

Key Points
Pediatric ALL relapses after allogeneic stem cell transplantation display highly diverse, dynamic and patient-individual genetic lesions.
Nine of 10 relapsing pediatric transplant recipients present with genetic alterations for which novel targeted therapies are available.

Abstract
Survival of patients with pediatric acute lymphoblastic leukemia (ALL) after allogeneic hematopoietic stem cell transplantation (allo-SCT) is mainly compromised by leukemia relapse, carrying dismal prognosis. As novel individualized therapeutic approaches are urgently needed, we performed whole-exome sequencing of leukemic blasts of 10 children with post–allo-SCT relapses with the aim of thoroughly characterizing the mutational landscape and identifying druggable mutations. We found that post–allo-SCT ALL relapses display highly diverse and mostly patient-individual genetic lesions. Moreover, mutational cluster analysis showed substantial clonal dynamics during leukemia progression from initial diagnosis to relapse after allo-SCT. Only very few alterations stayed constant over time. This dynamic clonality was exemplified by the detection of thiopurine resistance-mediating mutations in the nucleotidase NT5C2 in 3 patients’ first relapses, which disappeared in the post–allo-SCT relapses on relief of selective pressure of maintenance chemotherapy. Moreover, we identified TP53 mutations in 4 of 10 patients after allo-SCT, reflecting acquired chemoresistance associated with selective pressure of prior antineoplastic treatment. Finally, in 9 of 10 children’s post–allo-SCT relapse, we found alterations in genes for which targeted therapies with novel agents are readily available. We could show efficient targeting of leukemic blasts by APR-246 in 2 patients carrying TP53 mutations. Our findings shed light on the genetic basis of post–allo-SCT relapse and may pave the way for unraveling novel therapeutic strategies in this challenging situation.
Introduction
Acute lymphoblastic leukemia (ALL) represents the most prevalent pediatric cancer. Through the collaborative effort of large randomized multiinstitutional trials, approximately 90% of all pediatric patients with ALL survive for at least 5 years.1 Survival rates of patients with ALL who respond poorly to first-line treatment or who relapse are much less favorable. Thus, these children are eligible for allogeneic hematopoietic stem cell transplantation (allo-SCT) and display survival rates of 70% to 75% if allo-SCT is performed in complete remission.2,3
Although nonrelapse mortality could substantially be reduced via optimization of donor selection and supportive care, post–allo-SCT relapses still represent the major cause of treatment failure.4 A durable remission providing a realistic perspective for definitive cure is only rarely achieved, in particular when relapses occur early.5,6 Standard protocols for this situation do not exist, and treatment options in these heavily pretreated children are limited, in part because of cumulative toxicity. Thus, treatment remains a therapeutic challenge, and administered therapy represents individual case-based decisions, including immunotherapeutic approaches such as the CD19/CD3-bispecific antibody blinatumomab,7,8 the CD22-directed immunotoxin inotuzumab ozogamicin, or CD19-directed CAR-T cells.9,10
Second SCT has proven, among other options such as the above-mentioned CAR-T cells, to represent a curative treatment approach to some of these children with disease-free survival rates approximating 30% after 2 years.5,11 However, before proceeding to second SCT, complete morphologic remission or, even more challenging, a very low or negative minimal residual disease level should be achieved.5 Thus, novel individualized therapeutic approaches are urgently needed to achieve this goal. Because post–allo-SCT relapses escaped the combined chemotherapeutic and immunologic attack, they are presumed to display distinct genetic alterations compared with those ALL relapses after conventional chemotherapy.12
Two recent studies evaluated the landscape of genomic alterations in large cohorts of patients with pediatric cancer.13,14 In addition to defining distinct mutational patterns, these analyses revealed that nearly 50% of pediatric neoplasms harbor a potentially druggable genetic event. Moreover, several investigators have analyzed mutational profiles in pediatric ALLs focusing on initial disease and first relapses.15,,,,,-21 There remains, however, a lack of information on genetic alterations in pediatric post–allo-SCT relapses.
Here, it is of particular importance that the bone marrow comprises recipient-originating leukemic blasts and donor hematopoietic cells. Thus, to faithfully identify genetic lesions specific to the post–allo-SCT relapse, both donor and recipient genetic backgrounds must be considered (Figure 1A).
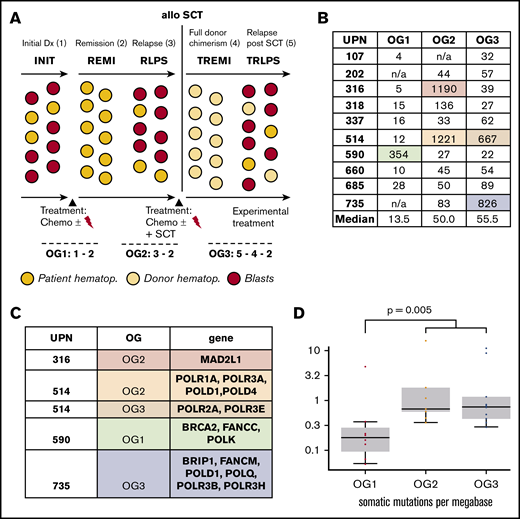
Large oncogenomes are characterized by acquired mutations in DNA repair genes. (A) Experimental setup including oncogenomes (OG). For patients 202 and 735, no material of the initial leukemia (INIT) was available; patient 107 did not relapse before allo-SCT. (B) Sizes of the individual oncogenomes. (C) Mutational loads across the oncogenomes. The increase from OG1 vs OG2/OG3 was statistically significant (P = .005). (D) Mutations in DNA polymerase/repair genes explaining the large oncogenomes. n/a, not available; REMI, first remission; RLPS, first relapse; TREMI, remission after allo-SCT; TRLPS, relapse after allo-SCT; UPN, unique patient identifier.
Large oncogenomes are characterized by acquired mutations in DNA repair genes. (A) Experimental setup including oncogenomes (OG). For patients 202 and 735, no material of the initial leukemia (INIT) was available; patient 107 did not relapse before allo-SCT. (B) Sizes of the individual oncogenomes. (C) Mutational loads across the oncogenomes. The increase from OG1 vs OG2/OG3 was statistically significant (P = .005). (D) Mutations in DNA polymerase/repair genes explaining the large oncogenomes. n/a, not available; REMI, first remission; RLPS, first relapse; TREMI, remission after allo-SCT; TRLPS, relapse after allo-SCT; UPN, unique patient identifier.
To address the urgent clinical need for identification of novel patient-individualized treatment approaches, we here performed whole-exome sequencing of leukemic blasts of 10 pediatric transplant recipients with post–allo-SCT relapses, and reveal substantial individuality and clonal dynamics of genetic alterations, selective pressure, and a high prevalence of mutations in such genes, for which nonstandard therapies with novel, approved agents are readily available.
Patients, materials, and methods
Patient samples
All patients were enrolled in multicenter pediatric ALL trials. The treatment studies and the concomitant research were all approved by the respective institutional review boards, and informed consent was obtained from all parents.
Exome library preparation and NGS
Nucleid acids were extracted using AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany). Exome library preparation was performed as detailed in Hoell et al.22 Two times 100 bp sequencing with a 6-bp index read was performed using the TruSeqTM SBS Kit v5+UTR on the HiSeq 2500 (Illumina, San Diego, CA). All sequencing data are available on request.
Sanger sequencing
Selected single nucleotide variants (SNVs) from exome sequencing (NT5C2: 514, 735; TP53: 514, 590, 660) were validated by Sanger sequencing (primer sequences in supplemental Methods) and were confirmed in all cases. See the supplemental Methods for details on the amplicon sequencing.
APR-246 sensitivity testing
Sensitivities of patient/xenograft leukemia samples to APR-246 (kindly provided by Aprea Therapeutics, Stockholm, Sweden) were assessed after incubation of ALL cells with increasing concentrations analyzing cell death by flow cytometry according to forward- and side-scatter criteria in triplicate measurements.
Bioinformatic analysis
Fastq files were generated using BcltoFastq 1.8.4 (Illumina). BWA version 0.6.1-r10423 with default parameters to align sequence data to the human reference genome (GRCh37). Conversion steps were carried out using Samtools,24 followed by removal of duplicate reads.25,26 Local realignment around indels, SNV-calling, annotation, and recalibration was facilitated by GATK.27 HapMap, OmniArray, and dbSNP135 datasets provided by The Broad Institute were used for recalibration. Resulting variant calls were annotated by Variant Effect Predictor,28 using the Ensembl database (v70) and imported into an in-house MySQL database to facilitate automatic and manual annotation, reconciliation, and data analysis by complex database queries. Loss of function prediction scores for PolyPhen229 and SIFT30 were extracted from the same Ensemble release.
To identify clones that follow a common clonal pattern during oncogenome progression, we applied a density-based clustering approach31 to the log-scaled frequencies of the somatic variants. Computing the Euclidean distance matrix on the logarithmic scaled frequencies allows the mitigation of the higher possible variance that occurs in high-frequency mutations compared with low-frequency mutations, which influences the Euclidian distances disproportionally. Frequencies were extracted using samtools from the alignments, using bases that exceeded the base quality of 13. Variants with a frequency of 0.0 were set to an artificial minimum of 10−4 for this scaling, 3 magnitudes smaller than the average read depth observed for the somatic mutations (79.23). We used the dbscan R package for the clustering.32 Alluvial diagrams of alternate allele fractions were generated using ggalluvial R package. Please see the supplemental Methods for details on the deep sequencing analysis and the computation of the oncogenomes.
Results
Cohort of 10 patients relapsing with ALL after SCT
All 10 patients were enrolled in multicenter pediatric ALL trials (ALL-BFM, CoALL, ALL-REZ BFM, ALL-SCT BFM 2003, TCRalpha/β-Haplo2010 trial). Mean age at first diagnosis was 4.6 years (range, 0.3-10.2 years), mean time from first diagnosis to relapse before allo-SCT was 2.2 years (range, 1.1-3.4 years), and mean time from initial diagnosis to post–allo-SCT relapse was 3.1 years (range, 0.8-5.1 years). Time from SCT to post–allo-SCT relapse was short (mean, 0.7 years; range, 0.2-1.7 years). Seven patients had received a matched-unrelated donor transplant, in 2 cases an HLA-identical sibling served as donor, and 1 patient was transplanted from his HLA-haploidentical mother (Table 1; supplemental Table 1). In 9 of 10 patients, signs of neither clinically significant acute nor chronic GvHD were reported to the study center (information missing for 1 patient). Because of the lack of standardized protocols, treatment after the post–allo-SCT relapse varied considerably and ranged from palliative care to second allo-SCT. At the time of this analysis, only 2 patients were still alive, reflecting the dismal prognosis of post–allo-SCT relapses.
Clinical characteristics of the 10 analyzed patients
| UPN | 107 | 202 | 316 | 318 | 337 | 514 | 590 | 660 | 685 | 735 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | F | F | M | M | M | M | F | M | F | M |
| Age at first disease, y | 0.3 | 5.7 | 1.0 | 5.3 | 2.8 | 2.5 | 7.3 | 5.1 | 5.7 | 10.2 |
| Immunophenotype | cALL | cALL | cALL | cALL | cALL | cALL | cALL | cort. T | pro-T | pB-ALL |
| Age at relapse, y | n/a | 7.5 | 3.3 | 7.7 | 5.9 | 5.0 | 10.7 | 6.4 | 6.8 | 12.3 |
| Time to relapse, y | n/a | 1.8 | 2.3 | 2.4 | 3.1 | 2.5 | 3.4 | 1.3 | 1.1 | 2.1 |
| Age at SCT, y | 0.8 | 8.1 | 3.9 | 8.0 | 6.3 | 5.5 | 11.2 | 6.7 | 7.0 | 12.8 |
| MRD before SCT | <1 × 10−4 | <1 × 10−3-1 × 10−4 | 1 × 10−6 | 1 × 10−4 | <1 × 10−4 | <1 × 10−4 | <1 × 10−5 | <1 × 10−4 | 1 × 10−3-1 × 10−4 | <1 × 10−4 |
| Time to relapse after SCT, y | 0.3 | 0.7 | 0.4 | 1.7 | 1.2 | 0.3 | 1.2 | 0.6 | 0.2 | 0.2 |
| Survival status | Alive | Dead | Dead | Alive | Dead | Dead | Dead | Dead | Dead | Dead |
| UPN | 107 | 202 | 316 | 318 | 337 | 514 | 590 | 660 | 685 | 735 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | F | F | M | M | M | M | F | M | F | M |
| Age at first disease, y | 0.3 | 5.7 | 1.0 | 5.3 | 2.8 | 2.5 | 7.3 | 5.1 | 5.7 | 10.2 |
| Immunophenotype | cALL | cALL | cALL | cALL | cALL | cALL | cALL | cort. T | pro-T | pB-ALL |
| Age at relapse, y | n/a | 7.5 | 3.3 | 7.7 | 5.9 | 5.0 | 10.7 | 6.4 | 6.8 | 12.3 |
| Time to relapse, y | n/a | 1.8 | 2.3 | 2.4 | 3.1 | 2.5 | 3.4 | 1.3 | 1.1 | 2.1 |
| Age at SCT, y | 0.8 | 8.1 | 3.9 | 8.0 | 6.3 | 5.5 | 11.2 | 6.7 | 7.0 | 12.8 |
| MRD before SCT | <1 × 10−4 | <1 × 10−3-1 × 10−4 | 1 × 10−6 | 1 × 10−4 | <1 × 10−4 | <1 × 10−4 | <1 × 10−5 | <1 × 10−4 | 1 × 10−3-1 × 10−4 | <1 × 10−4 |
| Time to relapse after SCT, y | 0.3 | 0.7 | 0.4 | 1.7 | 1.2 | 0.3 | 1.2 | 0.6 | 0.2 | 0.2 |
| Survival status | Alive | Dead | Dead | Alive | Dead | Dead | Dead | Dead | Dead | Dead |
cort., cortical; F, female; M, male; MRD, minimal residual disease; SCT, stem cell transplantation; UPN, unique patient identifier.
Addressing the 2-germline challenge to receive leukemia-specific SNVs after SCT
To address the question of how pediatric pre– and post–allo-SCT ALL relapses differ on a genetic level, we performed WES of 5 samples per patient (Figure 1A; supplemental Tables 2 and 3). Because post–allo-SCT relapses usually occur in a situation of hematopoietic chimerism, 2 genetic germline backgrounds (donor and recipient) were subtracted to uncover mutations specific to the post–allo-SCT relapse. To receive a comprehensive picture, we combined the outputs of 2 SNV callers, namely, MuTect33 and VarScan2,34 as prior reports had revealed that this approach provides a more complete overview of the oncogenomes in acute leukemia.35
To enable comparative analyses, we defined the following 3 “oncogenomes” (OGs): OG1 (initial leukemia, defined as SNVs in INIT minus those in REMI), OG2 (first relapse, defined as RLPS minus REMI), and OG3 (post–allo-SCT relapse, defined as OG3 = [TRLPS-REMI]ᴖ[TRLPS-TREMI]).
All SNVs from the first 4 patients in our series (514, 590, 660, 685) were validated in silico by manually checking all identified SNVs via assessment of sufficient sequencing depths in all patient samples and verifying B allele frequencies (BAF) in the mutated vs nonmutated samples with complete concordance in all instances.
Large oncogenomes are characterized by acquired mutations in DNA repair genes
Median numbers of leukemia-specific SNVs in OG1-3 were 13.5, 50, and 55.5, respectively. As can be seen in Figure 1B, SNV counts of individual oncogenomes varied substantially, but proved usually below 200. There was, however, 1 outlier in OG1 (590), 2 in OG2 (316, 514), and 2 in OG3 (514, 735; supplemental Table 4 for a full gene list).
Next, we investigated the 4 patients, who had unexpectedly large OGs. We found that all these oncogenomes carried newly acquired somatic SNVs in either DNA polymerases or DNA repair/Fanconi anemia genes, and all SNVs were exclusive to the respective large oncogenomes (Figure 1C; supplemental Table 5). Of note, no DNA polymerase/DNA repair gene mutations were detected in any of the other oncogenomes.
Looking at the therapy that these 4 patients received, there were no specific treatment elements that could be correlated with the occurrence of DNA polymerase/DNA repair gene mutations and high mutational load (supplemental Table 1).
Oncogenomes are exclusive to individual patients and relapse states
We detected a median of 0.18 mutations per megabase in OG1, 0.67 mutations per megabase in OG2, and 0.74 mutations per megabase in OG3. The increase in mutational load between initial leukemia and the relapses was statistically significant (P = .005; Figure 1D). Mutational load was comparable between OG2 and OG3; however, there was a profound shift on gene level, as most affected genes differed between OG2 and OG3 (Figure 2A).

Oncogenomes are exclusive to individual patients and relapse states. (A) Mutational spectra on SNV level in OG1, OG2, and OG3. (B) Recurrently mutated genes (≥3 patients) in OG2 (also see supplemental Table 6). UPNs as earlier. (C) Recurrently mutated genes (≥3 patients) in OG3 (also see supplemental Table 7). n.d., not done.
Oncogenomes are exclusive to individual patients and relapse states. (A) Mutational spectra on SNV level in OG1, OG2, and OG3. (B) Recurrently mutated genes (≥3 patients) in OG2 (also see supplemental Table 6). UPNs as earlier. (C) Recurrently mutated genes (≥3 patients) in OG3 (also see supplemental Table 7). n.d., not done.
Next, we analyzed which mutated genes were recurrently affected. In OG1, 5 genes carried SNVs in at least 2 patients (TTN, IGSF3, NOTCH1, CTBP2, NRAS), with NOTCH1 exhibiting SNVs in the 2 T-ALL patients (660, 685). In the case of OG2, 158 genes were recurrently mutated in at least 2 patients, of which 72 were only affected in the 2 patients (316, 514), who presented with exceptionally large oncogenomes. A total of 7 genes were affected 3 times (Figure 2B).
In OG3, 116 genes had SNVs in at least 2 patients, of which 58 genes were affected in both patients with large oncogenomes (514, 735) and 8 genes were affected in at least 2 patients excluding these 2 patients (Figure 2C; supplemental Tables 6 and 7).
Looking longitudinally at those 7 patients, from whom all 3 leukemia samples were available, only the minority of SNVs were recurrently detected in all leukemia states (Figure 2A). These 7 patients had a mean number of 7.1 genes (median, 6 genes; total, 50 genes in 7 patients) forming the core mutational sets in all 3 leukemias (supplemental Table 8). Of these, a mean number of 2 genes (median was also 2 genes) were known cancer genes. The only recurrent gene in the list of 50 genes was NOTCH1 in the T-cell leukemias.
In some cases (patients 316, 318, 660, 685), these “core genes“ likely represented driving leukemic mutations (including BCL6, ERG, KRAS, NOTCH1, NRAS). In the 3 other patients (337, 514, 590), the situation was less clear, as the genes found have so far not been described as classic leukemic mutations, opening the possibility that they represent bystander mutations of the dominant clone.
Thus, we identified profound clonal dynamics in the mutational landscapes, not only between initial diagnosis and relapse, but likewise between pre– and post–allo-SCT relapses.
Mutational clusters show substantial leukemic clonal dynamics from initial diagnosis across all relapses
As only the minority of affected genes overlapped between the 3 oncogenomes, which hinted at substantial leukemic clonal dynamics, we wished to assess leukemic evolution over time. To achieve this, we defined mutational clusters, which consisted of genes with similar mutational frequencies. We focused on those 7 patients (316, 318, 337, 514, 590, 660, 685) for which all 3 leukemias were available. Only the minority of patients (n = 3) carried clusters, which were present (BAF ≥ 0.1) without relevant changes in frequency across all 3 leukemias (supplemental Figure 1). The majority of patients carried at least 1 mutational cluster, which was present only in OG1, OG2, or OG3. In addition, 6 patients had clusters, which were first present in OG2 and continued to be detectable (at similar BAF levels) in OG3. Figure 3 shows pronounced dynamic clonal outgrowth of all mutational clusters (ie, clones) in the left column. The middle and right column show that this dynamic clonal behavior also affects pathogenetically relevant genes, thus underscoring that the most recent leukemia always has to be sequenced for a full picture of the mutational picture. Specifically, in most patients, at least 1 mutational cluster containing either known cancer and/or druggable genes newly arose in the post–allo-SCT relapse. Thus, as the mutational clusters containing cancer genes prove as dynamic as those with other genes, this hints at different oncogenic drivers for each leukemic state (Figure 3).


Mutational clusters show profound clonal dynamics. Shown are the 7 patients for whom information on all 3 oncogenomes was available (A: 316, 318, 337, 514; B: 590, 660, 685). Different mutational patterns can be observed, both within as well as across the patients. Individual clusters are indicated and marked in different colors.
Mutational clusters show profound clonal dynamics. Shown are the 7 patients for whom information on all 3 oncogenomes was available (A: 316, 318, 337, 514; B: 590, 660, 685). Different mutational patterns can be observed, both within as well as across the patients. Individual clusters are indicated and marked in different colors.
In addition, we analyzed which T-cell receptor (TCR) and immunoglobulin gene rearrangements were identified by minimal residual disease marker screening in the patient’s individual leukemia genomes before and after SCT, respectively. As shown in supplemental Table 9, we found gains (n = 3) or losses (n = 1) of immunoglobulin H/TCR-based minimal residual disease markers in 4 of 7 patients’ ALL relapses after allo-SCT.
Disappearance of NT5C2 mutations in post–allo-SCT relapses
Our overall hypothesis was that the selective pressure of administered therapies strongly affects the molecular makeup of leukemic blasts. Accordingly, we focused on the gene NT5C2, in which activating mutations were shown to drive 6-mercaptopurine (6-MP) and 6-thioguanine (6-TG) resistance.18,19 As our patients had only been treated with those nucleoside analogs during maintenance therapy (and also in blocks during the intensive phases of chemotherapy) before allo-SCT, tracking of NT5C2 alterations gave us a unique opportunity to evaluate the dynamic clonal outgrowth of leukemic blasts. In OG1, no NT5C2 SNVs were detected. However, we found a total of 4 nonsynonymously coding SNVs in NT5C2 (Figure 4A; supplemental Table 10) in the OG2 of 3 of 10 of our patients. One of the SNVs, which was detected in 2 patients (R367Q), is well-known in its pronounced enzyme-activating properties.18,19 The other 2 SNVs (K26E, D113N) have so far not been described. Notably, all NT5C2 SNVs that were detected in OG2 disappeared again in OG3 once selection pressure of maintenance chemotherapy employing nucleoside analogs had been withdrawn (Figure 4A). Only 1 very small subclone persisted in the post–allo-SCT relapse (clone size, 0.28%, patient 514/D113N), whereas the other cases proved negative with a high sensitivity of 0.1% to 0.2%, as determined by amplicon sequencing.

Clonal dynamics and selective pressure of NT5C2 and TP53 mutations. (A) Graphs indicate the variant allele frequencies in the SNVs in NT5C2 as detected by whole-exome sequencing (supplemental Table 9). Patient identifiers (UPN) as well as amino acid exchanges are indicated. Below each graph, the results of the amplicon sequencing (AS) are indicated. (B) SNVs detected in TP53 (also supplemental Table 10). The Li-Fraumeni patient (590) is not shown.
Clonal dynamics and selective pressure of NT5C2 and TP53 mutations. (A) Graphs indicate the variant allele frequencies in the SNVs in NT5C2 as detected by whole-exome sequencing (supplemental Table 9). Patient identifiers (UPN) as well as amino acid exchanges are indicated. Below each graph, the results of the amplicon sequencing (AS) are indicated. (B) SNVs detected in TP53 (also supplemental Table 10). The Li-Fraumeni patient (590) is not shown.
High frequency of TP53 mutations in post–allo-SCT relapses
Looking more specifically at recurrently mutated genes, we detected TP53 mutations in 5 of 10 patients (Figure 4B; supplemental Table 11) and, importantly, in 4 of 5 cases, at least 1 TP53 mutation was present in the post–allo-SCT relapse. One patient (590) had previously undiagnosed Li-Fraumeni syndrome. In the other 4 patients, 1 patient (514) had 1 unique SNV each in OG2 as well as OG3, 1 patient (735) only in OG2, and 2 patients had TP53 mutations only present in their OG3s (660, 685). One patient (685) acquired 3 different mutations in OG3. All identified TP53 mutations were predicted to be functionally relevant and were previously described in cancer and/or Li Fraumeni families (http://p53.iarc.fr/).
As with the NT5C2 mutations, the subclonal presence vs absence of the TP53 SNVs in preceding and subsequent samples was additionally analyzed via amplicon-based deep sequencing; in 3 of 7 analyzed cases a minor subclone was detected by deep sequencing (Figure 4B) in the prior relapse sample. Of particular interest, TP53-mutated minor subclones of 0.09% and 0.10% were found in the pre–allo-SCT relapse in 2 of 3 patients (514/R181H and 660/R248P), which then gave rise to clonal mutations in the post–allo-SCT leukemia. Time to post–allo-relapse could not be correlated to the presence of very low level TP53 mutations in the relapse preceding SCT; however, time to relapse was extremely short in all patients relapsing with TP53 mutations, highlighting the high aggressiveness of this leukemia (median time to post–allo-SCT relapse, 0.6 vs 0.2 years).
Cancer genes are recurrently mutated in OG3 and present in the core set of patient-individual mutations
Next, we analyzed which cancer-associated genes in addition to TP53 were mutated in the oncogenomes. Except for patient 514 in OG1, all other 26 oncogenomes showed mutations in at least 1 cancer-related gene (as determined by the cancer gene census; Figure 5A). Twenty-nine mutations were found in all OG1 combined, and 67 mutations in all OG2 combined. Regarding those mutated genes representing the core set of genetic alteration present in all leukemia states of an individual patient, we found at least 1 cancer-associated gene in 8 of 10 patients (supplemental Table 12).

Mutations in cancer genes and targetable genetic lesions in the post–allo-SCT relapses. (A) Mutated cancer genes across all patients and oncogenomes. (B) Details on those cancer genes, which were mutated in at least 2 patients. UPNs as earlier. (C) Targetable genes carrying mutations in the OG3 of the respective patients, with possible therapeutic options indicated. SNVs are indicated in red, copy number losses in orange, hypermutated oncogenomes (defined by ≥200 SNVs) in blue. (D) Sensitivity testing of patient cells/patient-derived xenografts against APR-246 in different concentrations (1-5-10-100 micro molar). neg. ctrl., negative control (primograft X116; TP53 wild-type); pos. ctrl., positive control (TP53 mutant primograft X172 carrying 2 TP53 mutations on different alleles).
Mutations in cancer genes and targetable genetic lesions in the post–allo-SCT relapses. (A) Mutated cancer genes across all patients and oncogenomes. (B) Details on those cancer genes, which were mutated in at least 2 patients. UPNs as earlier. (C) Targetable genes carrying mutations in the OG3 of the respective patients, with possible therapeutic options indicated. SNVs are indicated in red, copy number losses in orange, hypermutated oncogenomes (defined by ≥200 SNVs) in blue. (D) Sensitivity testing of patient cells/patient-derived xenografts against APR-246 in different concentrations (1-5-10-100 micro molar). neg. ctrl., negative control (primograft X116; TP53 wild-type); pos. ctrl., positive control (TP53 mutant primograft X172 carrying 2 TP53 mutations on different alleles).
Looking in more detail into OG3, 65 mutations were identified across all 10 post–allo-SCT relapses (Figure 5B).
Genetic lesions in 9 of 10 post–allo-SCT relapses can be targeted by nonstandard therapeutic agents
Next, based on a drug-target relationship recently employed by the German INFORM consortium,36 we searched for genetic alterations targeted directly (a certain gene) or indirectly (a certain pathway) by novel therapeutic agents already approved, and thus readily available in this clinically difficult situation of post–allo-SCT relapse.
Of particular note, 9 of 10 patients exhibited SNVs in genes and/or pathways, for which at least 1 additional targeted therapy is available (Figure 5C; supplemental Table 13). Those therapeutic agents comprised small-molecule inhibitors, tyrosine kinase inhibitors, and antibodies such as Cabozantinib, Canakinumab, Dasatinib, Erlotinib, Ibrutinib, Pazopanib, Olaparib, Sunitinib, and Trametinib. In addition, CDKN2A/B deletions as well as TP53 mutations are also targetable (Figure 5C).
Mutational cluster analysis containing druggable genes yet again revealed clusters, which proved highly variable, both between patients and between leukemic states of single patients (Figure 3).
As already described here, all TP53 mutations identified in post–allo-SCT relapses affect the DNA-binding domain. Emerging data indicate that ALL cell lines and primary ALL samples harboring this type of TP53 alteration are particularly sensitive to the small molecule APR-246, which induces restoration of p53 wild-type conformation in vitro and in vivo.37,,,-41 Thus, we performed in vitro drug sensitivity assays with leukemic blasts from the 2 patients 514 (R181H) and 660 (R248P) from the respective post–allo-SCT relapses. We could show sensitivity of blasts against APR-246 in both cases (Figure 5D).
Discussion
Our study provides the first comprehensive analysis of the exomic mutational landscape of pediatric post–allo-SCT relapses, including amplicon-based deep sequencing of NT5C2 and TP53 mutations for an in depth-analysis of their clonal dynamics. The main findings include high individuality and profoundly dynamic outgrowth of individual mutations.
The spectrum of somatic mutations in our patient cohort proved highly diverse and patient-specific, reflecting a mostly private set of specific genetic alterations found in each of the 10 children. Moreover, we identified profoundly dynamic mutational spectra in individual patient’s leukemias over time (Figure 3). Although we cannot exclude subclonal contributions, which fell below our threshold of detection, the observed individuality of the oncogenomes was not unexpected, as childhood ALL represents a genetically heterogeneous disease. On a molecular level, the pathogenesis of pediatric ALL is primarily determined by the lineage origin (B-cell or T-cell) and the presence of subtype-defining chromosomal translocations and specific somatic aberrations,42,43 and, thus, requires individual sequencing of the most recent leukemic samples.
As previously described in both relapsed pediatric B- and T-cell leukemias,44,-46 specific changes of relapsed leukemias include activating mutations in the gene NT5C2, which result in increased nucleotidase activity, conferring resistance to chemotherapy with 6-mercaptopurine and 6-thioguanine.19 We identified 1 known (R367Q) mutation in our study, as well as 2 not previously described mutations (K26E, D113N) in a total of 3 patients (prevalence 33%). Of interest, these mutations disappeared in the post–allo-SCT relapse, as confirmed by amplicon sequencing in all affected children once selection pressure of maintenance therapy (and blocks during the intensive phase of treatment) had been lifted. This finding demonstrating substantial clonal outgrowth and adaptivity is in line with the very recent observations that NT5C2 mutations while conferring resistance to maintenance chemotherapy come at the cost of impaired leukemia cell growth and leukemia-initiating cell activity, and thus decrease the fitness of the leukemic blasts.47
In our study, we identify TP53 mutations in 4 of 10 patients with post–allo-SCT relapse, which appears significantly more prevalent than in a previous study reporting TP53 alterations in 30 (11%) of 265 children with first ALL relapse (Fisher’s exact test P = .02).48,,-51 TP53 mutations are known to confer poor prognosis and are more common in relapsed pediatric ALL compared with initial disease52 ; however, the substantially increased frequency of TP53 alterations observed in our study for the post–allo-SCT relapses compared with the setting of first pre–allo-SCT relapses reflects selection induced by highly intensive treatment. This fits well with the recent observation that genotoxic stresses promote the clonal expansion of hematopoietic stem cells expressing mutant TP53, increasing the possibility of acquiring additional genetic and/or epigenetic changes.53
All TP53 mutations identified in ALL post–allo-SCT relapses in our study affect the DNA-binding domain. As it was previously reported that ALL samples harboring this type of mutation are sensitive to the small molecule APR-246, we performed drug sensitivity tests and could indeed show efficient targeting of leukemic blasts by this compound. Moreover, as TP53 mutations indicate tumor escape, and thus resistance to conventional chemotherapy, our findings point to a high demand for novel therapeutic targets in the setting of post–allo-SCT relapses.
The clinically most relevant finding was that in 9 of 10 children, we identified alterations in genes for which additional nonstandard therapies with approved drugs are available. This proportion was substantially higher than the recently published data from the INFORM pilot study, in which, although based on the same list of alterations and matched novel agents, a potentially druggable alteration was only detected in 50% of children.36
Five patients displayed NRAS/KRAS mutations, which were all in known hotspot positions (AA12/13/61). The MEK inhibitor Trametinib represents a promising therapeutic option in RAS mutated tumors and has already been used successfully in refractory myeloid malignancies.54,55 In addition, KRAS mutations were recently shown to be highly sensitive to the MEK inhibitor Selumetinib in mice xenografted with primary pediatric ALL blasts carrying the AA12 hotspot mutant,56 thus providing in vivo validation for this therapeutic target.
A previous study reported a mean of 0.5 mutations per megabase in ALL.57 Looking specifically at the few much larger oncogenomes in our study, newly acquired somatic SNVs were indeed found in DNA polymerases and DNA repair/Fanconi anemia genes in all cases. As mutational burden has recently emerged as a promising biomarker for clinical response to various immunotherapies, immune checkpoint inhibition58 represents another individualized therapeutic option for those patients with high mutational burden in the post–allo-SCT relapses.
Although the exact threshold for a hypermutator phenotype in pediatric ALL remains to be defined, 1 recent pan-pediatric cancer paper defined it as at least 2 coding mutations per MB (“pediatric high” in contrast to the adult “hypermutated” of at least 10 coding mutations per MB),13 whereas a second publication, which analyzed a larger number of pediatric lymphoblastic leukemias, defined it as at least 0.4 somatic mutations per MB (B-ALL) and at least 0.8 somatic mutations per MB (T-ALL), respectively.14 All large oncogenomes identified in our study are well above these thresholds.
As we had only leukemic DNA available, we were not able to analyze whether pathways that may influence immune function (such as the downregulation of MHC class II genes) were also affected in the post–allo-SCT setting, although on the DNA level, we did we did not identify any deletions spanning more than 1 of the following genetic loci: HLA-A, HLA-B, HLA-C, HLA-DPB1, HLA-DQB1, HLA-DRB1 (data not shown). Such dysregulations were recently shown in 50% of adult patients relapsing with AML after allo-SCT.59
Taken together, our study is the first to identify RAS pathway mutations, TP53 alterations, and hypermutator phenotypes as recurrent and potentially druggable genetic lesions in post–allo-SCT relapses. At the same time, mutational cluster analysis reveals highly dynamic clonal composition regarding both cancer-related and bystander mutations in individual patients over time, thus providing a cautionary note on leukemia escape to individualized therapies that rely on single-target approaches. Therapeutic targets identified in post–allo-SCT relapses are well amenable to combinatorial approaches, as they display nonoverlapping toxicities and hold promise to complement innovative approaches as CAR T-cell immunotherapy and bispecific T-cell engagers.
Acknowledgments
The authors thank Aprea Therapeutics (Stockholm, Sweden) for kindly providing APR-246 for the study.
Funding was received by the Düsseldorf School of Oncology (Comprehensive Cancer Center Düsseldorf/Deutsche Krebshilfe, Medical Faculty HHU Düsseldorf), Deutsche Forschungsgemeinschaft (DFG, HO 5456/3-1), Elterninitiative Kinderkrebsklinik e.V. (Düsseldorf), and the German Consortium for Translational Cancer Research (DKTK).
Authorship
Contribution: J.I.H., A.B., and R.M. designed research; J.I.H., S.G., A.K., U.F., A.C.M., S.K., J.E., A.B., R.T., and R.M. analyzed and interpreted data; J.I.H., M.K., A.B., and R.M. wrote the manuscript; M.G. and D.H. performed sequencing studies; S.D. and L.H.M. performed drug testing experiments; M. Stanulla, M. Schrappe, U.z.S., P.B., B.S., J.A., A.M., G.E., A.v.S., C.P., B.B., J.-P. B., F.B., F.S., and C.E., provided case information and vital clinical specimens; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: P.B. received research support, provided consultancy, and received honoraria (Novartis, Servier, Neovii Biotech, Riemser, Medac). C.P. provided consultancy and speakers bureau service (Medac, Novartis, Jazz Pharma, Amgen, Pfizer). M. Schrappe received research support and honoraria (Novartis, SigmaTau Rare Diseases, JAZZpharma, Amgen). The remaining authors declare no competing financial interests.
Correspondence: Jessica I. Hoell, Department of Pediatric Hematology and Oncology, Martin Luther University Halle-Wittenberg, Ernst-Grube-Str 40, 06120 Halle (Saale), Germany; e-mail: jessica.hoell@uk-halle.de.

