

Key Points
The ClinGen MM-VCEP has specified RUNX1-specific curation rules to address gene function, gene-specific domains, and phenotypic criteria.
RUNX1-specific criteria resulted in a reduction in CONF and VUS variants by 33%, emphasizing the need for expert variant curation.

Abstract
Standardized variant curation is essential for clinical care recommendations for patients with inherited disorders. Clinical Genome Resource (ClinGen) variant curation expert panels are developing disease-associated gene specifications using the 2015 American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) guidelines to reduce curation discrepancies. The ClinGen Myeloid Malignancy Variant Curation Expert Panel (MM-VCEP) was created collaboratively between the American Society of Hematology and ClinGen to perform gene- and disease-specific modifications for inherited myeloid malignancies. The MM-VCEP began optimizing ACMG/AMP rules for RUNX1 because many germline variants have been described in patients with familial platelet disorder with a predisposition to acute myeloid leukemia, characterized by thrombocytopenia, platelet functional/ultrastructural defects, and a predisposition to hematologic malignancies. The 28 ACMG/AMP codes were tailored for RUNX1 variants by modifying gene/disease specifications, incorporating strength adjustments of existing rules, or both. Key specifications included calculation of minor allele frequency thresholds, formulating a semi-quantitative approach to counting multiple independent variant occurrences, identifying functional domains and mutational hotspots, establishing functional assay thresholds, and characterizing phenotype-specific guidelines. Preliminary rules were tested by using a pilot set of 52 variants; among these, 50 were previously classified as benign/likely benign, pathogenic/likely pathogenic, variant of unknown significance (VUS), or conflicting interpretations (CONF) in ClinVar. The application of RUNX1-specific criteria resulted in a reduction in CONF and VUS variants by 33%, emphasizing the benefit of gene-specific criteria and sharing internal laboratory data.
Introduction
In 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) released a landmark document providing guidance on variant classification that has now been adopted by many international diagnostic laboratories. It was designed to have universal applicability to all Mendelian disorders, using several types of weighted and categorized evidence, and it therefore requires significant expertise as well as gene- and disease-specific knowledge to be correctly applied.1 Variable application of functional and domain-related evidence and inconsistent interpretation and use of the ACMG/AMP criteria are key contributors to incorrect classifications of variants, and significant discrepancies among laboratories highlight the utility of expert guidance.2,-4 A few studies have proposed approaches to one or more aspects of variant interpretation, such as quantitative criteria for cosegregation, use of population databases, adaptation of minor allele frequency (MAF), classes of evidence, and gene-level implications.2,,,,,-8 However, due to the unique characteristics of every gene and its disease correlates, along with the variability in the application of classification criteria and evidence interpretation, there is still a lack of comprehensive guidance for variant interpretation.
This need for expert involvement and gene-specific guidance has been addressed by the National Institutes of Health (NIH)–funded Clinical Genome Resource (ClinGen; https://clinicalgenome.org), which serves as a body for managing and centralizing clinically relevant genomic knowledge, providing guidance and tools for defining the clinical validity of gene and variant contributions to disease. Several working groups and expert panels were created within ClinGen, including gene- and disease-specific Variant Curation Expert Panels (VCEPs).9,10 Moreover, the ClinGen Sequence Variant Interpretation (SVI) Working Group (https://clinicalgenome.org/working-groups/sequence-variant-interpretation/) aims to provide general recommendations for the refinement and evolution of the ACMG/AMP guidelines, which are then specialized further by the gene-specific VCEP.
The publicly available ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), launched in 2013, serves as a valuable centralized resource for documenting the clinical significance of genetic variants submitted by clinical and research laboratories and databases such as OMIM (Online Mendelian Inheritance in Man) and GeneReviews.11 ClinVar uses the ACMG-recommended 5-level scoring system to indicate the level of evidence supporting the assertion of clinical significance of a variant. Human variant data curated by ClinGen expert panels are submitted to ClinVar with a 3-star status (reviewed by expert panel) including a designation that the ClinGen VCEP process has been recognized by the US Food and Drug Administration (FDA).
The general workflow of a VCEP is to define its leadership/membership and scope of focus as well as conflicts of interest.9 Once approved, this group develops disease-specific variant classification rules, based on curation of gene-specific features, published literature, and evidence thresholds that are consistent with gene–disease associations. ACMG and AMP have defined 28 codes that address specific evidence, including population data, segregation data, functional data, computational predictions, and allelic data.3 Each code is weighted according to the strength of the evidence: stand-alone, very strong, strong, moderate, or supporting. Codes are also designated as defining the direction of clinical significance: benign (BEN) or pathogenic (PATH). These evidence codes applied to variants are then combined to arrive at a single designation of clinical significance: PATH, likely pathogenic (LPATH), variant of uncertain significance (VUS), likely BEN (LBEN), or BEN. Once preliminary rules are specified, they are pilot tested on a collection of variants with existing assertions of clinical significance, and based on the results of this preliminary testing, the VCEP may adjust some of its rules to optimize variant classification. Once final rules have been approved, they are published and implemented, with VCEP-curated assertions disseminated via the ClinVar database.
A Myeloid Malignancy VCEP (hereafter referred to as MM-VCEP) was formed in 2018 as a collaboration between the American Society of Hematology and ClinGen. The MM-VCEP began adapting the ACMG/AMP framework for RUNX1 variant classification. Because it was the first germline predisposition syndrome identified for myeloid malignancies, there were many variants already deposited in the ClinVar repository. Germline PATH variants in RUNX1, first described in 1999, cause dominantly inherited familial platelet disorder with a predisposition to acute myeloid leukemia (FPD/AML), characterized by mild to moderate thrombocytopenia, functional and ultrastructural platelet defects, and a predisposition to myelodysplastic syndrome (MDS) and AML and less frequently to T-cell acute lymphoblastic leukemia (T-ALL).12,-14 In 2016, the revision of the World Health Organization classification of myeloid neoplasms and acute leukemia included myeloid malignancies arising from germline PATH variants in ANKRD26, ETV6, and RUNX1 in a new category defined as “myeloid neoplasms with germline predisposition and preexisting platelet disorder.”15 Reported inherited and de novo RUNX1 variants include missense, nonsense, and splice site single-nucleotide variants (SNVs), small in- or out-of-frame insertions and deletions (indels), as well as copy number variants (CNVs) such as intragenic or whole-gene deletions.16,-18 The prevalence of PATH RUNX1 germline variants is unknown but presumed to be rare. The disease shows high penetrance with variable expressivity and genotype/phenotype correlation, and the lifetime risk of hematologic malignancies is ∼44%, with an average age of onset of 33 years.19,-21 More than one-half of germline RUNX1 variants are reported in single probands/families,13 leading to a high allelic heterogeneity that restricts the collection of data from segregation analyses and functional analyses across several affected families. Individuals with a hematologic malignancy are often candidates for hematopoietic stem cell transplantation. The identification of patients with a PATH germline variant in RUNX1 and its correct classification of the variant are imperative to the selection of potential related donors, among other clinical implications.22,,,-26
Here, we present the RUNX1-specific guidelines generated by the MM-VCEP. The MM-VCEP adapted the ACMG/AMP framework for RUNX1 variant classification with the aim of improving consistency in variant classification and curating RUNX1 variants for 3-star submission to ClinVar. We used multiple lines of evidence, showing the rationale and data supporting each criterion’s modification, and the results from pilot testing the criteria on variants with BEN/LBEN, PATH/LPATH, VUS, and conflicting (CONF) ClinVar assertions. The application of rules for RUNX1 variant curation will serve as a model for the curation of variants in other genes that also cause inherited myeloid hematologic malignancies, such as ANKRD26, ETV6, DDX41, and GATA2. The ClinGen’s Web site contains the MM-VCEP variant classification recommendations and any subsequent modifications to these codes over time (https://www.clinicalgenome.org/affiliation/50034).
Methods
ClinGen MM-VCEP
The MM-VCEP is sponsored by the American Society of Hematology through its partnership with ClinGen and is described at https://clinicalgenome.org/affiliation/50034/. The MM-VCEP team comprises 34 professionals with expertise in key domains and includes clinical geneticists, genetic counselors, hematologists with professional training in genetics, laboratory and research scientists, and variant curation experts. Additional emphasis was placed on global representation, with 22 participating institutions in 6 countries: Australia, France, Italy, Sweden, the United Kingdom, and the United States. The MM-VCEP meets regularly via biweekly teleconferences and corresponds via e-mail on a regular basis. Approval of MM-VCEP is overseen by ClinGen and consists of 4 steps: (1) defining the group/members and scope of the VCEP; (2) developing gene/disease-specific classification rules; (3) optimization of rules using pilot variants; and (4) MM-VCEP approval by ClinGen, implementation of rules in the ClinGen Variant Curation Interface, and submission of curated variants to the ClinVar database. For step two, members were divided into 3 subgroups that focused on the modification of functional/computational/splicing criteria (Team F), population/phenotypic criteria (Team P), and segregation/allelic/de novo criteria (Team S). All members disclosed potential conflicts of interest as required by ClinGen.
ACMG/AMP specifications for RUNX1
MM-VCEP members proposed and discussed changes to the existing ACMG/AMP classifications for RUNX1 germline variants and arrived at consensus decisions via teleconference calls and e-mail. Criteria modifications included gene- or disease-specific modifications, strength-level adjustments, general recommendations, and certain criteria being deemed “not applicable.” Publicly available databases, predictive software, and published data obtained from relevant papers were used for criteria specifications. For BA1/BS1 RUNX1-specific population MAF, calculations were made assuming Hardy-Weinberg equilibrium using the recently published Whiffin/Ware online calculator.6 Additional efforts included identification of key functional domains and mutational hotspots within RUNX1, definition of informative functional assays, and characterization of phenotypic criteria. Recommendations for using ACMG/AMP criteria from the ClinGen’s SVI working group were also incorporated.27,-29 Preliminary and final ACMG/AMP specifications required complete consensus of the MM-VCEP.
Pilot variants
All pilot variants are annotated by using RefSeq IDs NM_001754.4 and NC_000021.9 (GRCh38/hg38). Variants submitted to ClinVar by a variety of clinical laboratories were prioritized for classification. Preliminary rules were refined by interpreting a set of 52 RUNX1 variants, which were selected to represent the spectrum of variants in RUNX1, covering various types of SNVs such as missense, nonsense, splice site, synonymous, and intronic variants; indels such as in-frame duplications and out-of-frame deletions; and CNVs such as intragenic deletions. Similarly, the pilot variants covered a diverse range of classifications in ClinVar, including discrepant assertions (12 BEN/LBEN, 14 VUS, 20 PATH/LPATH, 4 CONF, and 2 with no ClinVar assertions). The variant classification and rules applied were reviewed on conference calls to resolve discrepancies and reach consensus. Basic information regarding individual phenotypes and segregation with disease was obtained from the literature and ClinVar submitters. Statistical approaches for calculations of PS4 are available in the supplemental Methods. Further optimization of rules was performed, and a discussion with the entire MM-VCEP was triggered whenever members disagreed or raised concerns regarding the applicability of a given rule. Curators used ClinGen’s Variant Curation Interface (https://curation.clinicalgenome.org) to assess and document the applicable rules for each variant. Once the MM-VCEP was approved, the classified RUNX1 variants with the adapted evidence code framework applied to the variants were submitted to ClinVar and were designated with a 3-star evidence code and FDA recognition flag. The first 52 RUNX1 variant curations are now available in ClinVar and can be accessed at https://www.ncbi.nlm.nih.gov/clinvar/submitters/507107/.
Results
Summary of rule specifications
The final MM-VCEP ACMG/AMP specifications for RUNX1 were approved by ClinGen and are outlined in Table 1. Six of the original 28 ACMG/AMP criteria had general recommendations on the application of the rule (PM2, PP3, BS4, BP2, BP4, and BP7), 2 required gene- or disease-based specifications (BA1 and BS1), and 2 rules were adjusted in their level of strength (PS1 and PM5). Both gene- or disease-based and strength-level specifications were made to 9 rules (PVS1, PS2, PS3, PS4, PM1, PM4, PM6, PP1, and BS3). Five rules required exceptions for combinations with other rules (PS2, PS3, PM5, PM6, and PP3), and 9 rules were deemed not applicable (PM3, PP2, PP4, PP5, BS2, BP1, BP3, BP5, and BP6). One change to the ACMG/AMP combination of criteria for classification of clinical significance was made in the case of BS1, which can be used as a stand-alone criterion for LBEN classification in the absence of any supporting PATH evidence. The following section highlights the approaches and rationale behind key specifications such as phenotypic criteria, MAF thresholds, and validity of functional assays. Of note, germline material for patients with FPD/AML or patients with suspected inherited hematologic malignancies cannot include blood or bone marrow from these patients because this is the affected tissue harboring somatic mutations. We recommend using cultured skin fibroblasts as the gold standard, or alternatively DNA from hair roots or cultured mesenchymal stromal cells.19,25
MM-VCEP ACMG/AMP specifications for RUNX1 variants
| ACMG/AMP criteria code | Original ACMG/AMP rule summary | Specification | Stand-alone | Very strong | Strong | Moderate | Supporting | Comments |
|---|---|---|---|---|---|---|---|---|
| PVS1 | Null variant in a gene where LOF is a known mechanism of disease | Gene-specific, strength | NA | Per modified RUNX1 PVS1 decision tree for SNVs, indels, and CNVs and table of splicing effects | NA | RUNX1 LOF variants are a common mechanism of disease in FPD/AML. Three major isoforms (A, B, and C) are expressed by use of 2 promotors and alternative splicing. C-terminal variants not predicted to undergo NMD are classified as PVS1_strong, deletions of exons 2 and 3, presumably only affecting RUNX1 isoform 1C, meet PVS1_moderate | ||
| PS1 | Same AA change as a previously established PATH variant regardless of nucleotide change | Strength | NA | NA | Same AA change as a previously established PATH variant regardless of nucleotide change | Same AA change as a previously established, LPATH variant regardless of nucleotide change | NA | (1) RNA data or agreement in splicing predictors show no splicing effects (SSF and MES predict either increase in canonical splice site score or decrease in canonical splice score by no more than 10% and no putative splice sites are created). (2) The previously established variant must be asserted PATH/LPATH based on MM-VCEP rules for RUNX1 before this rule can be applied |
| PS2 | De novo (maternity and paternity confirmed) in a patient with the disease and no family history | Disease-specific, strength | NA | NA | NA | Two or more proven de novo occurrences (maternity and paternity confirmed) in patients with the RUNX1-phenotype | One proven de novo occurrence (maternity and paternity confirmed) in a patient with the RUNX1-phenotype | (1) No family history is defined as: absence of the variant and any of the RUNX1-phenotypic criteria in first- and/or second-degree relatives. (2) The proband must exhibit at least 1 phenotypic FPD/AML criterion. (3) The maximum allowable strength by combining PS2 and PM6 criteria is to apply 1 moderate or 2 supporting rules |
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect | Gene-specific, strength | NA | NA | Transactivation assays exhibiting altered transactivation (<20% of wt, and/or reduced to levels similar to well-established PATH variants such as R201Q or R166Q) and data from a secondary assay showing altered function. PS3 cannot be applied if the variant meets PVS1. If the variant meets criteria for PVS1_strong and PS3, we recommend either applying PVS1_strong and PS3_moderate or upgrading PVS1_strong to PVS1 without applying PS3 | Transactivation assays exhibiting altered transactivation (<20% of wt, and/or reduced to levels similar to well-established PATH variants such as R201Q or R166Q) or ≥2 secondary assays exhibiting altered function | Transactivation assays exhibiting enhanced transactivation (>115% of wt) | (1) Transactivation assays should include wt and known PATH controls, as well as coexpression with CBFβ. Promoter sequences of CSF1R (M-CSF-R), PF4, C-FMS, and GZMB, containing consensus RUNX1-binding sites have been used for transactivation assays. (2) The following secondary assays have been performed: EMSA and yeast hybrid assays (decreased DNA-binding affinity), co-IP, FRET, and affinity assays (diminished heterodimerization ability with CBFβ), IF and WB with cell fractionation (abnormal cellular localization), colony-forming assays (reduced colony-forming potential), and xenotransplantation experiments (abnormal function of mutant RUNX1 in vivo). (3) PS3 can also be applied for evidence of very low or abnormal mRNA/protein expression of the variant allele as a functional consequence of a null variant or incorrect mRNA/protein products |
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in control subjects | Disease-specific, strength | NA | NA | Four or more probands meeting RUNX1-phenotypic criteria | Two to 3 probands meeting RUNX1-phenotypic criteria | One proband meeting RUNX1-phenotypic criteria | The affected individual has to fit at least 1 of the RUNX1-phenotypic criteria and the variant has to be either absent from gnomAD (overall population) or only present once |
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain without BEN variation | Gene-specific, strength | NA | NA | NA | Variant affecting 1 of the following 13 hotspot residues: R107, K110, A134, R162, R166, S167, R169, G170, K194, T196, D198, R201, R204 | Variant affecting 1 of the other AA residues 105-204 within the RHD | The RHD (AA 77-204) has been established as a highly conserved DNA-binding domain without any BEN variation in ClinVar. No germline PATH variants have been reported in residues in the region (AA 77-104) to date. The AA range under PM1_supporting may be expanded in the future to other parts of the protein if more evidence emerges |
| PM2 | Absent from control subjects | General recommendation | NA | NA | NA | Per original ACMG/AMP guidelines | NA | Variant must be completely absent from all population databases. The mean coverage of RUNX1 in the population database used should be at least 20× |
| PM3 | For recessive disorders, detected in trans with a PATH variant | NA | FPD/AML is inherited in an autosomal dominant manner | |||||
| PM4 | Protein length changes due to in-frame deletions/insertions in a nonrepeat region or stop-loss variants | Gene-specific, strength | NA | NA | NA | In-frame deletion/insertion affecting at least 1 of the 13 hotspot residues (R107, K110, A134, R162, R166, S167, R169, G170, K194, T196, D198, R201, R204) | Other in-frame deletion/insertion affecting residues 105-204 within the RHD | See PM1 |
| PM5 | Missense change at AA residue where a different missense change determined to be PATH has been seen before | Strength | NA | NA | Missense change at the same residue where ≥2 different missense changes have previously been determined to be PATH. PM5_strong cannot be applied together with PM1 | Missense change at the same residue where a different missense change has previously been determined to be PATH | Missense change at the same residue where a different missense change has previously been determined to be LPATH | See PS1 |
| PM6 | Assumed de novo (but without confirmation of maternity and paternity) in a patient with the disease and no family history | Disease-specific, strength | NA | NA | NA | Four or more assumed de novo occurrences (without confirmation of maternity and paternity) in patients with the RUNX1-phenotype | Two or 3 assumed de novo occurrences (without confirmation of maternity and paternity) in patients with the RUNX1-phenotype | See PS2 |
| PP1 | Cosegregation with disease in multiple affected family members | Disease-specific, strength | NA | NA | Seven or more meioses observed within 1 family or across multiple families | Five or 6 meioses observed within 1 family or across multiple families | Three or 4 meioses observed within 1 family or across multiple families | (1) Affected individuals exhibit at least 1 of the RUNX1-specific phenotypic criteria. (2) Only genotype- and phenotype-positive individuals and obligate carriers are counted. (3) Demonstration of cosegregation in multiple families is not required because many RUNX1 variants are unique and only occur in 1 family |
| PP2 | Missense variant in a gene that has a low rate of BEN missense variation and where missense variants are a common mechanism of disease | NA | Missense constraint z score for RUNX1 is <3.09 | |||||
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product | General recommendation | NA | NA | NA | NA | Per original ACMG/AMP guidelines | (1) PP3 should be applied for missense variants with a REVEL score >0.75. (2) PP3 should be applied for missense or synonymous variants if the variant alters the last 3 bases of an exon preceding a donor splice site or the first 3 bases of an exon following a splice acceptor site and the predicted decrease in the score of the canonical splice site (measured by both MES and SSF) is at least 75% regardless of the predicted creation/presence of a putative cryptic splice site. (3) PP3 should also be applied for intronic variants (in introns 4-8) located in reference to exons at positions +3 to +5 for splice donor sites or −3 to −5 for splice acceptor sites for which the predicted decrease in the score is at least 75% (measured by both MES and SSF) regardless of the predicted creation/presence of a putative cryptic splice site. (4) PP3 cannot be applied for canonical splice site variants |
| PP4 | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology | NA | FPD/AML does not exhibit a highly specific phenotype, and there is substantial genetic heterogeneity | |||||
| PP5 | Reputable source recently reports variant as PATH, but the evidence is not available to the laboratory to perform an independent analysis | NA | According to SVI recommendations | |||||
| BA1 | Allele frequency is >5% in ESP, 1000G, or ExAC | Disease-specific | MAF ≥0.0015 (0.15%) | NA | NA | NA | NA | The variant is present in any general continental population dataset with a minimum number of 2000 alleles and variant present in ≥5 alleles |
| BS1 | Allele frequency is greater than expected for disorder | Disease-specific | NA | NA | MAF between 0.00015 (0.015%) and 0.0015 (0.15%) | NA | NA | (1) The variant is present in any general continental population dataset with a minimum number of 2000 alleles and variant present in ≥5 alleles. (2) Variant can be classified as LBEN based on BS1 alone if there is no contradictory evidence supporting pathogenicity |
| BS2 | Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age | NA | Patients with FPD/AML display incomplete penetrance, and the average age of onset of hematologic malignancies is 33 y | |||||
| BS3 | Well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing | Gene-specific, strength | NA | NA | (1) Transactivation assays exhibiting normal transactivation (80%-115% of wt); and (2) data from a secondary assay exhibiting normal function | NA | Transactivation assays exhibiting normal transactivation (80%-115% of wt) | See PS3 (1) and (2) |
| BS4 | Lack of segregation in affected members of a family | General recommendation | NA | NA | Applied when seen in ≥2 informative meioses | NA | NA | This code should only be applied for genotype-positive, phenotype-negative (with sufficient laboratory evidence) family members |
| BP1 | Missense variant in a gene for which primarily truncating variants are known to cause disease | NA | FDP/AML is caused by both PATH missense and truncating variants | |||||
| BP2 | Observed in trans with a PATH variant for a fully penetrant dominant gene/disorder or observed in cis with a PATH variant in any inheritance pattern | General recommendation | NA | NA | NA | NA | Per original ACMG/AMP guidelines | BP2 can also be applied if the variant is detected in a homozygous state |
| BP3 | In-frame deletions/insertions in a repetitive region without a known function | NA | RUNX1 does not contain a repetitive region without known function | |||||
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product | General recommendation | NA | NA | NA | NA | Per original ACMG/AMP guidelines | BP4 should be applied for missense variants if all of the following apply: (1) REVEL score <0.15; (2) SSF and MES predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10%; and (3) no putative cryptic splice sites are created. BP4 should also be applied for synonymous, intronic, and noncoding variants for which SSF and MES predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10% and no putative cryptic splice sites are created |
| BP5 | Variant found in a case with an alternate molecular basis for disease | NA | In rare circumstances, a patient can carry 2 variants in genes predisposing to hematologic malignancies | |||||
| BP6 | Reputable source recently reports variants as BEN, but the evidence is not available to the laboratory to perform an independent evaluation | NA | According to SVI recommendations | |||||
| BP7 | A synonymous variant for which splicing prediction algorithms predict no impact to the splice consensus sequence, nor the creation of a new splice site, and the nucleotide is not highly conserved | General recommendation | NA | NA | NA | NA | Per original ACMG/AMP guidelines. BP7 cannot be applied in combination with PP3 | Also applicable to intronic/noncoding variants at or beyond positions +7/–21 for which (1) SSF and MES predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10% and no putative cryptic splice sites are created; and (2) evolutionary conservation prediction algorithms predict the site as not conserved (eg, PhyloP score <0.1 or the variant is the reference nucleotide in 1 primate and/or 3 mammal species) |
| ACMG/AMP criteria code | Original ACMG/AMP rule summary | Specification | Stand-alone | Very strong | Strong | Moderate | Supporting | Comments |
|---|---|---|---|---|---|---|---|---|
| PVS1 | Null variant in a gene where LOF is a known mechanism of disease | Gene-specific, strength | NA | Per modified RUNX1 PVS1 decision tree for SNVs, indels, and CNVs and table of splicing effects | NA | RUNX1 LOF variants are a common mechanism of disease in FPD/AML. Three major isoforms (A, B, and C) are expressed by use of 2 promotors and alternative splicing. C-terminal variants not predicted to undergo NMD are classified as PVS1_strong, deletions of exons 2 and 3, presumably only affecting RUNX1 isoform 1C, meet PVS1_moderate | ||
| PS1 | Same AA change as a previously established PATH variant regardless of nucleotide change | Strength | NA | NA | Same AA change as a previously established PATH variant regardless of nucleotide change | Same AA change as a previously established, LPATH variant regardless of nucleotide change | NA | (1) RNA data or agreement in splicing predictors show no splicing effects (SSF and MES predict either increase in canonical splice site score or decrease in canonical splice score by no more than 10% and no putative splice sites are created). (2) The previously established variant must be asserted PATH/LPATH based on MM-VCEP rules for RUNX1 before this rule can be applied |
| PS2 | De novo (maternity and paternity confirmed) in a patient with the disease and no family history | Disease-specific, strength | NA | NA | NA | Two or more proven de novo occurrences (maternity and paternity confirmed) in patients with the RUNX1-phenotype | One proven de novo occurrence (maternity and paternity confirmed) in a patient with the RUNX1-phenotype | (1) No family history is defined as: absence of the variant and any of the RUNX1-phenotypic criteria in first- and/or second-degree relatives. (2) The proband must exhibit at least 1 phenotypic FPD/AML criterion. (3) The maximum allowable strength by combining PS2 and PM6 criteria is to apply 1 moderate or 2 supporting rules |
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect | Gene-specific, strength | NA | NA | Transactivation assays exhibiting altered transactivation (<20% of wt, and/or reduced to levels similar to well-established PATH variants such as R201Q or R166Q) and data from a secondary assay showing altered function. PS3 cannot be applied if the variant meets PVS1. If the variant meets criteria for PVS1_strong and PS3, we recommend either applying PVS1_strong and PS3_moderate or upgrading PVS1_strong to PVS1 without applying PS3 | Transactivation assays exhibiting altered transactivation (<20% of wt, and/or reduced to levels similar to well-established PATH variants such as R201Q or R166Q) or ≥2 secondary assays exhibiting altered function | Transactivation assays exhibiting enhanced transactivation (>115% of wt) | (1) Transactivation assays should include wt and known PATH controls, as well as coexpression with CBFβ. Promoter sequences of CSF1R (M-CSF-R), PF4, C-FMS, and GZMB, containing consensus RUNX1-binding sites have been used for transactivation assays. (2) The following secondary assays have been performed: EMSA and yeast hybrid assays (decreased DNA-binding affinity), co-IP, FRET, and affinity assays (diminished heterodimerization ability with CBFβ), IF and WB with cell fractionation (abnormal cellular localization), colony-forming assays (reduced colony-forming potential), and xenotransplantation experiments (abnormal function of mutant RUNX1 in vivo). (3) PS3 can also be applied for evidence of very low or abnormal mRNA/protein expression of the variant allele as a functional consequence of a null variant or incorrect mRNA/protein products |
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in control subjects | Disease-specific, strength | NA | NA | Four or more probands meeting RUNX1-phenotypic criteria | Two to 3 probands meeting RUNX1-phenotypic criteria | One proband meeting RUNX1-phenotypic criteria | The affected individual has to fit at least 1 of the RUNX1-phenotypic criteria and the variant has to be either absent from gnomAD (overall population) or only present once |
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain without BEN variation | Gene-specific, strength | NA | NA | NA | Variant affecting 1 of the following 13 hotspot residues: R107, K110, A134, R162, R166, S167, R169, G170, K194, T196, D198, R201, R204 | Variant affecting 1 of the other AA residues 105-204 within the RHD | The RHD (AA 77-204) has been established as a highly conserved DNA-binding domain without any BEN variation in ClinVar. No germline PATH variants have been reported in residues in the region (AA 77-104) to date. The AA range under PM1_supporting may be expanded in the future to other parts of the protein if more evidence emerges |
| PM2 | Absent from control subjects | General recommendation | NA | NA | NA | Per original ACMG/AMP guidelines | NA | Variant must be completely absent from all population databases. The mean coverage of RUNX1 in the population database used should be at least 20× |
| PM3 | For recessive disorders, detected in trans with a PATH variant | NA | FPD/AML is inherited in an autosomal dominant manner | |||||
| PM4 | Protein length changes due to in-frame deletions/insertions in a nonrepeat region or stop-loss variants | Gene-specific, strength | NA | NA | NA | In-frame deletion/insertion affecting at least 1 of the 13 hotspot residues (R107, K110, A134, R162, R166, S167, R169, G170, K194, T196, D198, R201, R204) | Other in-frame deletion/insertion affecting residues 105-204 within the RHD | See PM1 |
| PM5 | Missense change at AA residue where a different missense change determined to be PATH has been seen before | Strength | NA | NA | Missense change at the same residue where ≥2 different missense changes have previously been determined to be PATH. PM5_strong cannot be applied together with PM1 | Missense change at the same residue where a different missense change has previously been determined to be PATH | Missense change at the same residue where a different missense change has previously been determined to be LPATH | See PS1 |
| PM6 | Assumed de novo (but without confirmation of maternity and paternity) in a patient with the disease and no family history | Disease-specific, strength | NA | NA | NA | Four or more assumed de novo occurrences (without confirmation of maternity and paternity) in patients with the RUNX1-phenotype | Two or 3 assumed de novo occurrences (without confirmation of maternity and paternity) in patients with the RUNX1-phenotype | See PS2 |
| PP1 | Cosegregation with disease in multiple affected family members | Disease-specific, strength | NA | NA | Seven or more meioses observed within 1 family or across multiple families | Five or 6 meioses observed within 1 family or across multiple families | Three or 4 meioses observed within 1 family or across multiple families | (1) Affected individuals exhibit at least 1 of the RUNX1-specific phenotypic criteria. (2) Only genotype- and phenotype-positive individuals and obligate carriers are counted. (3) Demonstration of cosegregation in multiple families is not required because many RUNX1 variants are unique and only occur in 1 family |
| PP2 | Missense variant in a gene that has a low rate of BEN missense variation and where missense variants are a common mechanism of disease | NA | Missense constraint z score for RUNX1 is <3.09 | |||||
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product | General recommendation | NA | NA | NA | NA | Per original ACMG/AMP guidelines | (1) PP3 should be applied for missense variants with a REVEL score >0.75. (2) PP3 should be applied for missense or synonymous variants if the variant alters the last 3 bases of an exon preceding a donor splice site or the first 3 bases of an exon following a splice acceptor site and the predicted decrease in the score of the canonical splice site (measured by both MES and SSF) is at least 75% regardless of the predicted creation/presence of a putative cryptic splice site. (3) PP3 should also be applied for intronic variants (in introns 4-8) located in reference to exons at positions +3 to +5 for splice donor sites or −3 to −5 for splice acceptor sites for which the predicted decrease in the score is at least 75% (measured by both MES and SSF) regardless of the predicted creation/presence of a putative cryptic splice site. (4) PP3 cannot be applied for canonical splice site variants |
| PP4 | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology | NA | FPD/AML does not exhibit a highly specific phenotype, and there is substantial genetic heterogeneity | |||||
| PP5 | Reputable source recently reports variant as PATH, but the evidence is not available to the laboratory to perform an independent analysis | NA | According to SVI recommendations | |||||
| BA1 | Allele frequency is >5% in ESP, 1000G, or ExAC | Disease-specific | MAF ≥0.0015 (0.15%) | NA | NA | NA | NA | The variant is present in any general continental population dataset with a minimum number of 2000 alleles and variant present in ≥5 alleles |
| BS1 | Allele frequency is greater than expected for disorder | Disease-specific | NA | NA | MAF between 0.00015 (0.015%) and 0.0015 (0.15%) | NA | NA | (1) The variant is present in any general continental population dataset with a minimum number of 2000 alleles and variant present in ≥5 alleles. (2) Variant can be classified as LBEN based on BS1 alone if there is no contradictory evidence supporting pathogenicity |
| BS2 | Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age | NA | Patients with FPD/AML display incomplete penetrance, and the average age of onset of hematologic malignancies is 33 y | |||||
| BS3 | Well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing | Gene-specific, strength | NA | NA | (1) Transactivation assays exhibiting normal transactivation (80%-115% of wt); and (2) data from a secondary assay exhibiting normal function | NA | Transactivation assays exhibiting normal transactivation (80%-115% of wt) | See PS3 (1) and (2) |
| BS4 | Lack of segregation in affected members of a family | General recommendation | NA | NA | Applied when seen in ≥2 informative meioses | NA | NA | This code should only be applied for genotype-positive, phenotype-negative (with sufficient laboratory evidence) family members |
| BP1 | Missense variant in a gene for which primarily truncating variants are known to cause disease | NA | FDP/AML is caused by both PATH missense and truncating variants | |||||
| BP2 | Observed in trans with a PATH variant for a fully penetrant dominant gene/disorder or observed in cis with a PATH variant in any inheritance pattern | General recommendation | NA | NA | NA | NA | Per original ACMG/AMP guidelines | BP2 can also be applied if the variant is detected in a homozygous state |
| BP3 | In-frame deletions/insertions in a repetitive region without a known function | NA | RUNX1 does not contain a repetitive region without known function | |||||
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product | General recommendation | NA | NA | NA | NA | Per original ACMG/AMP guidelines | BP4 should be applied for missense variants if all of the following apply: (1) REVEL score <0.15; (2) SSF and MES predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10%; and (3) no putative cryptic splice sites are created. BP4 should also be applied for synonymous, intronic, and noncoding variants for which SSF and MES predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10% and no putative cryptic splice sites are created |
| BP5 | Variant found in a case with an alternate molecular basis for disease | NA | In rare circumstances, a patient can carry 2 variants in genes predisposing to hematologic malignancies | |||||
| BP6 | Reputable source recently reports variants as BEN, but the evidence is not available to the laboratory to perform an independent evaluation | NA | According to SVI recommendations | |||||
| BP7 | A synonymous variant for which splicing prediction algorithms predict no impact to the splice consensus sequence, nor the creation of a new splice site, and the nucleotide is not highly conserved | General recommendation | NA | NA | NA | NA | Per original ACMG/AMP guidelines. BP7 cannot be applied in combination with PP3 | Also applicable to intronic/noncoding variants at or beyond positions +7/–21 for which (1) SSF and MES predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10% and no putative cryptic splice sites are created; and (2) evolutionary conservation prediction algorithms predict the site as not conserved (eg, PhyloP score <0.1 or the variant is the reference nucleotide in 1 primate and/or 3 mammal species) |
Evidence codes are noted in bold font.
AA, amino acid; co-IP, coimmunoprecipitation; EMSA, electrophoretic mobility shift assay; FRET, fluorescence resonance energy transfer; IF, immunofluorescence; LOF, loss-of-function; MES, MaxEntScan; NA, not applicable; NMD, nonsense-mediated decay; RHD, Runt homology domain; SSF, Splice Site Finder; WB, western blot; wt, wild type.
Phenotypic criteria for FPD/AML
FPD/AML is characterized by mild to moderate thrombocytopenia, platelet functional and/or ultrastructural defects, and a predisposition to hematologic malignancies, most often AML and MDS, and less frequently T-ALL (Table 2). The penetrance is high; however, not all individuals carrying the PATH variant display the FPD/AML phenotype. Thrombocytopenia is the most common clinical presentation, followed by hematologic malignancies in ∼44% of these patients.19,-21 The MM-VCEP defined that in order to fit the FPD/AML phenotype, the patient must exhibit at least one of the following phenotypic criteria: (1) mild to moderate thrombocytopenia with normal platelet size and volume in the absence of other causative factors such as autoimmune (eg, antibodies against platelet surface antigens) or drug-related thrombocytopenia30 ; (2) platelet ultrastructural and/or functional defects, including platelet alpha31 or dense granule secretion defects30,32,33 or impaired platelet aggregation, particularly in response to collagen and epinephrine34,35 ; and (3) diagnosis of a hematologic malignancy, most commonly affecting the myeloid lineage causing AML or MDS, less frequently involving the lymphoid lineage and manifesting as T-ALL.26,30,36,37 There are rare case reports of patients with germline RUNX1 variants and mixed myeloproliferative syndromes/MDS such as chronic myelomonocytic leukemia,26,38 as well as case reports of patients with B-cell ALL39 and hairy-cell leukemia.40
FPD/AML phenotypic criteria
| Feature | Details | Lifetime risk |
|---|---|---|
| Thrombocytopenia | Mild to moderate, normal platelet size and volume, absence of other causes for thrombocytopenia | In most patients |
| Platelet ultrastructural and/or functional defects | Includes platelet alpha or dense granule secretion defects and impaired platelet aggregation (particularly in response to collagen and epinephrine) | Unknown |
| Hematologic malignancy | Most commonly AML or MDS, less frequently T-ALL. There are rare case reports of patients with germline RUNX1 mutations and mixed MPN/MDS such as CMML, as well as case reports of patients with B-cell ALL and hairy-cell leukemia | ∼44% |
| Feature | Details | Lifetime risk |
|---|---|---|
| Thrombocytopenia | Mild to moderate, normal platelet size and volume, absence of other causes for thrombocytopenia | In most patients |
| Platelet ultrastructural and/or functional defects | Includes platelet alpha or dense granule secretion defects and impaired platelet aggregation (particularly in response to collagen and epinephrine) | Unknown |
| Hematologic malignancy | Most commonly AML or MDS, less frequently T-ALL. There are rare case reports of patients with germline RUNX1 mutations and mixed MPN/MDS such as CMML, as well as case reports of patients with B-cell ALL and hairy-cell leukemia | ∼44% |
CMML, chronic myelomonocytic leukemia; MPN, myeloproliferative syndrome.
Population data (BA1, BS1, PM2, PS4, PS4_moderate, PS4_supporting, and BP2)
FPD/AML is a rare disorder. The prevalence of the disease-associated RUNX1 variants is unknown, with an estimated 5515 families worldwide based on a population incidence generated from a survey of centers with FPD/AML patients (A.L.B., written communication, 10 June 2019), which is likely an underestimate of the true prevalence. Among the 3 phenotypic features seen in individuals with germline RUNX1 variants (Table 2), thrombocytopenia is the most common. We conservatively estimated the prevalence of thrombocytopenia for use in the BA1/BS1 calculations. Most clinical laboratories establish their reference values for platelet counts by measuring samples from at least 120 healthy individuals and identifying the most outlying 5% of observed values. Most often, these outlying observations are split evenly between the ends of the test result distribution in the reference population (2.5% at each end of the distribution), resulting in a 2-sided reference interval.41 Using this approach, the prevalence of thrombocytopenia can be defined as 1 in 40. The penetrance in families with RUNX1 germline variant is high to near-complete, with 85% being the lowest penetrance reported to date13,19,-21 (S.K., written communication, 19 March 2019). Thus far, no founder variants in RUNX1 have been reported. De novo variants are rare but have been described.16,-18
The MM-VCEP modified BA1 using these conservative assumptions and corresponding values to account for the unknown prevalence and disease contribution of RUNX1. To obtain an RUNX1-specific population MAF threshold for BA1, we used the Whiffin/Ware calculator6 (http://cardiodb.org/allelefrequencyapp/) with a prevalence of 1 in 40, a conservative unascertained penetrance estimate of 85%, an allelic heterogeneity of 100%, and a maximum genetic heterogeneity of 10%. The MM-VCEP also adopted the SVI recommendation that the variant be present in any general continental population dataset with a minimum number of 2000 alleles and the variant present in ≥5 alleles.42 A 95% confidence interval was used to develop the thresholds. The threshold developed for application of BA1 as a stand-alone criterion is a MAF ≥0.0015 (0.15%). For BS1, a maximum genetic heterogeneity contribution of 1% (1 magnitude lower) was used, which led to a range of 0.00015 (0.015%) to 0.0015 (0.15%) for application of BS1. Because we used conservative values for the calculation, we allow a variant to reach an LBEN classification based on BS1 alone if there is no contradictory evidence supporting pathogenicity (as outlined in a recent SVI revision).29 For this work, the gnomAD population database was mostly used, although other databases with a minimum of 2000 alleles are also sufficient. However, we encourage the use of a large dataset such as gnomAD, ExAC, or ESP.
Because most RUNX1 variants are unique to probands or families,13 it was determined that the variant must be completely absent from all population databases to apply PM2. The MM-VCEP tested pilot PATH/LPATH variants with this rule and validated this determination. The MM-VCEP further recommends that the mean coverage of exome and genome sequencing data for RUNX1 in the population databases used should be at least 20×.
Criterion PS4 is based on the significantly higher prevalence of a variant in case cohorts vs control cohorts, which is considered strong evidence for pathogenicity. Ideally, published case-control studies are used as evidence. Given the rarity of FPD/AML, an existing case-control study for RUNX1 variants could not be identified. The original ACMG/AMP guideline states that the odds ratio (OR), measuring an association between a genotype and phenotype, can be used for Mendelian diseases. Accordingly, in the absence of a published case-control study, the MM-VCEP created a “quasi-case-control study” with the estimated number of probands worldwide and the overall gnomAD population as the control cohort. To apply this code, the proband has to meet at least one of the RUNX1-phenotypic criteria (Table 2), and the variant has to be either absent from gnomAD or only present once. This code has a sliding weight scale to account for the number of unrelated probands who meet the RUNX1-phenotypic criteria. PS4 is applied with ≥4 probands (OR, 100.6), PS4_moderate with 2 to 3 probands (OR, 50.3-75.5), and PS4_supporting with 1 proband (OR, 25.1) (supplemental Table 1).
BP2, supporting evidence for a BEN code, can be applied in the context of autosomal dominant FPD/AML when the variant is found in trans with a known PATH variant. Because there is no evidence in the literature of probands with a homozygous PATH RUNX1 variant, and lack of Runx1 is embryonically lethal in mice, the MM-VCEP recommends that BP2 also be applied when a variant is found in a confirmed homozygous state in population databases or internal laboratories.43,44
Segregation data (PP1_strong, PP1_moderate, PP1, and BS4)
Segregation with disease (PP1) is used as evidence for pathogenicity, and with increasing number of meioses, a stronger level of evidence can be applied. The MM-VCEP adopted the approach taken by various ClinGen expert panels,45,,-48 and supported by the SVI and others,49 that additional meioses support higher levels of evidence. Thus, based on a calculated logarithm of the odds score thresholds of 0.9, 1.5, and 2.1, respectively, 3 or 4 meioses fulfill criteria for PP1, 5 or 6 meioses for PP1_moderate, and ≥7 meioses for PP1_strong. Of note, only individuals well documented as having an RUNX1 phenotype (Table 2) and a positive genotype or obligate carriers are included when counting segregations. The phenotype of those individuals should be well described. We waived the ACMG/AMP recommendation for demonstrating cosegregation in >1 family, given that many RUNX1 variants are unique to a single family13 and have not been reported in other unrelated families, which would severely affect the utility of segregation data. We acknowledge that by waiving this recommendation, there is a possibility of the identified variant being in a linkage disequilibrium with a truly causative variant.
Lack of segregation in affected family members (BS4) can be used as a BEN criterion when an RUNX1 variant is present and nonsegregation with disease occurred in at least 2 or more informative meioses. BS4 should only be applied for genotype-positive, phenotype-negative family members, and there must be confidence that the family members do not meet any of our RUNX1-phenotypic criteria, taking into account ages of individuals.
De novo occurrence (PS2_moderate, PS2_supporting, PM6, and PM6_supporting)
De novo RUNX1 variants are rare but have been reported in the literature.16,-18 The 2 de novo criteria are applied when both maternity and paternity are confirmed (PS2) or assumed (PM6) and the variant has been assessed as de novo in a patient with the disease and no family history. The following specifications were added by our MM-VCEP: (1) no family history is defined by the absence of the FPD/AML-specific phenotype in first- and/or second-degree relatives; and (2) the proband must exhibit at least 1 phenotypic FPD/AML criterion (Table 2). PS2/PM6 were further specified by using the SVI recommendation of a point-based scoring system to determine the level of strength. The FPD/AML phenotype is not highly specific, and there is substantial genetic heterogeneity; the same phenotype can be caused by other underlying germline conditions such as PATH variants in ANKRD2650 or ETV6.51 We thus concluded that due to the lack of a highly specific phenotype and the presence of genetic heterogeneity, the maximum allowable value is 1 point contributing to the overall score. Due to this restriction, these 2 criteria do not have a strong or very strong level of evidence. PS2_moderate is reached with a score of 1 point (two or more proven de novo occurrences), and PS2_supporting is used when reaching a score of 0.5 point (one proven de novo occurrence). Likewise, PM6_moderate is met when 4 assumed de novo occurrences are present (score of 1), and PM6_supporting is applicable with 2 to 3 assumed de novo cases (score of 0.5). Combining these 2 criteria (eg, in the case of the same variant having both confirmed and assumed de novo evidence) is possible with the recognition that the maximum allowable value is still 1 point, which effectively leads to the application of 1 moderate or 2 supporting rules (supplemental Table 2).
Computational and predictive data (PVS1, PVS1_strong, PVS1_moderate, PS1, PS1_moderate, PM1, PM1_supporting, PM4, PM4_supporting, PM5_strong, PM5, PM5_supporting, PP3, BP4, and BP7)
RUNX1 germline variants have been well described as being dominant-negative, loss-of-function, or hypermorphic.13,31,52,53 Three major isoforms (A, B, and C) are expressed by the use of 2 promoters and alternative splicing (Figure 1B). Expression of the short human RUNX1A isoform has been shown to favor expansion of the hematopoietic stem cell pool, whereas expression of the full-length RUNX1B and RUNX1C isoforms, which only differ by 33 AAs at the N terminus of isoform C (exons 2-3 in NM_001754.4), function to promote hematopoietic differentiation.54,,,,,,-61 The differential function and expression of these isoforms in hematopoietic tissue are not fully understood. The MM-VCEP recommends using RUNX1 isoform C as the default transcript (NM_001754.4) because this is the isoform used for annotation by most clinical laboratories. The MM-VCEP decision tree for SNVs/indels (Figure 2; supplemental Table 3) and CNVs (supplemental Figure 1) refined the PVS1 criterion across all loss-of-function variant types previously reported for RUNX1 (nonsense, frameshift, canonical splice site variants, and single- or multi-exon deletions) by using the SVI recommendations27 and gene-specific adjustments. We recommend downgrading the strength level from very strong to strong for C-terminal variants that are not predicted to undergo nonsense-mediated decay but affect the transactivation domain, inhibitory domain, and/or the VWRPY motif (Figure 1).62,63 Nonsense-mediated decay is not predicted if the premature termination codon occurs in the 3′-most exon or within the 3′-most 50 nucleotides of the penultimate exon.64,65 Deletions of exon 2-3, presumably only affecting RUNX1 isoform C, have been reported in 4 families (L.A.G., A.L.B., P.B., and D.P., written communication, 1 July 2019),66 displaying a typical FPD/AML phenotype and segregation with disease. Although the functional effects of the exon 2-3 deletions on isoform C and potential effects on isoforms A and B require further investigation, we recommend applying PVS1_moderate according to the PVS1 CNV decision tree. The ClinGen CNV interpretation working group is currently developing a systematic framework for the clinical interpretation of CNVs, which will benefit the future curation of RUNX1 CNVs.
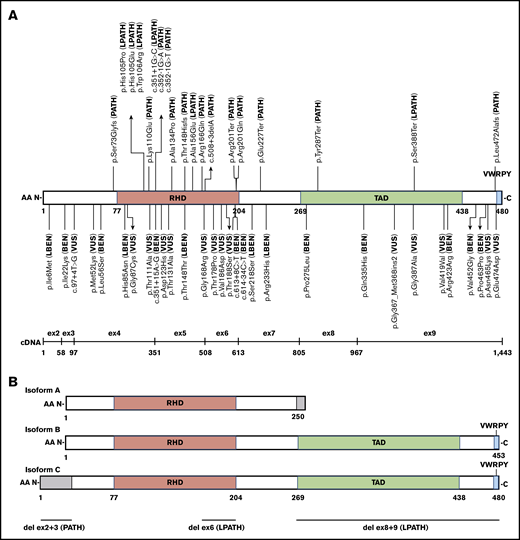
Schematic of RUNX1 exonic distribution, protein isoforms, and functional domain structure with all 52 pilot variants and their final MM-VCEP classification. (A) Isoform C with RHD, transactivation domain (TAD), and the VWRPY motif and location of all 49 single-nucleotide pilot variants with their final MM-VCEP classification. PATH and LPATH variants are shown at the top, and VUS, LBEN, and BEN variants are shown at the bottom. The exonic distribution of isoform C is displayed below. (B) Schematic of RUNX1 isoforms A, B, and C and their functional domains. Regions in gray are unique to 1 isoform. The 3 pilot CNVs are shown at the bottom, with the deletion of exons 2 and 3 exclusively affecting the N-terminal 33 AA of isoform C.
Schematic of RUNX1 exonic distribution, protein isoforms, and functional domain structure with all 52 pilot variants and their final MM-VCEP classification. (A) Isoform C with RHD, transactivation domain (TAD), and the VWRPY motif and location of all 49 single-nucleotide pilot variants with their final MM-VCEP classification. PATH and LPATH variants are shown at the top, and VUS, LBEN, and BEN variants are shown at the bottom. The exonic distribution of isoform C is displayed below. (B) Schematic of RUNX1 isoforms A, B, and C and their functional domains. Regions in gray are unique to 1 isoform. The 3 pilot CNVs are shown at the bottom, with the deletion of exons 2 and 3 exclusively affecting the N-terminal 33 AA of isoform C.
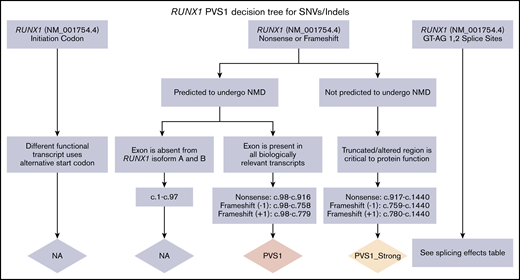
PVS1 decision tree for SNVs/indels. Application of different levels of strength for PVS1 depending on the prediction of nonsense-mediated decay (NMD), the location within a known critical protein domain, and the expression of alternative isoforms. The splicing effects table is given in supplemental Data.
PVS1 decision tree for SNVs/indels. Application of different levels of strength for PVS1 depending on the prediction of nonsense-mediated decay (NMD), the location within a known critical protein domain, and the expression of alternative isoforms. The splicing effects table is given in supplemental Data.
A variant affecting the same AA residue as a previously established PATH variant can either lead to the same AA change (PS1) or a different AA change (PM5). The MM-VCEP added the following recommendations for both rules: RNA data, or agreement in splicing predictors showing no splicing effects, which was defined as SSF and MES predicting either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10% and no putative cryptic splice sites are created. In addition, the previously established variant must be asserted PATH/LPATH based on MM-VCEP rules for RUNX1 before this rule can be applied. A strength modification was established for PS1 (same AA as previously established PATH variant) and PS1_moderate (same AA as previously established LPATH variant). Likewise, PM5_strong is applied when 2 or more different PATH missense changes have been detected previously at the same AA residue, PM5 is used when a different PATH missense change has been seen previously at the same residue, and PM5_supporting is used when one missense change at the same residue has previously been determined to be LPATH.
For in silico evaluation of missense variants, the MM-VCEP recommends using REVEL, a meta-predictor that combines 13 individual tools with high sensitivity and specificity and has recently shown the highest performance compared with any individual tool or other ensemble methods.67,68 For splicing predictions, we recommend using the SSF and MES, both of which have been shown to predict splicing effects with high accuracy.69,-71 PP3, defined as multiple lines of computational evidence supporting a deleterious effect, can be applied for missense variants with a REVEL score >0.75. It can also be applied for missense or synonymous variants if the variant alters the last 3 bases of an exon preceding a splice donor site or the first 3 bases of an exon following a splice acceptor site,69 and if the predicted decrease in the score of the canonical splice site (measured by both MES and SSF) is at least 75% regardless of the predicted creation/presence of a putative cryptic splice site. PP3 should be applied for intronic variants (in introns 4-8) located in reference to exons at positions +3 to +5 for splice donor sites or −3 to −5 for splice acceptor sites69,72 for which the predicted decrease in the score of the canonical splice site is at least 75% (measured by both MES and SSF) regardless of the predicted creation/presence of a putative cryptic splice site. PP3 cannot be used for canonical splice site variants.
The BEN criterion BP4 should be applied for missense variants if all of the following criteria apply: the variant’s REVEL score is <0.15, SSF and MES predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10%, and no putative cryptic splice sites are created. BP4 should also be applied for synonymous, intronic, and noncoding variants for which SSF and MES predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10%, and no putative cryptic splice sites are created.
The original PM1 code can be applied for variants affecting mutational hotspots and/or functional domains without BEN variation. The RHD, spanning from AA 77-204, has been established as a highly conserved DNA-binding domain without any BEN variation in ClinVar. Thirteen somatic and/or germline mutational hotspots within the RHD have been identified: R107, K110, A134, R162, R166, S167, R169, G170, K194, T196, D198, R210, and R204.12,20,73,74 The MM-VCEP recommends using PM1 for variants affecting these 13 AA residues. For variants in other parts of the RHD for which germline variants have been previously reported (AA 105-204), a reduced-strength level (PM1_supporting) is recommended. For other residues within the RHD (AA 77-104), no germline RUNX1 PATH variants have been reported to date. In the future, the AA range under PM1_supporting may be expanded to other parts of the protein if more evidence emerges. Analogous to PM1, PM4 (protein length changes due to in-frame deletions/insertions in a nonrepeat region or stop-loss variants) is applied to in-frame deletions/insertions affecting the same 13 AA residues (as listed earlier) and, likewise, PM4_supporting can be used for in-frame deletions/insertions affecting at least one of the other parts of the RHD in which germline variants have been previously reported (AA 105-204).
The MM-VCEP agreed to extend BP7 (synonymous variant with no splicing effect and position is not highly conserved) to apply to intronic/noncoding variants at or beyond positions +7/–21 for which SSF and MES predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10%, no putative cryptic splice sites are created, and the position is not conserved (eg, PhyloP score <0.175 ) or the variant is the reference nucleotide in 1 primate and/or 3 mammal species.76
Functional data (PS3, PS3_moderate, PS3_supporting, BS3, and BS3_supporting)
The evolutionarily conserved 128 AA RHD, present in most of the RUNX1 isoforms (Figure 1B), is involved in DNA binding and heterodimerization with core binding factor (CBF) β. Heterodimerization of RUNX1 with CBFβ promotes DNA binding by stabilizing the interaction of the complex with the DNA. RUNX1 regulates the activity of several important hematopoietic genes, such as the granulocyte-macrophage colony-stimulating factor,77,78 T-cell receptor,79,80 myeloperoxidase,81,82 and neutrophil elastase,82 by binding to a core sequence (TGTGGT) found in their promoters or enhancers.
Transactivation assays showing altered transactivation compared with wild type are often performed as functional studies to evaluate the pathogenicity of a RUNX1 variant. Promoter sequences of M-CSFR, PF4, C-FMS, and GZMB, containing consensus RUNX1 binding sites TGTGGT have been used for this purpose.31,83,,,-87 Data from secondary assays are frequently used to evaluate an altered function of mutant RUNX1. Electrophoretic mobility shift assays31,87,,-90 and yeast hybrid assays88,89 are performed to show decreased DNA-binding affinity, and coimmunoprecipitation assays,85,87,90 fluorescence resonance energy transfer assays,88 and affinity assays31 can illustrate the diminished heterodimerization ability of mutant RUNX1 with CBFβ. Abnormal cellular localization of mutant RUNX1 can be shown by immunofluorescence31,53,83 and cell fractionation with western blot.85,90 Sorted primary hematopoietic stem and progenitor cells can be used to show reduced colony-forming potential,53,91 and xenotransplantation experiments may reveal abnormal function of mutant RUNX1 in vivo.53
The MM-VCEP defined the strong PATH code PS3 as the combination of reduced transactivation (<20% of wild type and/or reduced to levels similar to well-established PATH variants such as R201Q or R166Q) and data from a secondary assay that show altered function of mutant RUNX1. The transactivation assay should include wild-type and known PATH controls as well as coexpression with CBFβ. PS3 can also be applied for evidence of very low or abnormal messenger RNA (mRNA)/protein expression of the variant allele as a functional consequence of a null variant or incorrect mRNA/protein products. The MM-VCEP further stipulates that PS3 cannot be applied if the variant meets PVS1. If the variant meets PVS1_strong and PS3, we recommend applying either PVS1_strong and PS3_moderate or upgrading PVS1_strong to PVS1 without applying PS3. PS3_moderate is applied when data from transactivation assays exhibit reduced transactivation (<20% of wild type and/or reduced to levels similar to well-established PATH variants such as R201Q or R166Q) or 2 or more secondary assays show altered function. PS3_supporting can be applied for transactivation assays exhibiting enhanced transactivation (>115% of wild type), as has been reported previously for the hypermorphic RUNX1 mutant, S388X.52
Likewise, the BS3 requirements (functional studies show no damaging effect on protein function) are a normal transactivation (80%-115% of wild type) and data from a secondary assay that exhibit normal function. BS3_supporting can be applied when there is evidence of normal transactivation (80%-115% of wild type); data from secondary assays are not required.
Rules deemed not applicable
Four rules of the PATH framework (PM3, PP2, PP4, and PP5) and 5 rules of the BEN framework (BS2, BP1, BP3, BP5, and BP6) were deemed not applicable. The reasoning behind the decision for each code is briefly explained here.
Because the FPD/AML phenotype is associated with autosomal dominant transmission, PM3 (detected in trans with a PATH variant in a recessive gene) cannot be applied for FPD/AML.
The recommended cutoff for PP2 (missense variant in a gene with low rate of missense variants) is a constraint z score ≥3.09,28 which was not met by RUNX1.
The phenotype observed in FPD/AML is rather nonspecific and can be caused by a number of other inherited predisposition syndromes, somatic variants, or environmental factors; this scenario makes the original ACMG/AMP rule PP4 for a highly specific phenotype not applicable to RUNX1.
Incomplete penetrance, an average age of onset of 33 years for hematologic malignancies,19,-21 and the lack of sufficient clinical data to exclude an RUNX1-related phenotype render BS2 (observed in a healthy individual with full penetrance at an early age) not applicable.
Both missense and truncating variants have been described as causative in FPD/AML, making BP1 (missense variant in a gene with primarily truncating variants) not applicable. Similarly, BP3 is not applicable, as RUNX1 lacks repetitive regions of unknown function.
BP5 can be applied when the variant is found in a case with an alternate molecular basis for disease. The MM-VCEP concluded that this rule is not applicable because in rare circumstances, a patient can carry variants in 2 genes predisposing to hematologic malignancies, as has been described in case reports. In addition, variants in other genes presenting as low-penetrance risk factors, modifier genes, and/or somatic mutations in hematopoietic stem and progenitor cells may contribute to the clinical presentation and complicate the search for the causative variant.92,93
Following recommendations from the SVI, the MM-VCEP agreed not to use the 2 variant classifications from reputable source evidence codes (PP5 and BP6) based on the published rationale.28
Performance of the MM-VCEP specifications in pilot variant classification
For pilot testing, 52 variants with a broad spectrum of ClinVar assertions (12 BEN/LBEN, 14 VUS, 20 PATH/LPATH, 4 CONF, and 2 variants with no ClinVar assertions) were selected. The MM-VCEP applied the RUNX1-modified ACMG/AMP criteria to all pilot variants. During testing, experts were able to provide feedback on the usability of the evidence codes, comment on the weight of certain lines of evidence, and suggest further modifications of the rule. A list of all pilot variants, the variant classification of the ClinVar submitters, and the classifications made by our MM-VCEP are presented in supplemental Table 4. Figure 3 compares the original ClinVar classifications vs our MM-VCEP classifications grouped according to PATH/LPATH, BEN/LBEN, VUS, and CONF variants. Of the 14 VUS, 2 were upgraded into the LPATH category. Of the 4 CONF variants, 1 was upgraded to PATH, and 3 were downgraded to LBEN. Two of 18 variants previously listed as PATH/LPATH in ClinVar were downgraded to VUS after applying the RUNX1-specific codes. MM-VCEP members with knowledge of the criteria applied by the ClinVar submitters were able to corroborate the VUS classifications. A detailed schematic of the RUNX1 gene and the newly classified pilot variants is shown in Figure 1A. Overall, applying the RUNX1 specifications to the VUS/CONF variants resulted in a reduction in VUS/CONF classifications of 33%. All of the 12 variants that were submitted in ClinVar as BEN/LBEN remained in this category, with most LBEN variants being downgraded to BEN and only 2 remaining LBEN. An overview of the frequency of PATH and BEN evidence codes applied is given in supplemental Figure 2. The test set received a final concordance of 92% with consensus ClinVar classifications (90% for the PATH/LPATH test set, 86% with the VUS test set, and 100% for the BEN/LBEN test set).
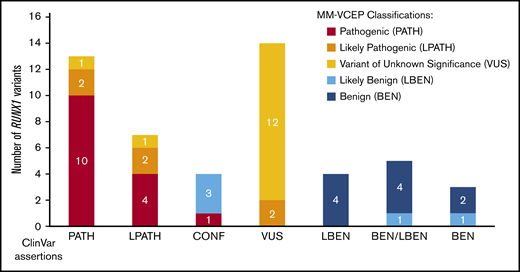
Comparison of ClinVar and MM-VCEP classifications. Fifty previously asserted and ClinVar-deposited RUNX1 variants are shown on the x-axis. Final MM-VCEP classifications are color-coded (see legend on the right). ClinVar variants with previous LPATH, CONF, and VUS assertions were most often reclassified by using MM-VCEP–specified rules for RUNX1.
Comparison of ClinVar and MM-VCEP classifications. Fifty previously asserted and ClinVar-deposited RUNX1 variants are shown on the x-axis. Final MM-VCEP classifications are color-coded (see legend on the right). ClinVar variants with previous LPATH, CONF, and VUS assertions were most often reclassified by using MM-VCEP–specified rules for RUNX1.
Discussion
RUNX1 is commonly mutated in hematologic malignancies with high rates of somatic variants in MDS/AML.73,94 Tumor-based next-generation sequencing panels covering RUNX1 among other genes are implicated in the molecular diagnostic process of MDS/AML in most treatment centers. Some of these somatic RUNX1 variants are subsequently determined to be germline.95 In addition, recent achievements such as the inclusion of inherited hematologic malignancies into the revised World Health Organization classification of myeloid neoplasms and acute leukemia15 and a more standardized evaluation of family history have raised awareness of these syndromes among physicians. This awareness will increase the identification of patients with FPD/AML. Accurate RUNX1 variant curation is fundamental for the appropriate clinical care of these patients, especially when considering a related donor for hematopoietic stem cell transplantation. In addition, FPD/AML with thrombocytopenia may be misdiagnosed as immune thrombocytopenic purpura, and the correlating dysmegakaryopoiesis in the bone marrow can be mistaken as an early-stage MDS, underscoring the importance of adequate RUNX1 variant curation.96,97
Our curation of pilot variants showed the impact of our proposed rules on improving variant classification, resulting in a reduction of VUS/CONF variants by 33%. Further use of these rules should continue to reduce the number of VUS and lead to fewer number of variants with VUS/CONF assertion within ClinVar. Being able to reclassify a variant from VUS/CONF assertions has a significant impact on patient care as it provides patients and physicians with the definitive data to guide treatment decisions, including donor selection among matched relatives. As we implement these RUNX1-specific rules, the variant annotation in ClinVar will contain a link to the specific version of the MM-VCEP RUNX1 evidence rules, a summary of the specific evidence codes used for that variant, and a link to the ClinGen evidence repository where all the evidence evaluated for that variant is found. Given these detailed expert-reviewed curations, MM-VCEP–curated variants will be submitted under a “3-star expert panel reviewed” FDA-recognized designation.
We expect that our RUNX1-specific rules will require further updating as additional data become available, or at a minimum every 2 years, and will address improved computational modeling, functional assays, and larger and more ethnically diverse population databases. Per ClinGen policy, RUNX1 VUS and LPATH variants will be reassessed by the expert panel every 2 years, and other variants may be re-curated if discrepancies in the variant classification or new evidence emerge over time. At any time, a link to the most up-to-date recommendations of RUNX1 evidence codes can be found on the MM-VCEP home page (https://www.clinicalgenome.org/affiliation/50034). Furthermore, ongoing general refinements to the ACMG/AMP guidelines made by the ClinGen SVI will need to be addressed, particularly for the curation of intragenic RUNX1 deletions and consensus rules for evaluation of splicing predictions. The next step of the MM-VCEP will be the curation of all current ClinVar-deposited RUNX1 variants. Further work will extend this study to other genes causing inherited hematologic malignancies.
Acknowledgments
The VCEP thanks the ClinGen SVI Working Group as well as the Executive Committee of the Hereditary Cancer Clinical Domain Working Group.
Results provided in this publication were generated by the American Society of Hematology in collaboration with Baylor College of Medicine and the University of North Carolina, NIH-funded Clinical Genome Resource grant award recipients. The NIH, National Human Genome Research Institute supported this work through U41HG009649 (X.L. and S.E.P.) and U41HG009650 (S.M. and J.E.R.); and the 2018 NIH/National Cancer Institute Leukemia SPORE DRP award (P50CA100632-16, project 00007529) (C.D.D).
Authorship
Contribution: All of the authors participated in the construction and pilot testing of the RUNX1 curation rules and edited the manuscript; and X.L., S.F., S.M., D.W., and L.A.G. participated in the majority of the manuscript writing.
Conflict-of-interest disclosure: S.E.P. is a member of the scientific advisory panel of Baylor Genetics Laboratories. L.A.G. is a member of the scientific advisory board for Invitae, Inc., and receives royalties from UpToDate, Inc. L.Z. received honoraria from Future Technology Research, LLC, Roche Diagnostics Asia Pacific, BGI, and Illumina. A family member of L.Z. has a leadership position and ownership interest in the Shanghai Genome Center. The remaining authors declare no competing financial interests.
Correspondence: David Wu, University of Washington, 825 Eastlake Ave E, G7800, Seattle, WA 98109; e-mail: dwu2@uw.edu; and Lucy A. Godley, The University of Chicago, 5841 S Maryland Ave, MC 2115, Chicago, IL 60637; e-mail: lgodley@medicine.bsd.uchicago.edu.
The full-text version of this article contains a data supplement.

