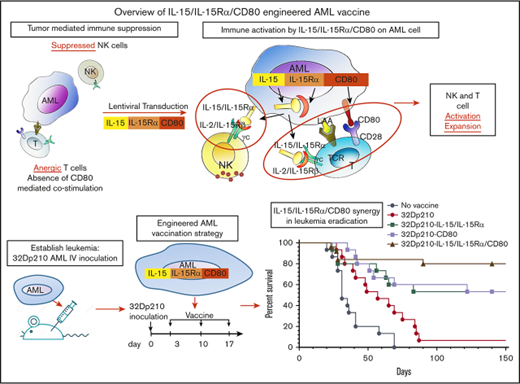
Key Points
Murine AML vaccines expressing CD80 and IL-15/IL-15Rα stimulate effective cytolytic activity despite leukemia-induced immune inhibition.
CD80 and IL-15/IL-15Rα coexpression on AML vaccine leads to superior survival and eradication of minimal residual disease in leukemic mice.

Abstract
Engineered autologous acute myeloid leukemia (AML) cells present multiple leukemia-associated and patient-specific antigens and as such hold promise as immunotherapeutic vaccines. However, prior vaccines have not reliably induced effective antileukemic immunity, in part because AML blasts have immune inhibitory effects and lack expression of the critical costimulatory molecule CD80. To enhance induction of leukemia-specific cytolytic activity, 32Dp210 murine AML cells were engineered to express either CD80 alone, or the immunostimulatory cytokine interleukin-15 (IL-15) with its receptor α (IL-15Rα), or heterodimeric IL-15/IL-15Rα together with CD80 and tested as irradiated cell vaccines. IL-15 is a γc-chain cytokine, with unique properties suited to stimulating antitumor immunity, including stimulation of both natural killer and CD8+ memory T cells. Coexpression of IL-15 and IL-15Rα markedly increases IL-15 stability and secretion. Non-tumor-bearing mice vaccinated with irradiated 32Dp210-IL-15/IL-15Rα/CD80 and challenged with 32Dp210 leukemia had greater survival than did mice treated with 32Dp210-CD80 or 32Dp210-IL-15/IL-15Rα vaccines, whereas no unvaccinated mice inoculated with leukemia survived. In mice with established leukemia, treatment with 32Dp210-IL-15/IL-15Rα/CD80 vaccination stimulated unprecedented antileukemic immunity enabling 80% survival, an effect that was abrogated by anti-CD8 antibody-mediated depletion in vivo. Because, clinically, AML vaccines are administered as postremission therapy, we established a novel model in which mice with high leukemic burdens were treated with cytotoxic therapy to induce remission (<5% marrow blasts). Postremission vaccination with 32Dp210-IL-15/IL-15Rα/CD80 achieved 50% overall survival in these mice, whereas all unvaccinated mice achieving remission subsequently relapsed. These studies demonstrate that combined expression of IL-15/IL-15Rα and CD80 by syngeneic AML vaccines stimulates effective and long-lasting antileukemic immunity.
Introduction
Older individuals with acute myelogenous leukemia (AML) have poor outcomes because of more frequent high-risk features and comorbidities.1 The improved survival achieved with allogeneic hematopoietic stem cell transplantation because of curative graft-versus-leukemia responses conferred by donor T cells provides evidence for the potential efficacy of immunotherapies (reviewed by Dombret and Gardin1 ). Because many older individuals are ineligible for transplants, there is an unmet need for novel therapeutic approaches.
Although immunotherapies for AML have been explored, to date none have reliably reduced relapse rates.2-5 In this context, autologous cell vaccines may have advantages for stimulating antileukemic immunity because responses are directed to multiple leukemia-associated antigens, some of which are patient specific. Immune responses generated against an autologous AML vaccine would obviate the problem of lack of a priori knowledge of the dominant antigens present in each patient’s leukemia. Previous trials with autologous cell vaccines have induced antileukemic immunity, but responses were variable.6 This is in part because AML blasts are ineffective in T-cell stimulation, because of their downregulation or absent expression of specific costimulators such as CD80,7,8 and because of their immune evasive effects including upregulation of checkpoint molecules and stimulation of inhibitory immune effectors (reviewed by Teague and Kline9 ). In older patients, immunotherapeutic efficacy may be further limited by a decline in T-cell responsiveness.10-13
Because engineering patient AML cells to express the missing costimulatory protein CD80 has shown promise,7,8,14,15 we engineered AML vaccines to express a novel combination of CD80 and the heterodimeric complex interleukin-15 (IL-15) and IL-15 receptor α (IL-15Rα) to improve the induction of antileukemic cytolytic responses. The IL-15/IL-15Rα heterodimer is the naturally occurring form of the cytokine and is a member of the γc cytokine family that engages a heterodimeric receptor comprising the IL-2Rβ/IL-15Rβ subunit (shared with the IL-2 receptor) and γc.16 Efficient IL-15 production requires coexpression of IL-15 and IL-15Rα in the same cell,17-22 which substantially increases IL-15 half-life and activity through the IL-2/IL-15R γc receptors.23-25
IL-15 has major advantages as an immune-stimulatory cytokine because in contrast to the effects of IL-2, previously used in immunotherapy, IL-15 reverses CD8+ T-cell unresponsiveness to tumor-associated antigens, renders T effector cells resistant to suppressive regulatory T cells (Treg’s), and participates in antiapoptotic signaling to effector T cells (reviewed by Waldmann 23 ). IL-15 also stimulates more effective induction of antigen-specific cytotoxic lymphocytes and more durable immunity through actions on memory T cells, and it has important roles in natural killer (NK) and NK T-cell activation, proliferation, and survival.23 Although systemic IL-15 administration has less toxicity than does high-dose IL-2 infusion, it does cause neutropenia, fever, and other side effects.26 Thus, local expression of IL-15 by genetically modified AML vaccines has the potential advantages of IL-15 immune stimulation, with reduced risk of systemic toxicities. Finally, local expression of membrane-bound and secreted heterodimeric IL-15/IL-15Rα together with costimulation by CD80 may mimic the interactions of professional antigen-presenting cells with lymphocytes, required for triggering effective cell-mediated immune responses.
In these studies, the leukemia-specific cytolytic activity stimulated by coexpression of IL-15/IL-15Rα and CD80 in lentivirally transduced irradiated AML cell vaccines was evaluated in 32Dp210 myeloid leukemia-bearing mice.27-30 We also developed a novel model of postremission minimal residual disease (MRD) in leukemic mice that in part recapitulates the clinical setting in which AML cell vaccines would be administered. Our studies show that in mice with prior high leukemic burdens, postremission therapy with 32Dp210-IL-15/IL-15Rα/CD80 vaccines can produce long-term survival and eradication of leukemia.
Methods
Cell lines
32Dp210 leukemia cells transformed by the p210 bcr abl transcript31 were provided by R. Arlinghaus (University of Texas MD Anderson Cancer Center). For longitudinal in vivo bioluminescence studies, 32Dp210 leukemia cells were transfected with a luciferase vector (Cat.VSL-0074; Capital Bioscience, Gaithersburg, MD). Other 32Dp210 cells were transduced with enhanced green fluorescent protein for flow cytometric analysis, or with the herpes simplex thymidine kinase (HSV-TK) suicide gene, to confer sensitivity to ganciclovir (GCV) and tumor-specific lethality after drug administration. For each experiment, 32Dp210 aliquots were thawed; expanded in RPMI, 10% fetal bovine serum (Gibco, Life Technology), and 1% glutamine; and then washed in phosphate-buffered saline and injected IV via lateral tail vein. 32Dp210-Luc+, 32Dp210-GFP+, and 32Dp210-Luc-HSV-TK+ cells all showed comparable growth in vivo.
Flow cytometric analyses
Antibodies to H-2DK (15-5-5), H-2Kk (36-7-5), H-2Kb (AF6-88.5), CD3 (2C11), CD4 (RM4-5), CD8 (53-6.7), CD25 (PC61), CD335 (29A-1.4), CD11b (M1/70), Ly-6G and Ly-6C (Gr-1), CD80 (16-10A1), IL-15 (16-7154-85), IL-15Rα (DNT15Ra), CD279 (J43), CD44 (IM7), CD62L (MEL-14), FoxP3 (FM23), interferon γ (IFN-γ; 14-4-4s) and Fc Block (2.4G2), PD-L1 (BV711 [Rat anti-mouse CD274, clone MIH5], matched BV711-conjugated isotype control [mAb; rat IgG 2a]), and PD-1 (BV421 [Hamster anti-mouse CD279, clone J43]) were purchased from BD Biosciences (San Jose, CA). Cultured and tissue-derived cells were suspended in phosphate-buffered saline, labeled with Fc block (BD Biosciences), and stained with the indicated antibodies. Intracellular staining for FoxP3 and IFN-γ was performed using Cytofix/Cytoperm-Fixation/Permeabilization Kit (Becton Dickinson), and data were analyzed using FlowJo software (Treestar, Ashland, OR).
Lentivirus construction and transduction
Lentiviral vectors were constructed by homologous recombination in the pMLV-mIL-2-Furin-CD80 backbone, containing the murine leukemia virus promoter, murine IL-2, and murine CD80.32 The murine IL-2-Furin-CD80 cassette in this vector was replaced either with mouse CD80 alone or with codon-optimized mouse IL-15 with the leader sequence from granulocyte-macrophage colony-stimulating factor to increase expression, and a RNA/codon-optimized, membrane associated, complete IL-15Rα.17,33 The IL-15 cassette was linked by a P2A sequence, to generate a self-cleaving peptide34 producing mouse IL-15-P2A-IL-15Rα. A tri-cistronic lentiviral vector was constructed by linking the murine IL-15-P2A-IL-15Rα to murine CD80 with an F2A sequence (coding for a second self-cleaving peptide)35 3′ to the IL-15/IL-15Rα cassette generating IL-15-P2A-IL-15Rα-F2A-CD80 (supplemental Figure 1). Lentivirus production and transduction was performed as described.36
Generation of 32Dp210-derived vaccines
32Dp210 cells were transduced with 8 µg/mL polybrene in 6-well plates. To achieve comparable levels of expression of mouse IL-15, IL-15Rα, and CD80 in 32Dp210 vaccines, transduced cells were first purified based on high-level and comparable CD80 expression. Then, purified 32Dp210-IL-15/IL-15Rα+ and 32Dp210-IL-15/IL-15Rα/CD80 cells with comparable expression of IL-15Rα were isolated and expanded.
Mice
Female C3H mice (8-10 weeks; Charles River, San Diego, CA) were housed under pathogen-free conditions at University of California, San Francisco. Experiments were conducted with supervision of University of California, San Francisco Institutional Animal Care and Use Committee according to approved protocol (AN108913-02A).
Vaccine protocols
Lentivirally transduced or untransduced 32Dp210-luc parental cells were irradiated with 40 Gy prior to subcutaneous administration. At lower doses of irradiation, occasional viable tumors were observed. Tumor progression and responses to vaccinations were analyzed in a Xenogen in vivo imaging system (IVIS), after intraperitoneal injection with 150 mg/kg luciferin and anesthesia with 3% isofluorane. Images were analyzed using Living Image software (Xenogen, Alameda, CA).
MRD model
Ten days after IV injection of 1 × 105 32Dp210-luc, all mice had detectable leukemia by IVIS and began GCV treatment (50 mg/kg/day × 14 days) (APP Pharmaceuticals LLC, Schaumburg, IL). To confirm that GCV had no effects on peripheral blood counts, counts were assessed after administration of 100 mg/kg per day × 14 days (supplemental Table 1).
In vivo cell depletion studies
In vivo cell depletion by intraperitoneal injection of antibodies was carried out on day 0 after inoculation of 1 × 104 32Dp210 cells. Antibodies (100 µL diluted rabbit anti-mouse anti-asialo-GM1, or 500 µg rat-anti-mouse CD4, or 500 µg rat anti-mouse CD8 [BioXCell, West Lebanon, NH]), produced >90% depletion of target cells 24 hours after injection, verified by staining for CD3, CD4, CD8, and NKp46 in blood and spleen.
Enzyme-linked immunospot (ELISpot) assays of IFN-γ production
The 96-well plates (MAIP S4510; Millipore) were coated with anti-IFN-γ antibody, washed, and blocked per manufacturers’ instructions (Mabtech). Splenocytes (3 × 105) from vaccinated mice were plated in duplicate with equal numbers of irradiated (100 Gy) 32Dp210 cells and incubated at 37°C and 5% CO2 for 20 hours. Plates were developed by incubation with biotinylated anti-IFN-γ antibody (Mabtech), and activity was detected using a colorimetric AP kit (Bio-Rad, CA) and counted (ELISpot reader, Cellular Technology Ltd.).
T-cell proliferation assay
Splenocytes from naïve mice were labeled with CellTrace Violet or carboxyfluorescein diacetate succinimidyl ester (CFSE); 2 × 106 cells were cocultured with an equal number of irradiated (100 Gy) 32Dp210-Luc, 32Dp210-Luc–IL-15/IL-15Rα, 32Dp210-Luc-CD80, or 32Dp210-Luc–IL-15/IL-15Rα/CD80 cells for 12 days. Splenocytes were then stained with anti-CD3, anti-CD4, and anti-CD8 antibodies, and proliferation was quantified by flow cytometric analyses.37
T-cell cytolytic assay
After 4 weekly vaccinations with 32Dp210, 32Dp210-IL-15/IL-15Rα, 32Dp210-CD80, or 32Dp210-IL-15/IL-15Rα/CD80, harvested splenocytes were restimulated in vitro with irradiated 32Dp210 parental cells for 5 days. These splenocytes were purified over Ficoll-Hypaque gradients and used as effectors. Target 32Dp210 cells were labeled with CellTrace 7-hydroxy-9H(1,3-dichloro-9,9-dimethylacridin-2-one) succinimidyl ester (DDAO-SE). Splenic effector cells were cocultured at the indicated ratios for 24 or 48 hours with (1 × 105) 32Dp210 target cells. In studies to assess whether responses to the human p210 breakpoint cluster region gene–Abelson murine leukemia viral oncogene homolog 1 (BCR-ABL) peptide were dominant, unmodified 32Dp210 cells, or BCR-ABL (GFKQSSKAL) peptide-loaded, or control (HYLSTQSALSK) peptide-loaded C3H splenocytes were used as targets (effector-to-target ratio = 10:1). The fractions of apoptotic 32Dp210 cells were assessed using the Active Caspase-3 apoptosis kit (BD Bioscience) and flow cytometry.
Intracellular IFN-γ assay
C3H mice were vaccinated weekly 4 times with 32Dp210-derived vaccines as described previously. Splenocytes (1 × 106) from immunized C3H mice were stimulated ex vivo for 20 hours with either (1) irradiated unmodified 32Dp210 (5 × 105), (2) PMA + ionomycin, or (3) C3H splenocytes from naïve mice loaded with 5 μg/mL BCR-ABL–derived peptide or with 5 μg/mL control peptide. Intracellular IFN-γ expression was quantified after staining and fluorescence-activated cell sorter analysis.
Measurement of IL-15
Secreted mouse IL-15 levels were measured by enzyme-linked immunosorbent assay (ELISA) using recombinant mouse IL-15 as a standard and polyclonal rabbit anti-mouse IL-15 antibody (H114; Santa Cruz Biotechnology Inc., Santa Cruz, CA).
Statistical analyses
Experiments were repeated at least 2 times unless indicated otherwise. Statistical analyses were performed using Prism 6 (GraphPad Software Inc., La Jolla, CA). Results are reported as the mean ± standard error of the mean (SEM) in independent experiments. The significance of differences was determined using Student paired t test.
Results
Murine 32Dp210 leukemia recapitulates features of clinical AML
Dose-finding studies with 32Dp210 leukemia demonstrated that the minimal cell dose that produced reliable leukemic engraftment, and adequate survival to allow induction of immunity (∼30-40 days38-40 ), was 104 cells administered IV (Figure 1A, upper panel). Because leukemia-associated stimulation of inhibitory immune responses poses challenges in the clinical setting,9 we assessed whether 32Dp210 leukemia has immunosuppressive effects comparable to those observed in patients. Fourteen days after leukemia inoculation, T cells increased the expression of the negative regulatory receptor PD-1, and 32Dp210 cells upregulated expression of the PD-1 ligand, PD-L1, on engrafted leukemia cells (Figure 1A lower panels). The numbers of CD3+CD4+ T cells, CD3+CD8+ T cells, and the frequency of CD3+CD8+CD44hi T cells were all decreased in leukemic mice, whereas the frequency of inhibitory CD4+FoxP3+ Treg’s and CD11b+Ly6G+ myeloid derived suppressor cells increased, demonstrating 32Dp210-mediated immunosuppression (supplemental Figures 2 and 3). 32Dp210 leukemia also reproduced early homing of blasts to the bone marrow compartment seen clinically (0.2% GFP+ cells at day 3) (Figure 1B).

The 32Dp210 murine leukemia model recapitulates features of clinical AML progression. (A) Progression of 32Dp210 leukemia. Upper panel: serial in vivo bioluminescence images of C3H mice on days 7, 14, 21, and 28 after IV inoculation of 1 × 104 32Dp210 murine leukemia cells on day 0. Lower panels: Left panel: level of PD-1 expression on T cells in naïve vs tumor-bearing mice detected by antibody staining and flow cytometric analysis (red bar, naïve non-leukemia-bearing mice; blue bar, leukemia-bearing mice). Middle panel: level of PD-1 expression on CD3+CD8+T cells in naïve vs leukemia-bearing mice (red bar, naïve mice; blue bar, tumor-bearing mice). Right panel: level of PD-L1 expression on 32Dp210-GFP+ cells in culture and after transplantation in vivo (red bar, 32Dp210 cells in ex vivo culture; blue bar, 32Dp210 cells harvested from leukemic mice after IV inoculation). Error bars are SEM. (B) 32Dp210-GFP+ leukemia cells selectively home to bone marrow compartment early after IV inoculation. The percentage of 32Dp210-GFP+ cells detected in peripheral blood, spleen, and bone marrow (depicted as bar graphs sequentially from left to right) was detected by antibody staining and flow cytometric analyses on days 1, 2, 3, 4, and 7 after IV injection of 1 × 104 32Dp210-GFP+ cells in C3H mice. The mean percentage of GFP+ cells in each tissue is indicated on the y-axis. Error bars are SEM.
The 32Dp210 murine leukemia model recapitulates features of clinical AML progression. (A) Progression of 32Dp210 leukemia. Upper panel: serial in vivo bioluminescence images of C3H mice on days 7, 14, 21, and 28 after IV inoculation of 1 × 104 32Dp210 murine leukemia cells on day 0. Lower panels: Left panel: level of PD-1 expression on T cells in naïve vs tumor-bearing mice detected by antibody staining and flow cytometric analysis (red bar, naïve non-leukemia-bearing mice; blue bar, leukemia-bearing mice). Middle panel: level of PD-1 expression on CD3+CD8+T cells in naïve vs leukemia-bearing mice (red bar, naïve mice; blue bar, tumor-bearing mice). Right panel: level of PD-L1 expression on 32Dp210-GFP+ cells in culture and after transplantation in vivo (red bar, 32Dp210 cells in ex vivo culture; blue bar, 32Dp210 cells harvested from leukemic mice after IV inoculation). Error bars are SEM. (B) 32Dp210-GFP+ leukemia cells selectively home to bone marrow compartment early after IV inoculation. The percentage of 32Dp210-GFP+ cells detected in peripheral blood, spleen, and bone marrow (depicted as bar graphs sequentially from left to right) was detected by antibody staining and flow cytometric analyses on days 1, 2, 3, 4, and 7 after IV injection of 1 × 104 32Dp210-GFP+ cells in C3H mice. The mean percentage of GFP+ cells in each tissue is indicated on the y-axis. Error bars are SEM.
Lentiviral transduction produces high-level IL-15, IL-15Rα, and CD80 expression
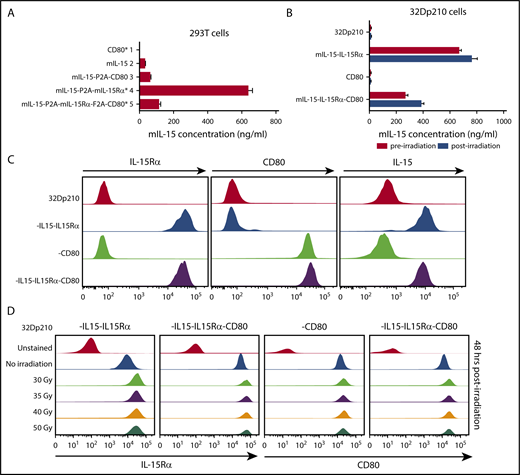
Because coexpression of IL-15 and IL-15Rα markedly increases the secretion, half-life, and bioactivity of IL-15, codon-optimized murine IL-15 and murine IL-15Rα constructs were both incorporated in vectors, with or without CD80 (supplemental Figure 1).17,33 As expected, IL-15 was barely detected in transduced 293T cell supernatants in the absence of IL-15Rα expression; however, coexpression of IL-15 and IL-15Rα resulted in high levels of IL-15 secretion (Figure 2A). Transduction with the IL-15/IL-15Rα/CD80 vector produced somewhat lower levels of IL-15 secretion both in 293T cells (Figure 2A) and in 32Dp210 cells, an effect of insert size (Figure 2B). Irradiation, required for vaccine preparation, did not adversely affect IL-15 secretion (Figure 2B) or cell surface expression of IL-15Rα, IL-15, or CD80 (Figure 2C-D).
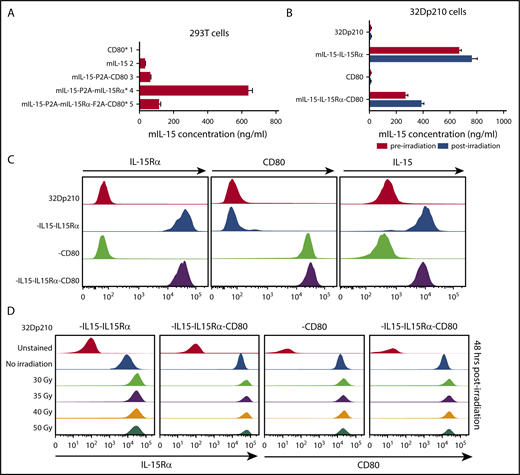
Appropriate cellular processing and expression of IL-15, IL-15Rα, and CD80 are observed after lentiviral transduction of 293T and 32Dp210 cell lines. (A) Levels of IL-15 secretion after lentiviral transduction of 293T cells. Murine IL-15 secretion was quantified by ELISAs of tissue culture supernatants 48 hours after transfection. On the left are designations of lentiviral vector constructs generated and used for transduction studies. Asterisks denote vectors that were used to generate the final 32Dp210 vaccines. Lanes 1 to 5 on the vertical axis depict IL-15 secretion detected as nanograms per milliliter as detected by ELISA for cells transduced with each of the designated constructs. Bars represent the mean cytokine concentration ± SEM. (B) Levels of IL-15 secretion after lentiviral transduction of 32Dp210 cells are comparable pre- and postirradiation. 32Dp210 cells transduced with lentiviral vectors containing mIL-15/IL-15Rα or CD80, or both IL-15/IL-15Rα and CD80, were expanded, and populations were selected that express similar levels of cell-surface IL-15Rα and/or CD80 by fluorescence-activated cell sorter. High-expressing populations were purified. Levels of IL-15 in culture supernatants (ng/mL) were measured before (red bars) and 48 hours after irradiation with 40 Gy (blue bars). 32Dp210, to the left of the uppermost 2 lanes, indicates IL-15 secretion in untransduced controls. As previously, designations to the left of the graph indicate the genes (IL-15, IL-15Rα, or CD80, or all 3 cassettes) contained in the lentiviral vector used to transduce the 32Dp210 cells analyzed. Levels of IL-15 detected in cell culture supernatants are indicated (ng/mL) on the horizontal axis. Bars represent the mean cytokine concentration ± SEM. (C) Transduced 32Dp210 cells exhibit high level cell-surface expression of IL-15Rα, CD80, and IL-15 after purification of transduced 32Dp210 populations. Lentivirally transduced, purified 32Dp210-derived cells were stained with anti-IL-15, anti-IL-15Rα, and anti-CD80 antibodies and subjected to flow cytometric analyses. 32Dp210 on the left indicates the untransduced parental cell line used as a control. The genes encoded by each lentiviral vector used to transduce the 32Dp210 cells are indicated to the left of the flow analysis, and the antibody used to stain cells is indicated above each flow plot. (D) Cell-surface expression of IL-15Rα and CD80 on 32Dp210 vaccines is stable 48 hours after irradiation with doses up to 50 Gy. Lentivirally transduced, purified 32Dp210 cells were stained with anti-IL-15Rα and anti-CD80 antibodies, and the effects of different doses of radiation on IL-15Rα and CD80 expression analyzed. The lentiviral vector used to transduce 32Dp210 cells is indicated above each plot, and the dose of irradiation indicated on the vertical axis. Antibodies used to analyze cell surface expression are indicated below the plots. Data showing cell surface IL-15Rα expression are shown in the left 2 graphs, and cell surface staining with anti-CD80 antibodies is presented in the right 2 graphs.
Appropriate cellular processing and expression of IL-15, IL-15Rα, and CD80 are observed after lentiviral transduction of 293T and 32Dp210 cell lines. (A) Levels of IL-15 secretion after lentiviral transduction of 293T cells. Murine IL-15 secretion was quantified by ELISAs of tissue culture supernatants 48 hours after transfection. On the left are designations of lentiviral vector constructs generated and used for transduction studies. Asterisks denote vectors that were used to generate the final 32Dp210 vaccines. Lanes 1 to 5 on the vertical axis depict IL-15 secretion detected as nanograms per milliliter as detected by ELISA for cells transduced with each of the designated constructs. Bars represent the mean cytokine concentration ± SEM. (B) Levels of IL-15 secretion after lentiviral transduction of 32Dp210 cells are comparable pre- and postirradiation. 32Dp210 cells transduced with lentiviral vectors containing mIL-15/IL-15Rα or CD80, or both IL-15/IL-15Rα and CD80, were expanded, and populations were selected that express similar levels of cell-surface IL-15Rα and/or CD80 by fluorescence-activated cell sorter. High-expressing populations were purified. Levels of IL-15 in culture supernatants (ng/mL) were measured before (red bars) and 48 hours after irradiation with 40 Gy (blue bars). 32Dp210, to the left of the uppermost 2 lanes, indicates IL-15 secretion in untransduced controls. As previously, designations to the left of the graph indicate the genes (IL-15, IL-15Rα, or CD80, or all 3 cassettes) contained in the lentiviral vector used to transduce the 32Dp210 cells analyzed. Levels of IL-15 detected in cell culture supernatants are indicated (ng/mL) on the horizontal axis. Bars represent the mean cytokine concentration ± SEM. (C) Transduced 32Dp210 cells exhibit high level cell-surface expression of IL-15Rα, CD80, and IL-15 after purification of transduced 32Dp210 populations. Lentivirally transduced, purified 32Dp210-derived cells were stained with anti-IL-15, anti-IL-15Rα, and anti-CD80 antibodies and subjected to flow cytometric analyses. 32Dp210 on the left indicates the untransduced parental cell line used as a control. The genes encoded by each lentiviral vector used to transduce the 32Dp210 cells are indicated to the left of the flow analysis, and the antibody used to stain cells is indicated above each flow plot. (D) Cell-surface expression of IL-15Rα and CD80 on 32Dp210 vaccines is stable 48 hours after irradiation with doses up to 50 Gy. Lentivirally transduced, purified 32Dp210 cells were stained with anti-IL-15Rα and anti-CD80 antibodies, and the effects of different doses of radiation on IL-15Rα and CD80 expression analyzed. The lentiviral vector used to transduce 32Dp210 cells is indicated above each plot, and the dose of irradiation indicated on the vertical axis. Antibodies used to analyze cell surface expression are indicated below the plots. Data showing cell surface IL-15Rα expression are shown in the left 2 graphs, and cell surface staining with anti-CD80 antibodies is presented in the right 2 graphs.
32Dp210-derived vaccines stimulate ex vivo T-cell proliferation in splenocytes from naïve mice
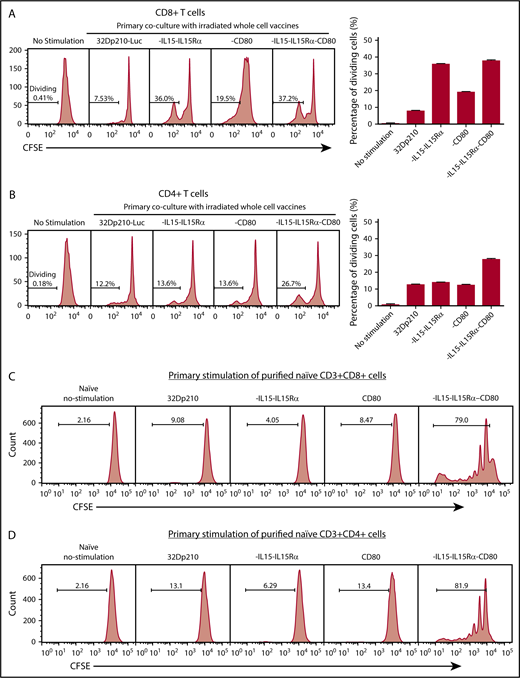
Primary stimulation of splenocytes from naïve mice with irradiated 32Dp210-IL-15/IL-15Rα or 32Dp210-IL-15/IL-15Rα/CD80 vaccines produced a fivefold increase in CD3+CD8+ T-cell proliferation compared with coculture with untransduced 32Dp210 cells (Figure 3A). Lower levels of proliferation were observed after stimulation with 32Dp210-CD80. In the case of CD3+CD4+ T cells, proliferation levels stimulated by coculture with either parent 32Dp210 cell vaccine, 32Dp210-CD80, or 32Dp210-IL-15/IL-15Rα were similar; however, the greatest responses were seen after 32Dp210-IL-15/IL-15Rα/CD80 vaccination (Figure 3B). To assess whether the combination of IL-15 and CD80 expression is synergistic in stimulating T cells in the absence of antigen presenting cells, and other coexisting populations in spleen, CD3+CD4+ and CD3+CD8+ T cells were purified and then stimulated with irradiated 32Dp210-derived cells. Consistent with our hypothesis, coexpression of IL-15/IL-15Rα and CD80 showed striking synergy in stimulating purified CD3+CD8+ (Figure 3C) and CD3+CD4+ T cells (Figure 3D).
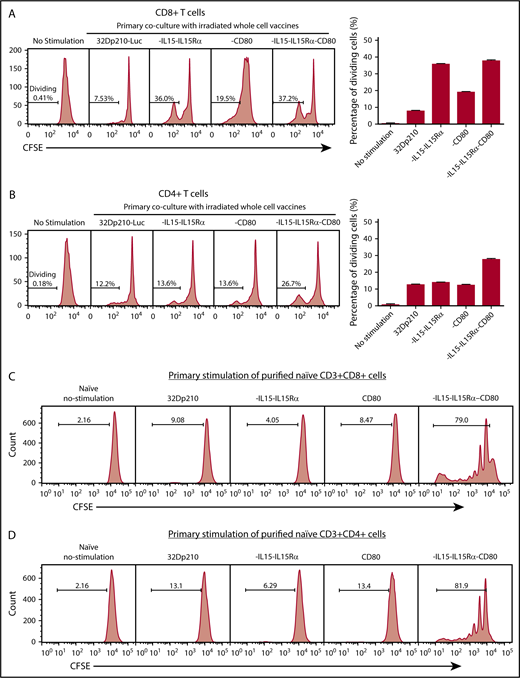
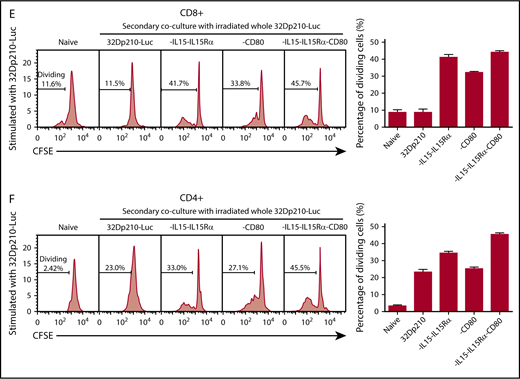
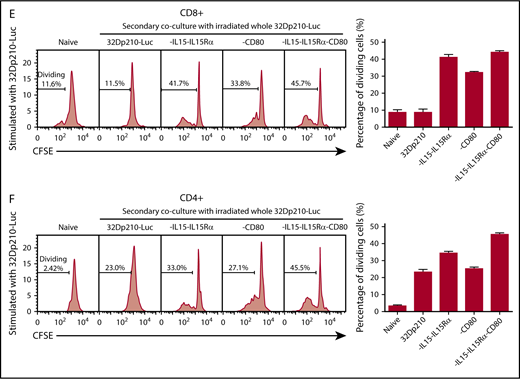
Primary coculture of splenocytes from normal naïve mice with transduced, irradiated 32Dp210-derived vaccines increases proliferation of CD3+CD8+and CD3+CD4+T cells. Whole splenocyte populations from normal naïve mice were labeled with CFSE and cultured alone (no stimulation), or in the presence of irradiated untransduced 32Dp210 cells, or with 32Dp210 cells transduced with lentiviral vectors carrying CD80, IL-15-IL-15Rα, or IL-15-IL-15Rα-CD80 (depicted above each graph) for 12 days. The percent dividing cells is indicated in each plot. After coculture, cells were stained with anti-CD3, anti-CD8, and anti-CD4 antibodies, and quantified by flow cytometric analyses. (A) Proliferation of CD3+CD8+ T cells. (B) Proliferation of CD3+CD4+ T cells. Bar graphs to the right of the flow plots depict the mean percentage of dividing cells from triplicate wells ± SEM. 32Dp210-luc indicates luciferase-positive parental leukemia cell line. The gene or genes expressed by each of the transduced 32Dp210 lines are indicated above each of the plots. (C and D) IL-15/IL-15Rα and CD80 expressed by the 32Dp210-IL-15/IL-15Rα/CD80 vaccine are highly synergistic in stimulating the proliferation of purified T cells from naïve normal mice. Splenocytes were harvested, and T cells were purified, labeled with CFSE, and cocultured with irradiated 32Dp210-derived vaccines in primary stimulation assays for 12 days. Thereafter, proliferation was quantified by flow cytometric analyses. (C) Proliferation of CD3+CD8+ T cells; (D) proliferation of CD3+CD4+T cells. The genes expressed by the 32Dp210-derived vaccines in each stimulation assay are indicated above each plot. The percent dividing cells is indicated in each plot. Controls include culture with media alone (no stimulation) and coculture with the untransduced parental 32Dp210 cells. (E-F) Secondary stimulation assays of splenocytes harvested from non-tumor-bearing mice treated with 32Dp210 vaccines and restimulated ex vivo with parental 32Dp210 cells exhibit enhanced proliferative capacity. Splenocytes were harvested from C3H mice previously vaccinated weekly 4 times with irradiated 32Dp210, 32Dp210-IL15-IL15Rα, 32Dp210-CD80, or 32Dp210-IL-15/IL15-Rα/CD80 cell vaccines. Cells were labeled with CFSE and cocultured for 12 days with untransduced 32Dp210 parent cells treated with 100 Gy γ-irradiation. Thereafter, cocultured splenocytes were stained with anti-CD3, CD4, and CD8 antibodies, and proliferation was quantified by flow cytometry. The percent dividing cells is indicated in each plot. (E) Proliferation of CD3+CD8+ T cells. (F) Proliferation of CD3+CD4+ T cells. Bar graphs to the right depict the mean percentage of dividing cells in triplicate wells ± SEM. Transgenes expressed by each vaccine cell line are indicated above each of the plots and below the corresponding bar graphs.
Primary coculture of splenocytes from normal naïve mice with transduced, irradiated 32Dp210-derived vaccines increases proliferation of CD3+CD8+and CD3+CD4+T cells. Whole splenocyte populations from normal naïve mice were labeled with CFSE and cultured alone (no stimulation), or in the presence of irradiated untransduced 32Dp210 cells, or with 32Dp210 cells transduced with lentiviral vectors carrying CD80, IL-15-IL-15Rα, or IL-15-IL-15Rα-CD80 (depicted above each graph) for 12 days. The percent dividing cells is indicated in each plot. After coculture, cells were stained with anti-CD3, anti-CD8, and anti-CD4 antibodies, and quantified by flow cytometric analyses. (A) Proliferation of CD3+CD8+ T cells. (B) Proliferation of CD3+CD4+ T cells. Bar graphs to the right of the flow plots depict the mean percentage of dividing cells from triplicate wells ± SEM. 32Dp210-luc indicates luciferase-positive parental leukemia cell line. The gene or genes expressed by each of the transduced 32Dp210 lines are indicated above each of the plots. (C and D) IL-15/IL-15Rα and CD80 expressed by the 32Dp210-IL-15/IL-15Rα/CD80 vaccine are highly synergistic in stimulating the proliferation of purified T cells from naïve normal mice. Splenocytes were harvested, and T cells were purified, labeled with CFSE, and cocultured with irradiated 32Dp210-derived vaccines in primary stimulation assays for 12 days. Thereafter, proliferation was quantified by flow cytometric analyses. (C) Proliferation of CD3+CD8+ T cells; (D) proliferation of CD3+CD4+T cells. The genes expressed by the 32Dp210-derived vaccines in each stimulation assay are indicated above each plot. The percent dividing cells is indicated in each plot. Controls include culture with media alone (no stimulation) and coculture with the untransduced parental 32Dp210 cells. (E-F) Secondary stimulation assays of splenocytes harvested from non-tumor-bearing mice treated with 32Dp210 vaccines and restimulated ex vivo with parental 32Dp210 cells exhibit enhanced proliferative capacity. Splenocytes were harvested from C3H mice previously vaccinated weekly 4 times with irradiated 32Dp210, 32Dp210-IL15-IL15Rα, 32Dp210-CD80, or 32Dp210-IL-15/IL15-Rα/CD80 cell vaccines. Cells were labeled with CFSE and cocultured for 12 days with untransduced 32Dp210 parent cells treated with 100 Gy γ-irradiation. Thereafter, cocultured splenocytes were stained with anti-CD3, CD4, and CD8 antibodies, and proliferation was quantified by flow cytometry. The percent dividing cells is indicated in each plot. (E) Proliferation of CD3+CD8+ T cells. (F) Proliferation of CD3+CD4+ T cells. Bar graphs to the right depict the mean percentage of dividing cells in triplicate wells ± SEM. Transgenes expressed by each vaccine cell line are indicated above each of the plots and below the corresponding bar graphs.
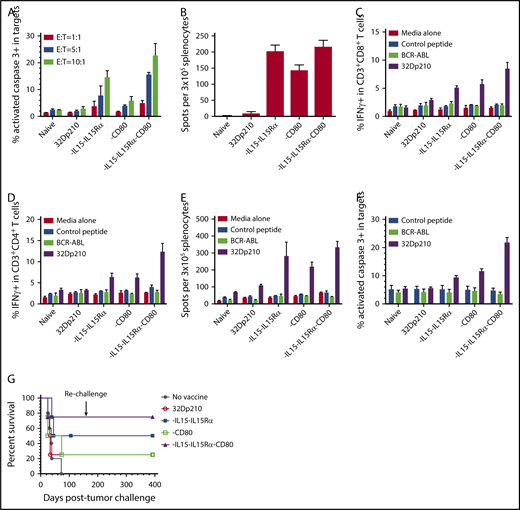
32Dp210-IL-15/IL-15Rα/CD80 vaccination stimulates robust T-cell proliferation and cytolytic activity
The immune-stimulatory effects of 32Dp210-based vaccination were then evaluated in vivo in normal, non-tumor-bearing mice where leukemia-mediated immunosuppression would not confound assessment of maximal induced immune responses. C3H mice were vaccinated with irradiated 32Dp210-CD80, 32Dp210-IL-15/IL-15Rα, or 32Dp210-IL-15/IL-15Rα/CD80 weekly for a month. Controls included age-matched, untreated C3H mice, and mice vaccinated with the irradiated 32Dp210 parent line. Thereafter, splenocytes were harvested, restimulated with 32Dp210 parent cells, and stained for flow cytometric analysis. The highest levels of CD3+CD8+ T-cell proliferation were observed in splenocytes from mice vaccinated with 32Dp210-IL-15/IL-15Rα and 32Dp210-IL-15/IL-15Rα/CD80 (Figure 3E). Prior stimulation with the 32Dp210-IL-15/IL-15Rα/CD80 vaccine also resulted in enhanced proliferation of CD3+CD4+ T cells (Figure 3F).
In ex vivo assays, leukemia-specific cytolytic activity stimulated by 32Dp210-derived vaccines against 32Dp210 targets was greatest in splenic T cells from 32Dp210-IL-15/IL-15Rα/CD80 vaccinated mice at all effector to 32Dp210 target cell ratios (Figure 4A). Vaccination with 32Dp210-IL-15/IL-15Rα also stimulated significant responses. ELISpot assays demonstrated increased frequencies of IFN-γ-expressing cells after secondary stimulation of splenocytes, consistent with activation and expansion of cytotoxic T cells (Figure 4B). In these assays, it was necessary to restrict the number of peripheral blood mononuclear cell to 3 × 105, as higher numbers of peripheral blood mononuclear cells produced too many spots for accurate quantification, because of the high magnitude of immune stimulation. Robust T-cell stimulation by vaccines was further supported by demonstration of increased intracellular IFN-γ expression in CD3+CD8+ and CD3+CD4+ T cells after 20 hours coculture with unmodified 32Dp210 cells. Again, highest levels of immune stimulation were induced by 32Dp210-IL-15/IL-15Rα/CD80 vaccination (Figure 4C-D).
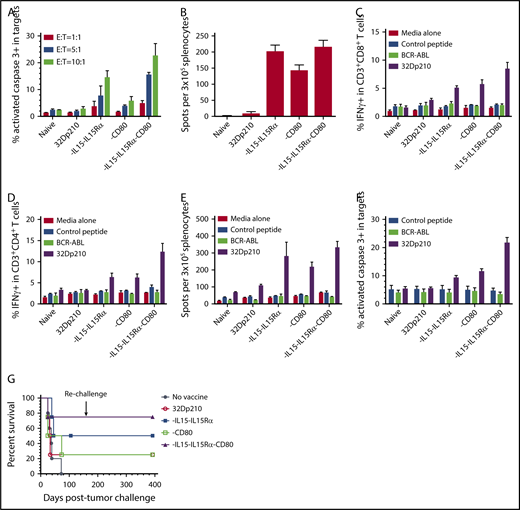
Serial vaccination with 32Dp210-derived whole cell vaccines in non-tumor-bearing mice stimulates robust antileukemic cytolytic activity. Naïve C3H mice were treated with 4 vaccinations with 2 × 106 irradiated 32Dp210-Luc, 32Dp210-mIL-15-IL-15Rα, 32Dp210-CD80, or 32Dp210-IL-15/IL-15Rα/CD80. In the first experimental group of mice, splenocytes were harvested and restimulated in vitro with irradiated (100 Gy) 32Dp210 parent cells for 5 days. Thereafter, cells were harvested and purified by Ficoll density gradient for ex vivo assays (A-F). In a parallel experimental group, vaccinated mice were challenged with IV inoculation of 32Dp210 leukemia and monitored for survival (G). (A) Cytolytic activity of splenocytes in ex vivo assays is greatest after stimulation by 32Dp210-IL-15/IL-15Rα/CD80 vaccine. Splenic effectors cells and 1 × 105 CellTrace DDAO-SE labeled 32Dp210 target cells were cocultured at different ratios (red bars, 1:1; blue bars, 5:1; green bars, 10:1) for 48 hours. X-axis: naïve: indicates splenocytes from uninjected C3H mice; 32Dp210: splenocytes from mice vaccinated with the parent 32Dp210 cell line; vaccine expressing IL-15/IL-15Rα, CD80, or IL-15/IL-15Rα/CD80 (below graph): depict assays with splenocytes from C3H mice treated with lentivirally transduced 32Dp210 vaccines expressing the indicated transgenes. The mean percentage of apoptotic cells, defined by detection of activated caspase-3 by antibody staining, is depicted on the y-axis (± SEM). (B) ELIspot assay of IFN-γ expression in splenocytes from vaccinated mice shows increased frequencies of cytotoxic cells after treatment with all transduced 32Dp210 vaccines. Splenocytes from unvaccinated mice (naïve), or from mice vaccinated with irradiated parent cells 32Dp210 cells, or with engineered 32Dp210 cells expressing each of the transgene cassettes, indicated below the bar graphs, were cocultured with irradiated 32Dp210 cells, and the frequency of IFN-γ positive cells was quantified. The mean number of spots per well per containing 3 × 105 cells in triplicate samples is depicted on the y-axis ± SEM. (C and D) Highest levels of intracellular IFN-γ expression are observed in CD3+CD8+ and CD3+CD4+ T cells from 32Dp210-IL-15/IL-15Rα/CD80-vaccinated mice after secondary stimulation, whereas stimulation with BCR-ABL-loaded splenocytes produces IFN-γ levels comparable to unstimulated controls. In panels C and D, the y-axis depicts the percent IFN-γ positive CD3+CD8+ T cells (C) and CD3+CD4+ T cells (D) in splenocytes from vaccinated non-tumor-bearing mice. X-axis: naïve = unvaccinated mice; 32Dp210 = vaccinated weekly, 4 times with unmodified irradiated 32Dp210 parent cells; -IL-15/IL-15Rα = vaccinated with 32Dp210-IL-15/IL-15Rα; -CD80 = vaccinated with 32Dp210-CD80; -IL-15/IL-15Rα/CD80 = vaccinated with 32Dp210-IL-15/IL-15Rα/CD80. Splenocytes were stimulated for 20 hours with either media alone, C3H splenocytes loaded with an irrelevant control peptide, or BCR-ABL peptide, or with unmodified 32Dp210 cells as indicated. (E) Comparative ELIspot assays of IFN-γ expression in splenocytes from vaccinated mice confirm increased frequencies of cytotoxic cells after treatment with all transduced 32Dp210 vaccines but no significant stimulation by BCR-ABL-loaded cells. ELIspot assays were performed as in panel A. C3H splenocytes were loaded with either BCR-ABL peptide or control peptide and cocultured with splenocytes from vaccinated animals as described in panel A, depicted on the x-axis. The mean number of spots per well per containing 3 × 105 cells in triplicate samples is depicted on the y-axis (± SEM). (F) Splenocytes from non-tumor-bearing mice treated with the 32Dp210-derived vaccines show high levels of lytic activity to 32Dp210 targets, but not to human BCR-ABL peptide loaded syngeneic C3H cells. Splenocytes from vaccinated non-tumor-bearing C3H mice, as described in panel A were stimulated ex vivo for 5 days with irradiated 32Dp210 cells and used as effectors. Unmodified 32Dp210 cells, BCR-ABL, or control peptide-loaded splenocytes from naïve C3H mice were used as targets. Ex vivo lytic activity was assayed by intracellular staining of active caspase-3 after coculture of effectors and targets at an effector-to-target ratio = 10:1 for 24 hours. X-axis depicts cells from different experimental vaccine groups as in panel A. The mean percentage of apoptotic cells, defined by detection of activated caspase-3 by antibody staining, is depicted on the y-axis (± SEM). (G) Treatment with transduced 32Dp210-derived vaccines confers greater survival after leukemia challenge than does administration of untransduced irradiated 32Dp210 cell vaccines. Mice were treated weekly 3 times with 32Dp210-derived vaccines as described previously. Thereafter, they were inoculated IV with 1 × 104 32Dp210-luc cells and longitudinally monitored by in vivo bioluminescence imaging for tumor progression and survival. Percent survival is depicted on the y-axis, and duration of survival on the x-axis. Animals surviving the initial leukemia challenge were rechallenged with a second IV inoculation of 32Dp210 leukemia (indicated by downward arrow), 150 days after the initial tumor challenge.
Serial vaccination with 32Dp210-derived whole cell vaccines in non-tumor-bearing mice stimulates robust antileukemic cytolytic activity. Naïve C3H mice were treated with 4 vaccinations with 2 × 106 irradiated 32Dp210-Luc, 32Dp210-mIL-15-IL-15Rα, 32Dp210-CD80, or 32Dp210-IL-15/IL-15Rα/CD80. In the first experimental group of mice, splenocytes were harvested and restimulated in vitro with irradiated (100 Gy) 32Dp210 parent cells for 5 days. Thereafter, cells were harvested and purified by Ficoll density gradient for ex vivo assays (A-F). In a parallel experimental group, vaccinated mice were challenged with IV inoculation of 32Dp210 leukemia and monitored for survival (G). (A) Cytolytic activity of splenocytes in ex vivo assays is greatest after stimulation by 32Dp210-IL-15/IL-15Rα/CD80 vaccine. Splenic effectors cells and 1 × 105 CellTrace DDAO-SE labeled 32Dp210 target cells were cocultured at different ratios (red bars, 1:1; blue bars, 5:1; green bars, 10:1) for 48 hours. X-axis: naïve: indicates splenocytes from uninjected C3H mice; 32Dp210: splenocytes from mice vaccinated with the parent 32Dp210 cell line; vaccine expressing IL-15/IL-15Rα, CD80, or IL-15/IL-15Rα/CD80 (below graph): depict assays with splenocytes from C3H mice treated with lentivirally transduced 32Dp210 vaccines expressing the indicated transgenes. The mean percentage of apoptotic cells, defined by detection of activated caspase-3 by antibody staining, is depicted on the y-axis (± SEM). (B) ELIspot assay of IFN-γ expression in splenocytes from vaccinated mice shows increased frequencies of cytotoxic cells after treatment with all transduced 32Dp210 vaccines. Splenocytes from unvaccinated mice (naïve), or from mice vaccinated with irradiated parent cells 32Dp210 cells, or with engineered 32Dp210 cells expressing each of the transgene cassettes, indicated below the bar graphs, were cocultured with irradiated 32Dp210 cells, and the frequency of IFN-γ positive cells was quantified. The mean number of spots per well per containing 3 × 105 cells in triplicate samples is depicted on the y-axis ± SEM. (C and D) Highest levels of intracellular IFN-γ expression are observed in CD3+CD8+ and CD3+CD4+ T cells from 32Dp210-IL-15/IL-15Rα/CD80-vaccinated mice after secondary stimulation, whereas stimulation with BCR-ABL-loaded splenocytes produces IFN-γ levels comparable to unstimulated controls. In panels C and D, the y-axis depicts the percent IFN-γ positive CD3+CD8+ T cells (C) and CD3+CD4+ T cells (D) in splenocytes from vaccinated non-tumor-bearing mice. X-axis: naïve = unvaccinated mice; 32Dp210 = vaccinated weekly, 4 times with unmodified irradiated 32Dp210 parent cells; -IL-15/IL-15Rα = vaccinated with 32Dp210-IL-15/IL-15Rα; -CD80 = vaccinated with 32Dp210-CD80; -IL-15/IL-15Rα/CD80 = vaccinated with 32Dp210-IL-15/IL-15Rα/CD80. Splenocytes were stimulated for 20 hours with either media alone, C3H splenocytes loaded with an irrelevant control peptide, or BCR-ABL peptide, or with unmodified 32Dp210 cells as indicated. (E) Comparative ELIspot assays of IFN-γ expression in splenocytes from vaccinated mice confirm increased frequencies of cytotoxic cells after treatment with all transduced 32Dp210 vaccines but no significant stimulation by BCR-ABL-loaded cells. ELIspot assays were performed as in panel A. C3H splenocytes were loaded with either BCR-ABL peptide or control peptide and cocultured with splenocytes from vaccinated animals as described in panel A, depicted on the x-axis. The mean number of spots per well per containing 3 × 105 cells in triplicate samples is depicted on the y-axis (± SEM). (F) Splenocytes from non-tumor-bearing mice treated with the 32Dp210-derived vaccines show high levels of lytic activity to 32Dp210 targets, but not to human BCR-ABL peptide loaded syngeneic C3H cells. Splenocytes from vaccinated non-tumor-bearing C3H mice, as described in panel A were stimulated ex vivo for 5 days with irradiated 32Dp210 cells and used as effectors. Unmodified 32Dp210 cells, BCR-ABL, or control peptide-loaded splenocytes from naïve C3H mice were used as targets. Ex vivo lytic activity was assayed by intracellular staining of active caspase-3 after coculture of effectors and targets at an effector-to-target ratio = 10:1 for 24 hours. X-axis depicts cells from different experimental vaccine groups as in panel A. The mean percentage of apoptotic cells, defined by detection of activated caspase-3 by antibody staining, is depicted on the y-axis (± SEM). (G) Treatment with transduced 32Dp210-derived vaccines confers greater survival after leukemia challenge than does administration of untransduced irradiated 32Dp210 cell vaccines. Mice were treated weekly 3 times with 32Dp210-derived vaccines as described previously. Thereafter, they were inoculated IV with 1 × 104 32Dp210-luc cells and longitudinally monitored by in vivo bioluminescence imaging for tumor progression and survival. Percent survival is depicted on the y-axis, and duration of survival on the x-axis. Animals surviving the initial leukemia challenge were rechallenged with a second IV inoculation of 32Dp210 leukemia (indicated by downward arrow), 150 days after the initial tumor challenge.
To determine whether expression of the human BCR-ABL oncogene expressed by 32Dp210 is the dominant antigen driving antileukemic responses, intracellular IFN-γ expression in T cells from vaccinated animals was assayed after stimulation with either (1) 32Dp210 or (2) naïve C3H splenocytes loaded with a BCR-ABL fusion peptide GFKQSSKAL shown to be targeted in other studies,41 or an irrelevant peptide. Intracellular IFN-γ expression in splenocytes from vaccinated mice after in vitro stimulation with BCR-ABL peptide-loaded cells was comparable to background levels seen after coculture with media alone or control peptide (Figure 4C-D). The absence of a dominant response to the BCR-ABL peptide was confirmed by ELISpot assays where BCR-ABL-loaded cells provided no significant stimulation (Figure 4E). The lack of a major response to BCR-ABL expressed by 32Dp210 supports our hypothesis that the potent antileukemic responses observed are induced by multiple leukemia-associated epitopes on 32Dp210 vaccines. Clinically, the magnitude of the protective response, elicited by a single peptide, is likely to be substantially lower than the aggregate response to a larger panel of patient specific and leukemia associated antigens, which collectively provide greater protective immunity. To further examine the role of BCR-ABL expression in vaccine responses, the ex vivo cytolytic activity of splenocytes against either unmodified 32Dp210, BCR-ABL-loaded, or control peptide-loaded C3H targets was compared. As previously, splenocytes from 32Dp210-IL-15/IL-15Rα/CD80 treated animals showed highest levels of cytolytic activity to 32Dp210 targets, whereas stimulation with either BCR-ABL or control HYLSTQSALSK peptide did not exceed background levels (Figure 4F).
In parallel experiments, serially vaccinated non-tumor-bearing mice underwent 32Dp210 tumor challenge. 32Dp210-IL-15/IL-15Rα/CD80-vaccinated mice had higher survival rates after IV 32Dp210 inoculation than mice treated with 32Dp210-IL-15/IL-15Rα or 32Dp210-CD80 vaccines (Figure 4G). Mice surviving 100 days after vaccination and initial tumor challenge demonstrated long-lasting antileukemic immunity as they were able to reject 32Dp210 leukemia after rechallenge with 32Dp210 leukemia at day 150.
32Dp210-IL-15/IL-15Rα/CD80 vaccination can eradicate disease in leukemic mice
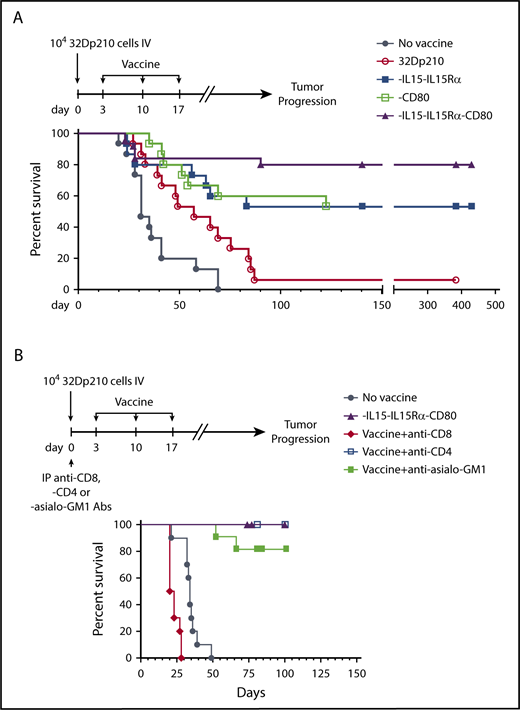
The therapeutic efficacy of 32Dp210-derived vaccines was then tested in mice with established leukemia. Mice received 3 weekly vaccinations, 3 days after injection of 1 × 104 32Dp210 cells, when leukemia has engrafted (Figure 1B). All unvaccinated leukemic controls died by day 70 (Figure 5A). Vaccination with parent 32Dp210 cells slightly prolonged survival, consistent with the previously described low-level immunogenicity of irradiated tumor cells.42,43 Treatment with any of the lenti-engineered vaccines produced greater survival after tumor challenge than did treatment with unmodified 32Dp210 cells (Figure 5A), but highest survival rates (80%) were observed after vaccination with 32Dp210-IL-15/IL-15Rα/CD80. These mice also rejected leukemia on rechallenge. Thus, coexpression of CD80, and IL-15/IL-15Rα, induces highly effective antileukemic activity, even in the context of leukemia-associated immunosuppression.
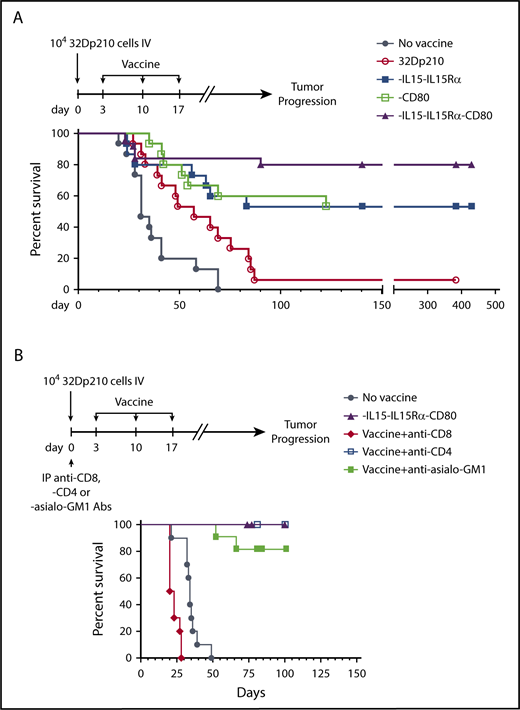
Administration of lentivirally engineered 32Dp210 vaccines in 32Dp210 leukemia-bearing mice confers greater progression-free and overall survival. (A) Overall survival of mice with 32Dp210 leukemia is greatest after serial vaccination with 32Dp210-IL-15/IL-15Rα/CD80: mice were inoculated with 1 × 104 32Dp210 leukemia cells, and vaccination was initiated 3 days later (depicted in the schema above the graph). Experimental groups included mice inoculated with tumor that received no further treatment (no vaccine, filled gray circles, n = 15) and mice vaccinated with irradiated parental 32Dp210 cells (open red circles, n = 15), 32Dp210-IL-15/IL-15Rα (filled blue squares, n = 15), 32Dp210-CD80 (open green squares, n = 15), or 32Dp210-IL-15/IL-15Rα/CD80 vaccines (filled purple triangles, n = 25). Percent survival is depicted on the y-axis, and duration of survival (days), beginning on day 0 with tumor inoculation, is shown on the x-axis. Data represent the results of 3 independent experiments. (B) In vivo, antibody-mediated depletion of CD8+ cells abrogates 32Dp210-IL-15/IL-15Rα/CD80 vaccine effects on overall survival in leukemic mice. After tumor inoculation on day 0, mice underwent 3 weekly vaccinations with 32Dp210-IL-15/IL-15Rα/CD80. Filled purple triangles depict vaccination without antibody depletion. Filled gray circles depict unvaccinated control mice inoculated with 32Dp210 leukemia. Antibody-mediated in vivo depletion of CD8+ cells (filled red diamonds, n = 10), CD4+ cells (open blue squares, n = 5), and asialo GM1+ cells (filled green squares, n = 10) are shown according to the schema at the top of the figure. Percent survival is depicted on the y-axis, and duration of survival (days) on the x-axis.
Administration of lentivirally engineered 32Dp210 vaccines in 32Dp210 leukemia-bearing mice confers greater progression-free and overall survival. (A) Overall survival of mice with 32Dp210 leukemia is greatest after serial vaccination with 32Dp210-IL-15/IL-15Rα/CD80: mice were inoculated with 1 × 104 32Dp210 leukemia cells, and vaccination was initiated 3 days later (depicted in the schema above the graph). Experimental groups included mice inoculated with tumor that received no further treatment (no vaccine, filled gray circles, n = 15) and mice vaccinated with irradiated parental 32Dp210 cells (open red circles, n = 15), 32Dp210-IL-15/IL-15Rα (filled blue squares, n = 15), 32Dp210-CD80 (open green squares, n = 15), or 32Dp210-IL-15/IL-15Rα/CD80 vaccines (filled purple triangles, n = 25). Percent survival is depicted on the y-axis, and duration of survival (days), beginning on day 0 with tumor inoculation, is shown on the x-axis. Data represent the results of 3 independent experiments. (B) In vivo, antibody-mediated depletion of CD8+ cells abrogates 32Dp210-IL-15/IL-15Rα/CD80 vaccine effects on overall survival in leukemic mice. After tumor inoculation on day 0, mice underwent 3 weekly vaccinations with 32Dp210-IL-15/IL-15Rα/CD80. Filled purple triangles depict vaccination without antibody depletion. Filled gray circles depict unvaccinated control mice inoculated with 32Dp210 leukemia. Antibody-mediated in vivo depletion of CD8+ cells (filled red diamonds, n = 10), CD4+ cells (open blue squares, n = 5), and asialo GM1+ cells (filled green squares, n = 10) are shown according to the schema at the top of the figure. Percent survival is depicted on the y-axis, and duration of survival (days) on the x-axis.
In vivo depletion of CD8+ populations abrogates 32Dp210-IL-15/IL-15Rα/CD80 vaccine efficacy
To define immune effectors mediating leukemia-specific responses, in vivo antibody-mediated depletion studies were performed with either anti-CD4 or anti-CD8 or anti-asialo-GM1 (AS-GM1) antibodies. The AS-GM1 antibody, which binds to NK cells and also to some activated T cells44,45 and to basophils,46 was used instead of anti-NK1.1 antibody, as the NK1.1 antigen (CD161b/CD161c) is not expressed in C3H mice.47,48 While depletion with anti-CD4+ or anti-AS-GM1 antibodies had minimal effects on survival outcomes, depletion of CD8+ cells completely abrogated the effects of 32Dp210-IL-15/IL-15Rα/CD80 vaccination (Figure 5B).
The 32Dp210-IL-15/IL-15Rα/CD80 vaccine has efficacy in eradicating postremission MRD
The clinical application of an autologous AML vaccine strategy would be as postremission immunotherapy. To recapitulate this clinical setting, we developed a murine model of MRD for testing our vaccine. Several chemotherapeutic regimens previously shown to induce transient responses in 32Dp210 leukemia were tested.49 However, advanced 32Dp210 leukemia proved to be highly chemorefractory as dose-finding studies with cytosine arabinoside (AraC), and/or high dose dasatinib to target the BCR-ABL tyrosine kinase, were either ineffective or resulted in toxicity (data not shown). We therefore adopted an alternative strategy for achieving postremission MRD in mice with high leukemia burdens by engineering 32Dp210 cells to express the HSV-TK suicide gene.36 Treatment with GCV could induce remission without the confounding immune and cytopenic effects of conventional chemotherapy.50 Because effects of GCV are primarily restricted to suppression of marrow proliferation, but not inhibition of immune function, treatment should not alter leukemia-associated immune deviation, except as associated with reduction of tumor burden.51 Blood samples confirmed that there was no significant effect on counts after 2 weeks of GCV administration (supplemental Table 1).
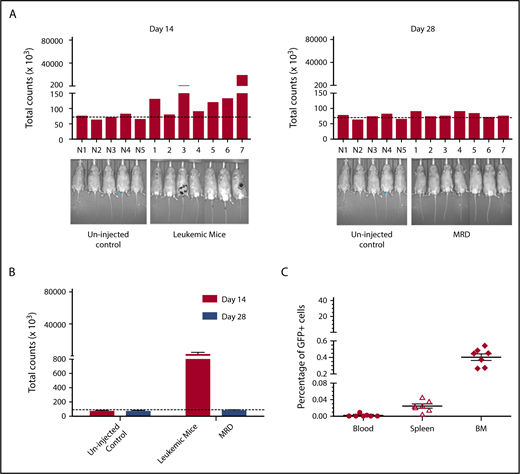
To validate this model, mice were treated daily with GCV once a significant leukemia burden was evident by IVIS. Although expression of HSV-TK has sometimes been reported to be immunogenic,52 HSV-TK expression did not affect leukemic engraftment/progression (Figure 6). Remission was arbitrarily defined as a level of bioluminescence comparable to background levels in luciferin-injected non-tumor-bearing mice. (Figure 6A). When GCV treatment began on day 14 after tumor inoculation, 7/10 mice showed remission at day 28 (Figure 6A). These mice had comparable leukemic burdens (Figure 6B), and GCV treatment decreased 32Dp210-GFP+/HSV-TK+ cells to background levels (Figure 6C). All GCV-treated leukemic mice exhibiting background levels of in vivo bioluminescence, and by definition clinical remission, also showed pathological remission (<5% blasts in bone marrow), but persistence of MRD (<0.5% GFP+ cells in BM, n = 7/7) (Figure 6C).
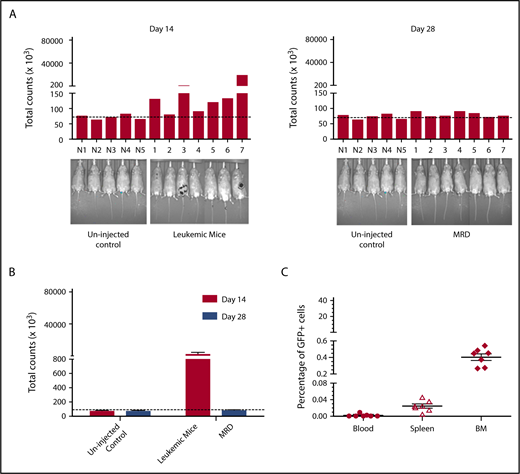
Development of an MRD model in 32Dp210 leukemia. (A) In vivo bioluminescence analyses demonstrate efficacy of GCV treatment in mice inoculated with 32Dp210-luc-HSV-TK+ leukemia. Mice were inoculated IV with 32Dp210-luc-HSV-TK+ leukemia on day 0 and began treatment with GCV (50 mg/kg) beginning at day 14 for 2 weeks. Left panel: day 14 after tumor inoculation prior to GCV treatment; right panel: day 28 after tumor inoculation and day 14 of GCV treatment. Leukemic burden as measured by total photon counts per mouse is depicted on the y-axis. Lanes N1 to N5: normal non-tumor-bearing mice injected with luciferin as background controls; lanes 1-7 depict in vivo bioluminescence assays of 32Dp210-luc-HSV-TK+ leukemia-bearing mice after 14 days of GCV administration. (B) Bioluminescence studies demonstrate induction of remission in 32Dp210-luc-HSV-TK+ tumor-bearing mice with background levels of bioluminescence after GCV treatment at day 28. Remission was arbitrarily defined as a level of bioluminescence comparable to background levels in luciferin-injected non-tumor-bearing mice (n = 5) (uninjected controls, left). Fourteen days after tumor inoculation, the mean photon counts ± SEM in 32Dp210-luc-HSV-TK+ leukemic mice were plotted on the y-axis after imaging (leukemic mice, red bar on right). Background controls for MRD in normal mice 2 weeks after GCV administration (blue bar, left). MRD (blue bar, far right) indicates the level of in vivo bioluminescence in leukemic mice after 2 weeks of GCV treatment (n = 7). (C) GCV-mediated remission induction demonstrated by in vivo bioluminescence studies correlates with induction of pathological remission. The frequency of 32Dp210-GFP+HSVTK+ cells (MRD) in peripheral blood, spleen, and bone marrow of GCV-treated, responding mice was quantified by flow cytometric analyses after 14 days of GCV administration. The percentage of GFP+ cells in blood (filled circles), spleen (open triangles), and bone marrow (filled diamonds) from each mouse achieving remission (as defined by bioluminescence studies above) are indicated on the y-axis. The mean for each group is designated by a horizontal line for each group ± SEM. Mice with remission, as defined by in vivo bioluminescence analyses that showed background levels of photon counts/subject, had detectable leukemia (MRD), but <5% leukemic cells in all tissues examined, consistent with the clinical/pathological definition of leukemic remission.
Development of an MRD model in 32Dp210 leukemia. (A) In vivo bioluminescence analyses demonstrate efficacy of GCV treatment in mice inoculated with 32Dp210-luc-HSV-TK+ leukemia. Mice were inoculated IV with 32Dp210-luc-HSV-TK+ leukemia on day 0 and began treatment with GCV (50 mg/kg) beginning at day 14 for 2 weeks. Left panel: day 14 after tumor inoculation prior to GCV treatment; right panel: day 28 after tumor inoculation and day 14 of GCV treatment. Leukemic burden as measured by total photon counts per mouse is depicted on the y-axis. Lanes N1 to N5: normal non-tumor-bearing mice injected with luciferin as background controls; lanes 1-7 depict in vivo bioluminescence assays of 32Dp210-luc-HSV-TK+ leukemia-bearing mice after 14 days of GCV administration. (B) Bioluminescence studies demonstrate induction of remission in 32Dp210-luc-HSV-TK+ tumor-bearing mice with background levels of bioluminescence after GCV treatment at day 28. Remission was arbitrarily defined as a level of bioluminescence comparable to background levels in luciferin-injected non-tumor-bearing mice (n = 5) (uninjected controls, left). Fourteen days after tumor inoculation, the mean photon counts ± SEM in 32Dp210-luc-HSV-TK+ leukemic mice were plotted on the y-axis after imaging (leukemic mice, red bar on right). Background controls for MRD in normal mice 2 weeks after GCV administration (blue bar, left). MRD (blue bar, far right) indicates the level of in vivo bioluminescence in leukemic mice after 2 weeks of GCV treatment (n = 7). (C) GCV-mediated remission induction demonstrated by in vivo bioluminescence studies correlates with induction of pathological remission. The frequency of 32Dp210-GFP+HSVTK+ cells (MRD) in peripheral blood, spleen, and bone marrow of GCV-treated, responding mice was quantified by flow cytometric analyses after 14 days of GCV administration. The percentage of GFP+ cells in blood (filled circles), spleen (open triangles), and bone marrow (filled diamonds) from each mouse achieving remission (as defined by bioluminescence studies above) are indicated on the y-axis. The mean for each group is designated by a horizontal line for each group ± SEM. Mice with remission, as defined by in vivo bioluminescence analyses that showed background levels of photon counts/subject, had detectable leukemia (MRD), but <5% leukemic cells in all tissues examined, consistent with the clinical/pathological definition of leukemic remission.
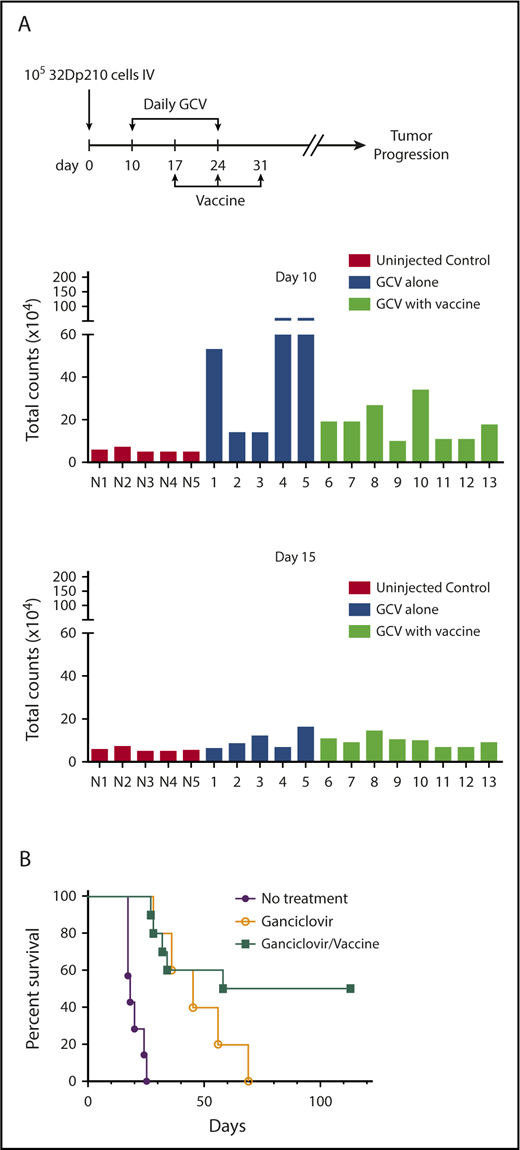
The efficacy of postremission vaccination with 32Dp210-IL-15/IL-15Rα/CD80 was then tested in mice with GCV-induced MRD. A 10-fold larger dose of 32Dp210-luc-HSV-TK+ cells (1 × 105) was administered to ensure that all mice had evidence of leukemia on day 10 by IVIS, when GCV treatment started (Figure 7A, upper panel). By day 5 of GCV administration (day 15), most leukemic mice responded (Figure 7A, lower panel). All animals that received no further therapy after achieving remission with 14-day GCV, relapsed; however, postremission treatment with 32Dp210-IL-15/IL-15Rα/CD80 was curative, eliminating MRD in 50% of mice, despite the persistence of leukemia-mediated immune suppression (Figure 7B).
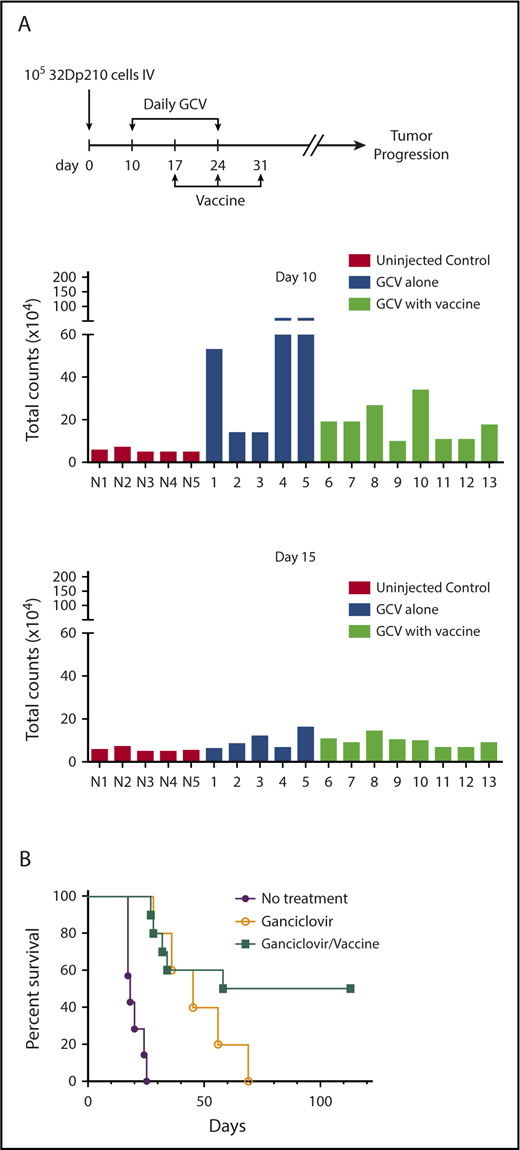
32Dp210-IL-15/IL-15Rα/CD80 vaccines administered postremission induction have efficacy in eradicating MRD. (A) Leukemic burden of groups 32Dp210-luc-HSV-TK leukemic mice before and after 5 days of GCV treatment destined for postremission vaccination with 32Dp210-IL-15/IL-15Rα/CD80 or no further therapy. Mice were inoculated with 1 × 105 32Dp210 cells on day 0 as indicated by the schema at the top of the figure. On day 10, mice underwent in vivo bioluminescence imaging, and average photon counts per animal were plotted (upper set of bar graphs). N1 to N5 represent normal, non-tumor-bearing mice injected with luciferin on the same day, to establish background luminescence levels. Lanes 1 to 13 indicate mice inoculated with leukemia and subsequently treated with GCV according to the schema at the top of the figure. Blue bars in lanes 1 to 5 indicate analyses of mice that would receive 14 days GCV treatment and no further intervention. Green bars in lanes 6 to 13 depict analyses of mice treated with 14 days of GCV treatment that would undergo serial vaccination with irradiated 32Dp210-IL-15/IL-15Rα/CD80 cells beginning at day 17. Lower panel of bar graphs depicts semiquantitative IVIS studies after 5 days of GCV administration (day 15) in the same animals analyzed in the upper panel of bar graphs. (B) Survival of 32Dp210-luc-HSV-TK leukemia bearing mice after remission induction with GCV treatment is predicated on administration of irradiated 32Dp210-IL-15/IL-15Rα/CD80 vaccine. Purple filled circles: leukemia inoculation with no treatment (n = 5); open circles: 14 days GCV treatment beginning day 10 after leukemia injection with no additional treatment (n = 5); filled green squares: GCV-mediated remission induction followed by serial vaccination with irradiated 32Dp210-IL-15/IL-15Rα/CD80 cells (n = 10).
32Dp210-IL-15/IL-15Rα/CD80 vaccines administered postremission induction have efficacy in eradicating MRD. (A) Leukemic burden of groups 32Dp210-luc-HSV-TK leukemic mice before and after 5 days of GCV treatment destined for postremission vaccination with 32Dp210-IL-15/IL-15Rα/CD80 or no further therapy. Mice were inoculated with 1 × 105 32Dp210 cells on day 0 as indicated by the schema at the top of the figure. On day 10, mice underwent in vivo bioluminescence imaging, and average photon counts per animal were plotted (upper set of bar graphs). N1 to N5 represent normal, non-tumor-bearing mice injected with luciferin on the same day, to establish background luminescence levels. Lanes 1 to 13 indicate mice inoculated with leukemia and subsequently treated with GCV according to the schema at the top of the figure. Blue bars in lanes 1 to 5 indicate analyses of mice that would receive 14 days GCV treatment and no further intervention. Green bars in lanes 6 to 13 depict analyses of mice treated with 14 days of GCV treatment that would undergo serial vaccination with irradiated 32Dp210-IL-15/IL-15Rα/CD80 cells beginning at day 17. Lower panel of bar graphs depicts semiquantitative IVIS studies after 5 days of GCV administration (day 15) in the same animals analyzed in the upper panel of bar graphs. (B) Survival of 32Dp210-luc-HSV-TK leukemia bearing mice after remission induction with GCV treatment is predicated on administration of irradiated 32Dp210-IL-15/IL-15Rα/CD80 vaccine. Purple filled circles: leukemia inoculation with no treatment (n = 5); open circles: 14 days GCV treatment beginning day 10 after leukemia injection with no additional treatment (n = 5); filled green squares: GCV-mediated remission induction followed by serial vaccination with irradiated 32Dp210-IL-15/IL-15Rα/CD80 cells (n = 10).
Discussion
Prior AML vaccines have not reliably increased relapse-free survival, because of insufficient induction of antileukemic cytolytic activity; however, the promise of IL-15 in cancer immunotherapy provided a compelling rationale for combining IL-15 and IL-15Rα, with CD80-mediated costimulation to improve vaccine efficacy.
In ex vivo assays, treatment with the 32Dp210-IL-15/IL-15Rα/CD80 vaccine stimulated the highest levels of cytolytic activity when compared with effects of vaccination with 32Dp210-IL-15/IL-15Rα, 32Dp210-CD80, or unmodified 32Dp210 cells. The effects of CD80 costimulation and IL-15/IL-15Rα were additive in this context and correlated with induction of cytotoxic lymphocytes with increased IFN-γ expression. Major responses most likely reflect responses to multiple leukemia-associated antigens and were not targeted to the human BCR-ABL antigen as demonstrated by the lack of responses after secondary stimulation with the BCR-ABL peptide GFKQSSKAL.41
The 32Dp210 leukemia recapitulates many features of human AML including early homing to bone marrow and negative effects on host immunity. Although treatment with all of the engineered vaccines increased survival in leukemic mice, 32Dp210-IL-15/IL-15Rα/CD80 vaccination produced the highest survival rates, consistent with the effects of combined IL-15/IL-15Rα and CD80 expression in stimulating cytolytic activity, also shown ex vivo. Tumor-specific cytotoxic treatment with GCV in HSV-TK-expressing 32Dp210 leukemia induced remission in the majority of mice, as defined pathologically, and by IVIS. However, similar to the clinical setting, MRD persisted after remission induction as evidenced by (1) the presence of MRD (<5% leukemia in bone marrow) in all mice meeting remission criteria by IVIS and (2) the universal relapse in mice that achieved remission with GCV treatment but no further treatment. Postremission 32Dp210-IL-15/IL-15Rα/CD80 vaccination was curative in 50% of GCV-treated subjects, while depletion of CD8+ cells, but not CD4+ cells or asialo-GM1+ NK cells, abrogated vaccine efficacy. Although it is possible that differential levels of in vivo depletion could account for the modest effects of anti-CD4 or anti-AS-GM1 antibodies, dependence on CD8+ T cells for immunotherapeutic efficacy is consistent with recent studies with the IL-15/IL-15Rα/Fc superagonist ALT-803 in a myeloma model,53 and with effects of IL-15 in a transgenic adenocarcinoma of the mouse prostate (TRAMP-C2) cancer model.54
Other studies have also demonstrated the superiority of the IL-15/IL-15Rα heterodimer in stimulating cell-mediated antitumor responses.53,55-57 Thus, a 10-fold decrease in breast cancer metastases was observed in tumor-bearing mice treated with IL-15/IL-15Rα, compared with outcomes in untreated mice.58 Addition of IL-15 and IL-15Rα during ex vivo T-cell expansion for adoptive immunotherapy stimulated T cells responding to lower peptide concentrations that lysed targets at lower effector-to-target ratios, than did cells without IL-15 treatment.59
Although IL-15 has been shown to play an important role in NK cell proliferation and homeostasis,23 the role of NK cells in some studies of IL-15-mediated antitumor immunity has been less clear. For example, treatment with IL-15- and IL-12-expressing tumor cell vaccines resulted in similar effects on tumor rejection in normal vs NK-depleted mice.60,61 In contrast, the efficacy of immunotherapy in murine lymphoma62 and neuroblastoma63 with IL-15 and IL-12 required the presence of both CD8+ T cells and NK cells. In our studies, NK depletion by in vivo administration of anti-As-GM antibodies during 32Dp210-derived vaccination did not have a major effect. This could in part be because of the persistence of some tissue-resident NK cells, recently shown to be depleted by administration of anti-NK1.1 antibodies in C57Bl/6 mice, but not by anti-asialo-GM antibodies.48 As stated previously, NK1.1 is not expressed in the C3H strain and therefore could not be tested in our model.
The feasibility of using IL-15 to enhance stimulation of autologous antileukemic immunity has been demonstrated with T cells from leukemia patients, previously shown to have an exhausted phenotype.64 Patient-derived T cells, collected at presentation, were expanded and then stimulated in vitro with IFN-γ-treated autologous AML cells weekly for 3 weeks, in combination with IL-15 and agonistic anti-CD28 antibodies. AML-reactive, leukemia-specific T cells were generated in 5/8 patients.64 The availability of clinical grade IL-15/IL-15Rα, and of an Fc fusion to a mutated form of the heterodimer (ALT803), has allowed the ongoing clinical evaluation of the protein in cancer patients (www.clinicaltrials.gov; #NCT02452268, #NCT01670994). ALT 803 produced clinical responses with CD8+ and NK cell activation and expansion, without stimulatory effects on Treg’s, in patients relapsing after allo-transplant (#NCT01885897).65 Although significant toxicity was observed with systemic administration of ALT803, these effects were mitigated by subcutaneous administration.
Our studies indicate that AML cell vaccines may enable induction of leukemia-specific immunity by amplification of responses to subdominant leukemia antigens, even when those against the immune dominant antigens are lost. There is now evidence from preclinical studies and recently published phase 1 trials with engineered autologous AML vaccines showing that they (1) are readily generated as cells can be reliably collected, cryopreserved, and engineered7,66 ; (2) can overcome tumor-related immunosuppression; and (3) stimulate leukemia-specific cytolytic immune responses when administered either after remission/consolidation chemotherapy or in immunosuppressed patients early after hematopoietic stem cell transplantation.66,67 The potent immune-stimulatory combination of heterodimeric IL-15/IL-15Rα and CD80 expression in autologous leukemia cells provides a promising and universally applicable approach to generation of personalized leukemia vaccine therapy that could improve progression-free survival.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank William J. Murphy (University of California, Davis), Lewis L. Lanier (University of California, San Francisco), Michael Rosenblum (University of California, San Francisco), Kenneth I. Weinberg (Stanford University), and Donald B. Kohn (University of California, Los Angeles) for helpful discussions, and Ralph B. Arlinghaus (University of Texas MD Anderson Cancer Center) for providing the 32Dp210 cells.
This work was supported by grants from the National Cancer Institute, National Institutes of Health (1 R21 CA177284-01A1) (K.M.L.G.); University of California, San Francisco–King’s Health Partners Faculty Fellowship Travel Grant (K.M.L.G.); University of California, San Francisco/Clinical and Translational Science Institute Pilot Project (K.M.L.G.); and a Leukemia and Lymphoma Society Translational Research Grant (6532-18) (K.M.L.G.).
Authorship
Contribution: K.M.L.G., F.F., and Y.S. designed the experiments and discussed the results; Y.S., L.D.-V., J.P.F., and C.E.A. performed or participated in experimental work; K.M.L.G. wrote the manuscript; F.F. and G.N.P. edited the manuscript; G.N.P. and C.B. provided IL-15 and IL-15Rα plasmid constructs; and F.F. provided CD80 plasmids.
Conflict-of-interest statement: The authors declare no competing financial interests.
Correspondence: Karin M. L. Gaensler, Department of Medicine, Division of Hematology/Oncology, University of California San Francisco School of Medicine, 502 Parnassus Ave, M1286, Box 1270, San Francisco, CA 94143; e-mail: karin.gaensler@ucsf.edu.