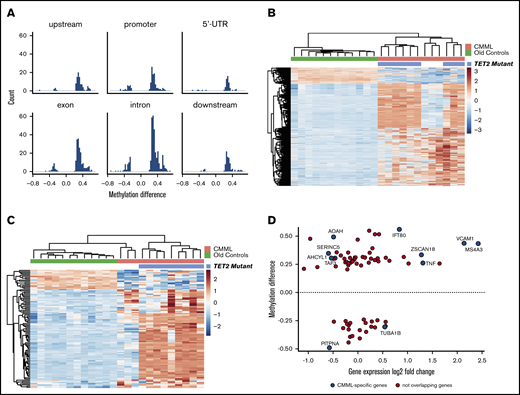
DNA methylation analysis of CD14+monocytes from CMML patients and age-matched healthy controls. (A) Comparison of methylation between CMML and old controls. Most regions are hypermethylated. (B) Unsupervised clustering of the 909 DMRs identified in CD14+ monocytes from CMML patients vs healthy age-matched controls, using a P value cutoff of .05. The heat map represents average β-values pseudocolored blue (hypomethylated) to red (hypermethylated). TET2 mutational status is shown. (C) Intersection of methylation differences (909 DMRs in the CMML vs old contrast) and log2 expression fold changes (2480 CMML-specific genes) identified 99 DMRs (94 unique genes, since some genes have 2 or more DMRs). The heat map represents β-values from the 99 DMRs. (D) A total of 124 DMRs localized near gene promoters, and 74 of these genes were also detected in RNA sequencing comparison in monocytes from CMML patients vs old controls. The graphic shows methylation differences in the promoter regions of those 74 DMRs vs fold changes in gene expression. Only 10 of the genes were specific to CMML signature (shown in blue), whereas the remainder represented age-related changes.
DNA methylation analysis of CD14+monocytes from CMML patients and age-matched healthy controls. (A) Comparison of methylation between CMML and old controls. Most regions are hypermethylated. (B) Unsupervised clustering of the 909 DMRs identified in CD14+ monocytes from CMML patients vs healthy age-matched controls, using a P value cutoff of .05. The heat map represents average β-values pseudocolored blue (hypomethylated) to red (hypermethylated). TET2 mutational status is shown. (C) Intersection of methylation differences (909 DMRs in the CMML vs old contrast) and log2 expression fold changes (2480 CMML-specific genes) identified 99 DMRs (94 unique genes, since some genes have 2 or more DMRs). The heat map represents β-values from the 99 DMRs. (D) A total of 124 DMRs localized near gene promoters, and 74 of these genes were also detected in RNA sequencing comparison in monocytes from CMML patients vs old controls. The graphic shows methylation differences in the promoter regions of those 74 DMRs vs fold changes in gene expression. Only 10 of the genes were specific to CMML signature (shown in blue), whereas the remainder represented age-related changes.
As previous studies in unselected CMML blood or bone marrow cells have attributed alterations in DNA methylation to mutations in epigenetic regulators, we initially sought to correlate somatic mutation status with methylation patterns in our cohort.56 TET2 is one of the most frequently mutated genes in CMML,57 and TET2 mutations were detected in 9 of 12 (75%) of our CMML patients. Unsupervised clustering analysis of the 909 DMRs revealed that DNA methylation patterns among all CMML patients differentiate between CMML patients with TET2 mutations and patients with wild-type TET2 (Figure 4B), with most of the regions significantly hypermethylated in CMML monocytes. IPA showed significant enrichment of DMRs in the sonic hedgehog signaling, dopamine-DARPP32 feedback in cAMP signaling, gap junction signaling, and adipogenesis pathways (supplemental Figure 3). Next, we cross-referenced methylation changes to mRNA expression, to identify those genes with coordinated regulation. Using the 909 DMRs and the 2480 CMML-specific genes, we identified 99 shared regions in 94 unique genes, including several genes with 2 or more DMRs. To determine whether the methylation status of this selected gene set would be sufficient to distinguish between CMML and controls, we performed unsupervised clustering analysis using the β-values from the 94 DMRs. The resulting CMML-specific methylation patterns completely separated CMML and normal monocytes (Figure 4C). The 3 TET2 wild-type CMML cases had a methylation profile closer to that of the old controls than to TET2-mutated CMML, resembling a hybrid between old normal and CMML. This is consistent with previously reported methylation patterns of CMML bone marrow mononuclear cells that separated TET2 wild-type from TET2 mutant cases.58
Methylation of CpG islands in gene promoters is a well-established mechanism of transcriptional silencing in cancer.59 In CMML, hypomethylating agents can induce demethylation of CpG islands within promoters, thereby leading to reexpression of silenced tumor-suppressor genes.55 Therefore, although most of the CMML-specific DMRs are intronic (supplemental Figure 4), we focused specifically on DMRs within genes promoters to understand which CMML-specific genes may be controlled by promoter methylation. We observed significantly coordinated changes in CMML-specific mRNA transcripts and DNA methylation promoter regions for 10 genes (Figure 4D). However, only half of these genes followed the predicted regulation pattern: AOAH, SERINC5, TAF3, and AHCYL1 were hypermethylated and downregulated; TUBA1B was hypomethylated and upregulated. In contrast, MS4A3, TNF, VCAM1, and IFT80 were hypermethylated and upregulated, and PITPNA was hypomethylated and downregulated. Thus, gene expression in CMML monocytes is controlled by complex mechanisms, which may or may not include promoter methylation.
Discussion
CMML is a genetically heterogeneous MDS/MPN overlap disorder with the unifying phenotypic characteristic of persistent monocytosis.39 Given the steep increase of CMML incidence with advanced age, the myelomonocytic differentiation bias of aged bone marrow, and the overlap of genes mutated in CMML with those mutated in ARCH, CMML could be regarded as the malignant conversion of ARCH.60 However, it is unknown which characteristics of CMML monocytes reflect ageing rather than the disease state of CMML, and the available RNAseq data are limited to 1 small study that provided little detail.61 Gene expression profiles of CMML monocytes and non–age-matched healthy controls using gene expression arrays have been described.62 Identifying features specific for CMML monocytes is critical from the perspective of understanding their biology and for therapeutically targeting CMML over normal monocytes. To approach this question, we performed RNAseq on highly purified blood monocytes from CMML patients, old controls, and young controls. Approximately one fourth of the genes differentially expressed between normal young and old monocytes overlapped with those differentially expressed between CMML and aged-matched monocytes, underscoring the importance of our 3-way comparison to isolate the transcriptional signature of CMML monocytes (Figure 2A). IPA of the CMML monocyte transcriptome identified multiple inflammatory signaling pathways, most of which were dominated by upregulated genes (Figure 3). Specifically, 4 pathways related to the TNF family signaling were enriched in CMML monocytes, including lymphotoxin-β receptor, IL-17A signaling in fibroblasts, NFκB activation by viruses, CD40, and CD27. High-mobility group protein B1 (HMGB1) and IL-6 signaling were additional highly proinflammatory pathways identified in the transcriptome of the CMML monocytes. The latter is consistent with recent reports of increased IL-6 expression in Tet2−/− mouse monocytes and macrophages and with our previous work on cytokine expression in CMML vs normal bone marrow.50,-52 HMGB1 is a nuclear protein that acts as a danger associated molecular pattern (DAMP) and extracellular trigger of inflammation through binding to Toll-like receptors 2 and 4.63 Another proinflammatory pathway characteristic of CMML monocytes is IL-17 signaling (Figure 3). This observation is consistent with our report of increased IL-17 expression in CMML vs normal bone marrow.50 IL-17 has been implicated as a master regulator of pathologic inflammation in psoriasis and other autoimmune diseases, and stimulates the production of proinflammatory cytokines from monocytes (TNFα, IL-1B, IL-6, GM-CSF, and G-CSF).64,65 A second cluster of enriched pathways in CMML monocytes involves cell cycle regulation and DNA damage, with pololike kinases as the most significantly enriched pathway overall, and a third cluster involves proliferative signaling (JAK/STAT and lipopolysaccharide-stimulated MAPK signaling), the latter linking inflammation to proliferation. At the single cytokine level, we also found elevated IL-1A and -1B expression in CMML monocytes, again consistent with data from Tet2−/− mouse monocytes.51,52 In aggregate, these data show that cytokine/chemokine expression patterns in CMML monocytes are highly aberrant. More work is needed to identify the regulatory circuits governing expression of these inflammatory cytokines.
Recent reports have suggested that most myeloid neoplasms, including CMML, originate from age-related precursor lesions that initially manifest as small populations of mutated blood cells in individuals with normal blood counts.17,,,,-22 The term CHIP is typically used in case of mutations with a variant allele frequency of >2%, the sensitivity limit of many next-generation sequencing assays, whereas ARCH refers to the presence of mutated cells at any level.66 When highly sensitive and specific assays are used, DNMT3A and TET2 mutations are detectable at low frequency in almost 100% of healthy middle-aged women.67 It is conceivable that mutated cells are more resistant to an inflammatory environment, providing them with a selective advantage over normal competitors. Indeed, several recent reports support this concept in the case of Tet2−/− mouse hematopoietic stem and progenitor cells.26,68,69 Similarly, we have previously shown that TNFα promotes expansion of JAK2V617F expressing cells in myelofibrosis.70,71 Current thinking holds that multiple mechanisms promote an inflammatory state in the aging bone marrow that manifests as a myelomonocytic differentiation bias (reviewed by Elias et al72 ). Unexpectedly, we found that expression of several inflammatory cytokines such as IL-16 and -32 was higher in young compared with old control monocytes, suggesting that not all aged-related transcriptional changes in monocytes are proinflammatory and that additional cell types are involved in promoting age-associated inflammation. Our study is limited by the lack of clinical information and mutational profiling for the normal controls, and as such we cannot exclude the presence of inflammatory conditions and/or the presence of CHIP/ARCH in the control population. This may explain why 3 of the normal controls grouped with CMML monocytes on unsupervised clustering. Alternatively, mutant cells may promote their own expansion by releasing proinflammatory cytokines into their microenvironment, consistent with previous reports of complex interactions between various CMML cell types. For instance, dysplastic granulocytes generated by the CMML clone secrete large amounts of α-defensin 1 to 3 peptides, which in turn inhibit CSF1-driven differentiation of peripheral blood monocytes into macrophages.47 Mapping the interactions between mutant and residual normal hematopoietic stem and progenitor cells, and mature cells, including monocytes, are critical for understanding their roles in clonal selection. The consequences of CMML-driven inflammation may reach beyond the hematopoietic system. Kyoto Encyclopedia of Genes and Genomes pathway analysis of the CMML-specific deregulated genes identified increased systemic lupus erythematosus, type 1 diabetes mellitus, and Toll-like receptor signaling, whereas the primary immunodeficiency cluster was reduced compared with age-matched controls (supplemental Figure 5). This suggests that monocytes may contribute to the increased incidence of autoimmune disease in patients with CMML.73,-75 In our cohort, 4 CMML patients had history of inflammatory and autoimmune diseases (supplemental Table 5).
CMML is one of the most aggressive of the MDS/MPN overlap disorders, with a median overall survival (OS) of only 20 to 32 months.76,-78 Current therapy is based on hypomethylating agents such as 5-azacytidine, with response rates of ∼50%, but no certain prolongation of OS.79 Although transformation to acute myeloid leukemia and complications of cytopenias are important contributors to poor survival, many patients succumb to comorbid conditions.80 The inclusion of somatic mutations in prognostic scoring systems has failed decisively to improve prognostication, suggesting that additional leukemia and/or host factors influence prognosis.9,76 In contrast to other subtypes of MPN and MDS, accumulation of monocytes is a hallmark of CMML, and our data show that these monocytes are highly proinflammatory compared with normal age-matched controls. As CHIP with a relatively small number of mutated monocytes is sufficient to increase cardiovascular risk, including mortality from ischemic heart disease, it is conceivable that the massive expansion of proinflammatory monocytes in CMML constitutes a considerable vascular risk factor.19,51,52,81 In support of this, a retrospective study found a twofold increased risk for cardiovascular events in CMML compared with patients with chronic lymphocytic leukemia.27 Another study reported a positive correlation between blood monocyte counts and end organ damage in CMML patients, including chronic kidney disease and cardiovascular events, consistent with the reported role of blood monocytes in atherosclerosis and the positive correlation between absolute monocyte counts and cardiovascular risk.82,83 Based on this, we compared the transcriptional signature of atherosclerosis reported from the MESA cohort and our CMML gene signature and identified a set of overlapping genes, including the atherosclerosis regulator ARID5B, suggesting pathogenetic similarities.40,53,84,,-87
Profound changes in DNA methylation contribute to CMML pathogenesis. These changes are mostly genotype agnostic and correlate with response to 5-azacytidine and prognosis.11 Most differentially methylated regions were intragenic (supplemental Figure 4), consistent with published data.11 However, as cross-referencing of CpG island methylation in promoter regions of genes with CMML-specific gene expression changes revealed relatively little overlap and no consistent regulation pattern (Figure 4D), mechanisms other than promoter methylation must account for the bulk of the CMML-specific transcriptional dysregulation, including the dysregulation of cytokine expression.10,54
In summary, we report the first systematic analysis of CMML monocyte transcriptional profiles that uses aged-matched and young controls to separate CMML-specific changes from aged-related changes. However, our study comprises a relatively low number of patients (the majority CMML-0 subtype), and a direct comparison with a reactive monocytosis control group is absent. Further validation with larger number of patients and functional studies are needed. Although our cohort is not large enough to fully account for the genetic complexity of CMML, our findings identify CMML monocytes as highly proinflammatory compared with controls. The detrimental effects of proinflammatory monocytes on extrahematopoietic organs, specifically the cardiovascular system, raise the question of whether even asymptomatic and low risk CMML patients would benefit from monocyte-reducing and/or anti-inflammatory therapy such as the IL-1β antibody canakinumab.88
Acknowledgments
The authors thank Claire Davis, who provided the original illustrations for the visual abstract.
Research reported in this publication used the High-Throughput Genomics and Bioinformatic Analysis Shared Resource at Huntsman Cancer Institute at the University of Utah and was supported by the National Institutes of Health (NIH), National Cancer Institute Grant P30CA042014. The research was also supported by NIH, National Cancer Institute Grant R01CA178397 (M.W.D. and T.O.) and the V Foundation for Cancer Research (M.W.D. and T.O.). J.S.K. was and D.Y. is supported by a Special Fellow Award from the Leukemia and Lymphoma Society.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: A.F. supervised the study, performed experiments and computational analyses, and cowrote the manuscript; A.D.P supervised collection of the samples (CMML patients, old and young donors), supervised sample purification and analysis, prepared figures and edited the manuscript; D.Y. designed and performed experiments; J.S.K. designed and performed experiments; S.K.T. collected and provided the patients’ clinical information; H.T. contributed to data collection and interpretation; J.M.A. performed the qPCR validation experiments; T.O. supervised the study; and M.W.D initiated and supervised the study and cowrote the manuscript.
Conflict-of-interest disclosure: M.W.D. is a consultant or advisory board member for Novartis, Pfizer, Galena Biopharma, Takeda Pharmaceutical Company, Blueprint Medicines, and Incyte. M.W.D.’s laboratory receives grant funding from Novartis and Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Michael W. Deininger, Huntsman Cancer Institute, The University of Utah, 2000 Circle of Hope, Salt Lake City, UT 84112; e-mail: michael.deininger@hci.utah.edu.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE135902).
The full-text version of this article contains a data supplement.