

Key Points
CD19+ and negative ASCs share a development path and are both capable of providing long-lasting immune memory.
Most ASCs are targets of early chimeric antigen receptor–T-cell therapy; knowing this may help to improve current cancer treatments.

Abstract
Long-lived antibody-secreting cells (ASCs) are critical for the maintenance of humoral immunity through the continued production of antibodies specific for previously encountered pathogen or vaccine antigens. Recent reports describing humoral immune memory have suggested the importance of long-lived CD19− bone marrow (BM) ASCs, which secrete antibodies recognizing previously encountered vaccine antigens. However, these reports do not agree upon the unique contribution of the CD19+ BM ASC subset toward humoral immunity. Here, we found both CD19+ and negative ASCs from human BM were similar in functional capacity to react to a number of vaccine antigens via ELISpot assays. The CD19+ cells were the predominant ASC population found in lymphoid tissues, and unlike the CD19− ASCs, which were found only in spleen and BM, the CD19+ ASCs were found in tonsil and blood. CD19+ ASCs from the BM, spleen, and tonsil were capable of recognizing polio vaccine antigens, indicating the CD19+ ASC cells play a novel role in long-lasting immune defense. Comparative gene expression analysis indicated CD19+ and negative BM ASCs differed significantly by only 14 distinct messenger RNAs and exhibited similar gene expression for cell cycle, autophagy, and apoptosis control necessary for long life. In addition, we show identical CDR-H3 sequences found on both BM ASC subsets, indicating a shared developmental path. Together, these results provide novel insight for the distribution, function, genetic regulation, and development of long-lived ASCs and may not only impact improved cell therapies but also enhance strategies for vaccine development.
Introduction
Long-lived antibody-secreting cells (ASCs) are differentiated B cells that play the central role in humoral immunity as they actively secrete antibodies, which provide a first line of defense against infection by identifying and neutralizing foreign antigens. Long-lived ASCs develop in response to immune challenges such as bacterial or viral infection whereby they are essential to immune protection and memory for a lifetime.1-3 As ASCs develop and mature, they exhibit altered morphology and adopt a genetic program initiated by a group of transcription factors that are exclusive and antagonistic to those required for B cells. ASCs virtually switch lineage from B cells through coordinated chromatin remodeling4 whereby their new genetic program favors survival over proliferation.5 The newly formed ASCs exhibit a progressive loss of common B-cell lineage markers,6 but the 2 markers that have been the most consistently expressed on primary human ASCs are CD38 and CD27.7-12 It is commonly thought that CD20 and CD19, the most widely expressed B-cell antigens, are lost upon terminal differentiation into long-lived ASCs.13,14 Furthermore, it is believed that only when the ASCs migrate to the bone marrow (BM) are they able to fully develop and terminally differentiate into long-lived ASCs able to secrete antibodies for a lifetime.15,16
The examination of ASCs in the BM has been the focus of recent efforts to elucidate the mechanisms of humoral memory, which were characterized through responses to vaccine antigens. These studies described BM ASCs as comprising 2 main subsets distinguished by their expression of CD19. Halliley et al17 concluded that the CD19− ASCs were the sole subset contributing to humoral immune memory, based on their reactivity to measles and mumps experienced as a childhood infection. This rationale was supported by the finding that CD19− ASCs were exclusively able to secrete immunoglobulins specific for viral antigens to which the subjects had not been exposed for >40 years, whereas the CD19+ BM ASCs did not have this capability. Mei et al18 described a CD19− ASC subset enriched in BM and not found in any other normal tissues. These BM cells exhibited fewer immunoglobulin gene rearrangements, a distinct genotype and phenotype, and increased in vitro survival compared with CD19+ BM ASCs. The differential expression of cytoplasmic Ki67 and cyclin-D2 gene expression was described as an explanation for the improved in vitro survival of the CD19− ASCs relative to CD19+. CD19− BM ASC-derived long-term immune memory was shown by Bhoj et al19 through the measurement of blood serum antibody levels in individuals before and after undergoing B-cell depletion with CD19-directed chimeric antigen receptor (CAR) T-cell therapy. Cumulatively, these works suggested that CD19− BM ASCs fit the description of the BM cells long suspected of maintaining serum antibody levels in the blood.20-22 However, none of the recent works fully addressed or agreed upon the functional capacity the CD19+ BM ASC subset. The observation that the CD19+ ASCs were depleted by cell therapy underscored the need to explore the biology of CD19+ ASCs in the BM and other tissues.
To gain insight into the importance of, and clarify the differences between, ASC subsets, we performed flow cytometry (FCM) immunophenotyping, fluorescence-activated cell sorting (FACS) for cell subset isolation, ELISpot assays detecting secreted antibodies against vaccine-derived antigens, comparative gene expression analyses, and immunoglobulin sequencing. Fundamentally, we found a common phenotype shared by all ASCs in blood, tonsil, BM, and spleen, which we used to further determine the functional and genetic differences between CD19+ and negative ASCs. Our results show that CD19 expression is found on most ASCs, and unlike CD19− ASCs, CD19+ ASCs were not restricted to the BM as previously thought.17-22 Furthermore, we show that both ASC subsets in the BM contained clones with identical antigen binding sequences, indicating a shared developmental path. Importantly, the CD19+ ASCs in both BM and spleen had the capacity to provide immune memory similar to the CD19− BM ASCs. Our results may have implications for vaccine development, the treatment of autoimmune diseases, and B-cell malignancies with CD19-targeted therapies.
Materials and methods
Further descriptive information is provided in the supplemental Material.
Preparation of human tissues
Our experiments examined maintenance levels of humoral immunity, because they were conducted without immunization records, but presumed that our donors received immunizations consistent with government recommendations in the United States.
Human blood was collected from healthy donors according to the MedImmune Institutional Research Specimen Collection Program and informed consent policy. Human BM cells were purchased from Lonza Group Ltd. Tonsil and spleen specimens were obtained through an institutional agreement with the National Disease Research Interchange.
Tissues were minced using sterile scissors and forceps and passed through a 40-μm sterile strainer (BD Falcon). Four to 5 tubes of blood per donor were collected in Vacutainer CPT (BD Biosciences) and processed following the manufacturer’s protocol. All cell suspensions were filtered and treated with ACK lysing buffer (ThermoFisher) following the manufacturer’s protocol (∼10-15 minutes at room temperature) as needed. Cells were suspended at 1 to 100 × 106/mL depending on assay.
FCM immunophenotyping
Cells amounting to 1 × 106 to 5 × 106 per test were stained, incubated, and washed once with Dulbecco’s phosphate-buffered saline, and further incubated with fixable live/dead blue (ThermoFisher) per manufacturer’s protocol. For detection of cytoplasmic antigens, cells were fixed with Fix-Perm buffers (eBioscience) following manufacturer protocols and incubated with antibodies to cytoplasmic antigens. Antibody and clone fluorophore information is available in the supplemental Methods, and optimization of our FCM method was described by Carrell and Groves.23 Due to scarcity of ASCs, we used 95% Poisson statistical cutoff as our method for acquisition of rare cell subset data.24 For example, for our BM phenotyping data, we measured a median ASC (CD27+CD38high) frequency of 0.238% (n = 17), where the median number of total events measured was 501 187, with gated-event ASC median event number of 842, which fulfills the Poisson statistical criteria of <5% coefficient of variation essential for accurate rare event detection. All FCM assays were performed on either BD Biosciences LSRII or Fortessa cytometers, followed by analysis using FlowJo.
Cell sorting
For sorting cells into ELISpot plates, we used rigorous sorting methods described by Evans et al25 with minor modifications. Cell sorting for microarray was performed on a BD Biosciences FACSAria IIu, FACSFusion, and/or Influx platforms as we previously described.26,27 Briefly, the sorter drop delay profile was tested and validated using fluorescent particles (Flow-Check Fluorospheres, Beckman Coulter, Inc) sorted onto slides and visually counted under a fluorescent microscope before and after each sort to verify sorting accuracy and efficiency. Accurate well deposition was optimized daily with fluorescent particles sorted onto the lid of a covered 96-well ELISpot plate.
Detecting immunoglobulin secretion by Elispot
Mabtech (Mariemont, OH) human immunoglobulin G (IgG), IgM, and IgA kits were used for assessing functional immunoglobulin-secreting capacity of ASCs following the manufacturer’s protocol. Briefly, ELISpot wells were prewet using 100 μL of 70% ethanol for 2 minutes. Plates were washed with distilled deionized water (200 μL per well, 5 times) using a plate washer (BioTek, Winooski, VT). Anti-IgG, anti-IgA, or anti-IgM capture antibody was diluted to 15 μg/mL with sterile phosphate-buffered saline (pH 7.4) and added to wells at 100 μL per well. In some experiments, anti-IgG, IgA, and IgM were combined within the same wells to assess the efficiency of ELISpot total immunoglobulin detection. Detection of antigen-specific immunoglobulins was performed as described by Sasaki et al28 with modifications. Briefly, vaccine antigens were precoated onto an ELISpot plate as above, and live cells from BM, spleen, or tonsil were sorted directly into the wells. Plates were read using an AID ELISpot Reader (Strassberg, Germany).
Flow cytometric detection of RNA
ASCs and B-cell–associated RNAs were detected by FCM using the PrimeFlow (ThermoFisher) and strictly following the manufacturer’s recommended protocol. In brief, cells were first stained with fluor-conjugated monoclonal antibodies to surface antigens to allow selective identification of B-cell subsets and plasma cells (CD19-PE, CD20-BV421, CD27-BV786, IgD-FITC, and CD38-PECy7), followed by washing, fixation, and permeabilization. Cells were incubated with gene-specific probes and incubated for 2 hours at 40°C for hybridization. Bound probes were detected with further hybridization reactions at 40°C for 1.5 hours with PrimeFlow complementary preamplifier and AlexaFluor-647–labeled amplifier probes.
Microarray and gene expression analysis
BM was processed similar to that described by Karnell et al.26 Briefly, of viable cells (4′,6-diamidino-2-phenylindole–negative, Sigma Aldrich), ASCs were defined as CD38highCD27+CD138+. BM was sorted into 4 populations composed of CD19− or CD19+ ASCs, and CD19+ non-ASC B-cell populations were composed of CD20+ or negative. The 2 ASC fractions were placed on the outside collection when performing 4-way cell sorting in order to reduce the possibility of contamination. All sorted fractions were collected in FACS buffer and centrifuged, and the resulting cell pellet was suspended in RNA lysis buffer (Ambion).
Total RNA was extracted using PAXgene Blood RNA kit (Qiagen) and was quantified spectrophotometrically to ensure that the absorbance at 260 nm/280 nm was >1.9. RNA quality was assessed on an Agilent 2100 Bioanalyzer using an RNA 6000 Nano LabChip and prepared and run on Affymetrix Human Genome U133 Plus 2.0 GeneCHip array as described by Streicher et al.27 Group comparisons were performed using a Bayesian linear model–based approach (Limma package in R). Genes with expression difference of fold change ≥2 and a false discovery rate of ≤0.05 were defined as significant. All whole-genome microarray data have been deposited into the Gene Expression Omnibus (accession number GSE107683).
B-cell V(D)J sequencing methods
B-cell immunoglobulin repertoires were profiled at the single-cell level using the 10× Genomics Single Cell V(D)J Reagent Kit (10× Genomics, Pleasanton, CA). BM from 6 donors was prepared and separated by FACS sorting as described above for microarray analysis.
BCR libraries were quantified on the QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA) using the KAPA Library Quantification Kit for Illumina Platforms with ROX Low qPCR Mastermix (Kapa Biosystems, Wilmington, MA). Libraries were diluted to 4 nM and pooled in equal volumes. The pool was diluted to 1 pM with 1% PhiX spike-in and sequenced on the NextSeq 500 System (Illumina, San Diego, CA) with 150-bp paired-end reads.
Cell Ranger (v2.1, 10× Genomics) was used for fastq generation, V(D)J gene alignment, and annotation. For each sample, CDR-H3 and CDR-L3 were annotated and extracted to count for unique clones. Clones with the same CDR-H3 across samples were considered as identical clones, and pairwise clonotype overlap was calculated as the number of identical clones between 2 given samples. Heat map visualization of the overlap was plotted using pheatmap.
Results
CD19+CD38highCD27+ ASCs are present in major human lymphoid tissues
CD38highCD27+ gating7-11,23,26,27 was used for our characterization of ASCs from human BM, peripheral blood (PB), tonsil, and spleen. Based on that phenotype, we showed a clearly identifiable population found in all tissues examined (Figure 1A). The frequency of ASCs ranged from 0.2% to 1.23% of gated CD3−/CD14−/CD15− leukocytes (Figure 1B). CD38 was plotted against CD27, CD19, CD20, Ki67, IgG, IgM, IgA, and HLA-DR, which provided a clearly distinguished ASC cell phenotype. The levels of surface CD19 expression on the BM, tonsil, and spleen ASC subsets were comparable to those on mature B cells. CD20 was negative on all ASCs,29,30 but we found CD20 to be expressed at low levels on ASCs in the tonsil, which could be consistent with a newly formed ASC population31 also observed in autoimmune disease.32 Cytoplasmic Ki67 was negative in all tissues except in the PB, where it was positively expressed on most ASCs. ASC subsets contained cytoplasmic immunoglobulins of either IgG, IgM, or IgA isotypes. HLA-DR was negative on BM and spleen ASCs, but was found both positive and negative on ASCs in tonsil and PB. Most ASCs were CD19+ in all tissues examined, except for a very clear but low-frequency population found in both the BM and spleen. The frequency of CD19+ ASCs was nearly threefold greater than the number of CD19− ASCs in both the BM (P < .0001, n = 16) and spleen (P < .0001, n = 8) (Figure 1C).
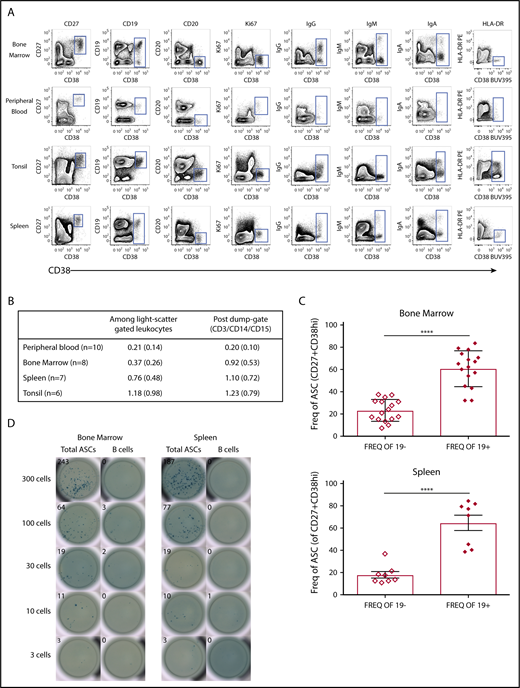
Most CD38highCD27+ASCs in human tissues express CD19. (A) Contour plots revealed a rare cell subset with high expression of CD38 and positive for CD27, which showed a clear cytoplasmic IgG, IgM, or IgA positive expression and was found in all human tissues (BM, PB, tonsil, and spleen) indicative of ASCs. (B) ASC (CD38highCD27+) frequencies in human tissues: mean, %, and standard deviation (SD). (C) CD19+ ASCs were statistically more abundant that CD19− ASCs in the BM (upper graph, unpaired Student t test, ****P < .0001, mean ± standard error of the mean (SEM) 23.14 ± 2.477 and 60.74 ± 3.994, both n = 16, median ± SD age 32 ± 7.713) and spleen (lower graph, unpaired Student t test, ****P < .0001, mean ± SEM 17.70 ± 2.983 and 64.7 ± 2.983, both n = 8, median ± SD age 51 ± 14.125). (D) CD38highCD27+ ASCs or CD19+CD20+ non-ASC B cells from BM were sorted directly onto total IgG/IgM/IgA precoated ELISpot plates. BCs, B cells; Freq., frequency.
Most CD38highCD27+ASCs in human tissues express CD19. (A) Contour plots revealed a rare cell subset with high expression of CD38 and positive for CD27, which showed a clear cytoplasmic IgG, IgM, or IgA positive expression and was found in all human tissues (BM, PB, tonsil, and spleen) indicative of ASCs. (B) ASC (CD38highCD27+) frequencies in human tissues: mean, %, and standard deviation (SD). (C) CD19+ ASCs were statistically more abundant that CD19− ASCs in the BM (upper graph, unpaired Student t test, ****P < .0001, mean ± standard error of the mean (SEM) 23.14 ± 2.477 and 60.74 ± 3.994, both n = 16, median ± SD age 32 ± 7.713) and spleen (lower graph, unpaired Student t test, ****P < .0001, mean ± SEM 17.70 ± 2.983 and 64.7 ± 2.983, both n = 8, median ± SD age 51 ± 14.125). (D) CD38highCD27+ ASCs or CD19+CD20+ non-ASC B cells from BM were sorted directly onto total IgG/IgM/IgA precoated ELISpot plates. BCs, B cells; Freq., frequency.
We performed an immunoglobulin ELISpot assay with freshly isolated FACS-sorted BM and spleen mononuclear cells from healthy donors. Only the sorted ASC BM and spleen cells produced spots, whereas the sorted non-ASC CD19+CD20+ B-cell fraction did not secrete immunoglobulin. Furthermore, we detected immunoglobulin spots in decreasing sorted events down to 3 cells per well, demonstrating that our cell sorting was efficient and accurate (Figure 1D).
CD19+ ASCs are the most frequent immunoglobulin-secreting cells in normal human BM and spleen
CD19+ and negative ASCs, delineated by quadrants, contained detectable levels of cytoplasmic IgG, IgA, and IgM in the BM and spleen (Figure 2A). IgG was the primary cytoplasmic isotype in both ASC subsets (Figure 2B), and there was no significant difference in immunoglobulin isotype determination between CD19+ and negative ASCs (unpaired Student t test, n = 8). The discrepancy between our results and those reported by Mei et al18 could be the result of donor-dependent factors, the average efficiency immunoglobulin detection, or the reagents used. Among total ASCs, the relative frequency of cytoplasmic IgG and IgM from the CD19+ compared with CD19− ASCs in BM was significantly greater (IgG P < .0002, n = 6, IgM P < .043, n = 6). In the spleen, all isotypes from the CD19+ ASCs were more frequent (IgG P < .0001, IgM P < .0179, IgA P < .0035, all n = 6) (Figure 2C). We found no significant difference in immunoglobulin secretion via ELISpot for any isotype between ASC subsets in the BM or spleen (Figure 2D). Representative ELISpots for 300 BM and 300 spleen CD19+ and negative ASC subsets sorted directly into precoated ELISpot plates are shown in Figure 2E.
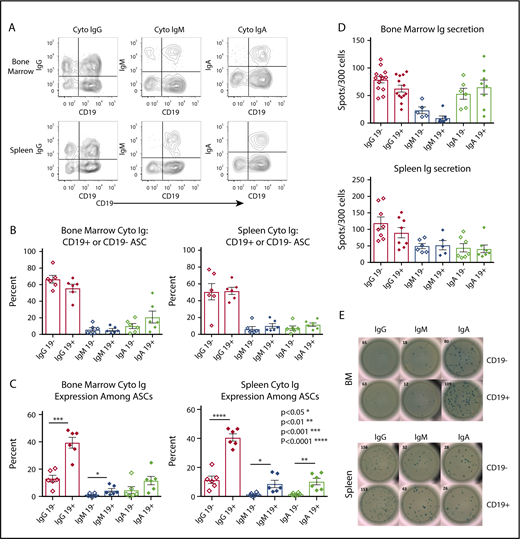
CD19+ASCs are the most common immunoglobulin-secreting cells in BM and spleen. (A) Cytoplasmic immunoglobulin immunophenotyping gated on CD38highCD27+ ASCs. Representative plots showed CD19+ and CD19− vs cytoplasmic immunoglobulin in BM and spleen. (B) Relative frequency of immunoglobulin-secreting isotypes among ASC CD19+ or CD19− from BM (n = 8) and spleen (n = 7). (C) The frequency between CD19+ and CD19− ASCs for immunoglobulin isotype secretion via cytoplasmic immunoglobulin FCM in BM (upper, unpaired Student t test, IgG P < .0002, CD19+ mean ± SEM 39.58 ± 3.952, CD19− mean ± SEM 13.18 ± 2.473, n = 6, IgM P < .043, CD19+ mean ± SEM 4.393 ± 1.359, CD19− mean ± SEM 1.095 ± 0.4234, n = 6) and spleen (lower, Student unpaired Student t test, IgG P < .0001, CD19+ mean ± SEM 40.78 ± 2.48, CD19− mean ± SEM 11.54 ± 2.515, n = 6, IgM P < .0179, CD19+ mean ± SEM 8.665 ± 2.491, CD19− mean ± SEM 1.515 ± 0.4246, n = 6, IgA P < .0035, CD19+ mean ± SEM 10.3 ± 2.242, CD19− mean ± SEM 1.686 ± 0.3156, n = 6) immunoglobulin isotype testing via cytoplasmic immunoglobulin FCM. (D) Immunoglobulin secretion by ELISpot showed no significance between CD19+ and negative ASCs in BM (upper, IgG n = 13, IgM n = 6, IgA n = 6) or spleen (lower, IgG n = 8, IgM n = 6, IgA n = 7). (E) Representative ELISpots are shown for both BM (upper) and spleen (lower). Cyto, cytoplasmic; Ig, immunoglobulin.
CD19+ASCs are the most common immunoglobulin-secreting cells in BM and spleen. (A) Cytoplasmic immunoglobulin immunophenotyping gated on CD38highCD27+ ASCs. Representative plots showed CD19+ and CD19− vs cytoplasmic immunoglobulin in BM and spleen. (B) Relative frequency of immunoglobulin-secreting isotypes among ASC CD19+ or CD19− from BM (n = 8) and spleen (n = 7). (C) The frequency between CD19+ and CD19− ASCs for immunoglobulin isotype secretion via cytoplasmic immunoglobulin FCM in BM (upper, unpaired Student t test, IgG P < .0002, CD19+ mean ± SEM 39.58 ± 3.952, CD19− mean ± SEM 13.18 ± 2.473, n = 6, IgM P < .043, CD19+ mean ± SEM 4.393 ± 1.359, CD19− mean ± SEM 1.095 ± 0.4234, n = 6) and spleen (lower, Student unpaired Student t test, IgG P < .0001, CD19+ mean ± SEM 40.78 ± 2.48, CD19− mean ± SEM 11.54 ± 2.515, n = 6, IgM P < .0179, CD19+ mean ± SEM 8.665 ± 2.491, CD19− mean ± SEM 1.515 ± 0.4246, n = 6, IgA P < .0035, CD19+ mean ± SEM 10.3 ± 2.242, CD19− mean ± SEM 1.686 ± 0.3156, n = 6) immunoglobulin isotype testing via cytoplasmic immunoglobulin FCM. (D) Immunoglobulin secretion by ELISpot showed no significance between CD19+ and negative ASCs in BM (upper, IgG n = 13, IgM n = 6, IgA n = 6) or spleen (lower, IgG n = 8, IgM n = 6, IgA n = 7). (E) Representative ELISpots are shown for both BM (upper) and spleen (lower). Cyto, cytoplasmic; Ig, immunoglobulin.
The CD19+ ASCs produce IgG antibodies to vaccine antigens and are found in BM, spleen, and tonsil
We compared the relative frequency of CD19+ and negative ASCs that secreted IgG antibodies, which recognized vaccine antigens. Antigens that could generally be thought of as newly exposed (influenza), exposed within 8 years (tetanus), or exposed as a child/over a lifetime (polio) were tested. We sorted 3000 BM CD19+ and negative ASCs directly into vaccine precoated ELISpot plates and detected for IgG (Figure 3A). We did not observe a statistically significant difference between CD19+ and negative BM ASCs, which secrete IgG antibodies to Daptacel, but our results showed a significant increase in frequency of CD19− ASCs specific for Fluzone (P < .0327, n = 10). Representative IgG ELISpots are shown in Figure 3B. We observed secretion of IgG antibodies that bound vaccine antigens from both CD19+ and negative ASCs in the spleen (Figure 3C). We did not have enough number of spleen replicates for statistical analyses; however, we detected ELISpots for IgG specific to MMRII (n = 2), IPOL (n = 3), and Varivax (n = 2) vaccines in addition to Daptacel (n = 4) and Fluzone (n = 5). Representative IgG ELISpots are shown in Figure 3D. We sorted 10 000 cells per well of the CD19+ ASCs from BM, spleen, and tonsil for Fluzone and Daptacel, and 30 000 cells per well for IPOL and found that the CD19+ ASCs in all tissues contained cells that secreted IgG able to identify all tested vaccine antigens; B-cell controls were negative (Figure 3E). Our results were similar to those observed in mice, wherein a subpopulation of ASCs remained in the spleen following infection or vaccination.33,34
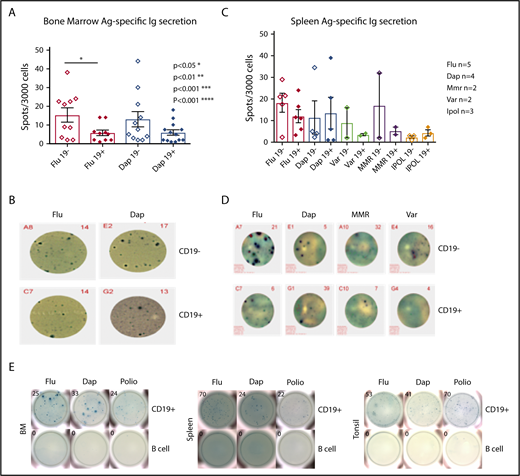
Vaccine-specific IgG are produced by both the CD19+and the CD19−ASC subsets. (A) BD CD19+ and CD19− ASC antigen-specific ELISpot results for Daptacel and Fluzone (unpaired Student t test, P < .0327, CD19+ mean ± SEM 5.810 ± 1.498, n = 10, CD19− mean ± SEM 15.29 ± 3.814, n = 10). (B) Representative ELISpot wells from 3000 CD19+ and CD19− FACS sorted ASCs from BM. (C) 3000 CD19+ and CD19− ASCs from spleen were sorted onto Daptacel (n = 4), Fluzone (n = 5), MMR (n = 2), Varicella (n = 2), or IPOL (n = 3) vaccine antigen precoated ELISpot plates and detected for total IgG. (D) Representative ELISpot wells showing sorted ASCs from either CD19+ or negative spleen subsets. (E) 10 000 (Flu/Dap) and 30 000 (polio) CD19+ ASCs or B cells were FACS sorted from BM, spleen, and tonsil, which indicated antigen-specific CD19+ ASCs existed in all tissues. Ag., antigen; Dap., Daptacel; Flu., Fluzone; MMR, measles, mumps, and rubella; Var., Varicella.
Vaccine-specific IgG are produced by both the CD19+and the CD19−ASC subsets. (A) BD CD19+ and CD19− ASC antigen-specific ELISpot results for Daptacel and Fluzone (unpaired Student t test, P < .0327, CD19+ mean ± SEM 5.810 ± 1.498, n = 10, CD19− mean ± SEM 15.29 ± 3.814, n = 10). (B) Representative ELISpot wells from 3000 CD19+ and CD19− FACS sorted ASCs from BM. (C) 3000 CD19+ and CD19− ASCs from spleen were sorted onto Daptacel (n = 4), Fluzone (n = 5), MMR (n = 2), Varicella (n = 2), or IPOL (n = 3) vaccine antigen precoated ELISpot plates and detected for total IgG. (D) Representative ELISpot wells showing sorted ASCs from either CD19+ or negative spleen subsets. (E) 10 000 (Flu/Dap) and 30 000 (polio) CD19+ ASCs or B cells were FACS sorted from BM, spleen, and tonsil, which indicated antigen-specific CD19+ ASCs existed in all tissues. Ag., antigen; Dap., Daptacel; Flu., Fluzone; MMR, measles, mumps, and rubella; Var., Varicella.
CD19+ and negative BM ASCs gene expression and sequence identity indicates a common differentiation path
We examined fresh human BM with probes specific for genes that were differentially expressed between ASCs and normal B cells. IL4R, TNFRSF7 (CD27), CD19, MS4A1 (CD20), XBP1, PRDM1 (Blimp-1), SDC1 (CD138), CD38, and TNFRSF17 (BCMA) RNA expression was measured in BM cells by the PrimeFlow (ThermoFisher). Higher levels of expression of CD27 (TNFSF7) and CD38 on ASCs (red) were measured and compared with B cells (blue) and fluorescence minus one control (no probe, shaded) controls, which correlated with positive cell surface phenotype (Figure 4A). The CD19 RNA level was found to be low but clearly higher than background, whereas CD20 (MS4A) level was very low or negative on ASCs, but positive on normal B cells.35,36 High expression of XBP-1, SDC1, and PRDM1 was found only on ASCs, consistent with genes known to be required for ASC development and survival.15,37,38 Component analyses revealed that both BM CD19 ASC subsets were tightly clustered (Figure 4B) compared with BM B-cell subsets. Overall, the CD19+CD20+ B-cell population showed 7810 probe sets for 4780 genes, which were differentially regulated between the CD19− ASCs, and 7671 probe sets for 4604 genes regulated between the CD19+ ASCs. Similarly, the CD19+CD20− B-cell population showed 8188 probe sets for 4832 genes, which were differentially regulated between the CD19− ASCs, and 8382 probe sets for 5001 genes regulated between the CD19+ ASCs. The CD19+CD20+ B-cell population differed in gene expression with 992 probe sets for 659 genes, from the CD19+CD20+ B-cell population. Of the 54 000 gene probes examined, only 21 probe sets recognizing 14 distinct genes were significantly altered between the CD19+ and negative BM ASCs (Figure 4C). All genes from each FCM-sorted BM cell population were plotted in paired combinations where red points indicate genes with significant expression change and gray points correspond to genes without significant expression (Figure 4D). A heat map of the whole genome profiling displays differential gene expression between ASC cell subsets (Figure 4E). Pairwise comparison heat map showed common CDRH3 sequences shared between CD19+ and negative ASCs from 6 BM donors (Figure 4F). The CDR-H3 is a highly diversified sequence, and we found a low frequency of identical sequences, suggesting that CD19+ and negative ASCs can be clonally related.
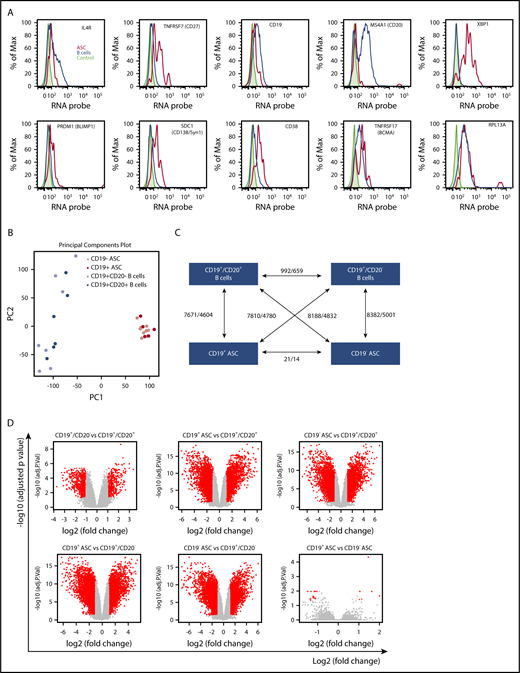
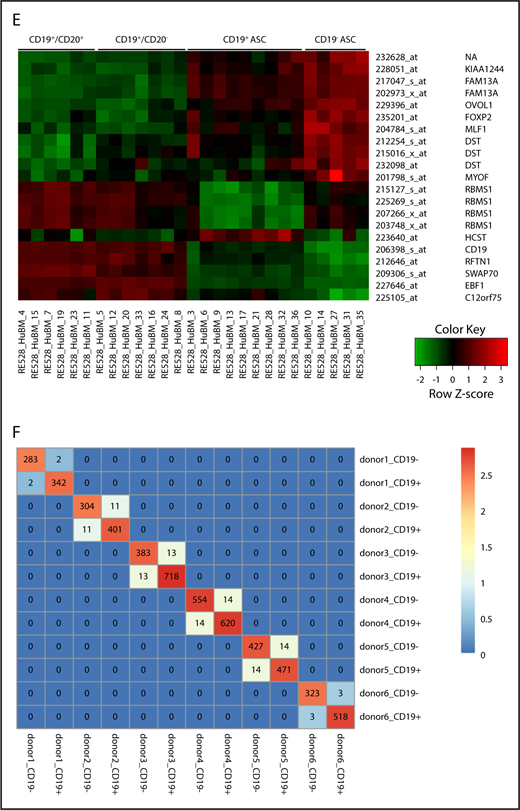
CD19+and negative BM ASC gene expression and sequence identity indicates a common differentiation path. (A) Known ASC genes (XBP-1, PRDM-1, SDC-1), B-cell genes (IL-4R, CD19, MS4A1), and phenotype genes (TNFRSF7, CD38) were assayed from freshly isolated BM. CD38highCD27+ gated cells showed increased expression of ASC genes and low levels of expression of B-cell genes by PrimeFlow. (B) Principal component (PC) analysis was performed on all genes based on relative expression from Affymetrix microarray (U133) for all 4 sorted cell populations from human BM. (C) Graphical representation showing the number of differentially regulated probes (upregulated/downregulated) for each cell population combination. (D) A volcano plot showing log2 (fold change) vs −log10 (adjusted P value) for each paired BM population comparison. Red dots indicate genes with significant expression change; gray dots indicate genes with no significant expression change. (E) Six genes with increased expression on CD19+ ASC and 8 uncharacterized genes with increased expression specific to CD19− ASCs. (F) Heat map of a pairwise comparison of common CDR-H3 sequences between donors. CD19+ and CD19− ASCs were profiled for 6 donors, and the sequences with identical CDR-H3 were considered as identical clones. The number in the heat map describes the number of common clones between 2 samples, and the number in the diagonal line showed the total number of CDR-H3 sequences in the sample. Max., maximum.
CD19+and negative BM ASC gene expression and sequence identity indicates a common differentiation path. (A) Known ASC genes (XBP-1, PRDM-1, SDC-1), B-cell genes (IL-4R, CD19, MS4A1), and phenotype genes (TNFRSF7, CD38) were assayed from freshly isolated BM. CD38highCD27+ gated cells showed increased expression of ASC genes and low levels of expression of B-cell genes by PrimeFlow. (B) Principal component (PC) analysis was performed on all genes based on relative expression from Affymetrix microarray (U133) for all 4 sorted cell populations from human BM. (C) Graphical representation showing the number of differentially regulated probes (upregulated/downregulated) for each cell population combination. (D) A volcano plot showing log2 (fold change) vs −log10 (adjusted P value) for each paired BM population comparison. Red dots indicate genes with significant expression change; gray dots indicate genes with no significant expression change. (E) Six genes with increased expression on CD19+ ASC and 8 uncharacterized genes with increased expression specific to CD19− ASCs. (F) Heat map of a pairwise comparison of common CDR-H3 sequences between donors. CD19+ and CD19− ASCs were profiled for 6 donors, and the sequences with identical CDR-H3 were considered as identical clones. The number in the heat map describes the number of common clones between 2 samples, and the number in the diagonal line showed the total number of CDR-H3 sequences in the sample. Max., maximum.
CD19+ and negative BM ASCs exhibit similar genetic mechanisms for survival
A heat map of the whole genome profiling displaying differential gene expression between 1 of the 4 cell subsets showed nearly identical genetic regulation within ASC subsets from the BM (Figure 5A). Clustered ASCs compared with B-cell gene expression suggested that both ASC subsets shared similar genetic mechanisms for survival with the following gene families: caspases, bcl-2, and cyclins. We showed that the expression of the caspase genes CFLAR and CASP10 were significantly elevated in ASCs compared with B cells in BM. In addition, CASP6 and CASP8AP2 showed a decrease in expression on the ASC subsets (Figure 5B). Expression of antiapoptotic bcl-2 family genes: MCL-1, BCL2L11, and BNIP, were significantly increased in ASCs. BIK was also enhanced in ASCs, but this difference did not reach statistical significance. BCL10 was significantly decreased in ASCs compared with B cells (Figure 5C). We observed a significant reduction in relative expression of CCNA2, CCNB1, CCNB1IP1, and CCNE2 on ASCs in a similar fashion compared with B-cell populations in BM. CCND2 was reduced in the CD19− ASCs but not significantly lowered in our results as those reported by Mei et al18 (Figure 5D).
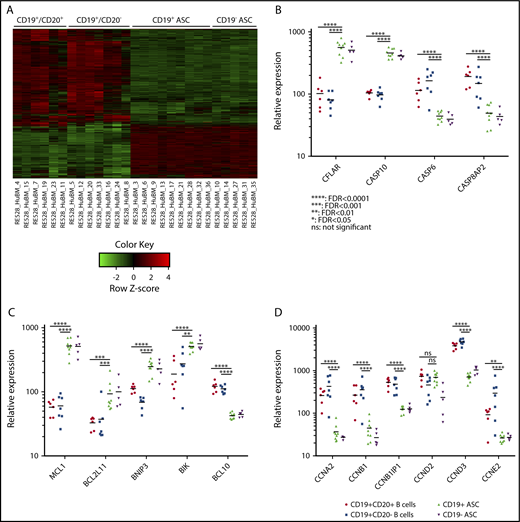
CD19+and CD19−BM ASCs exhibit similar genetic mechanisms for survival. (A) A heat map showing expression of genes with differential expression between 1 of any 2 BM cell populations. Most gene expression appears similar between ASCs and distinct from BM B-cell populations. (B) Scatter plot of 4 caspase-family relative gene expression between BM CD19+CD20+ B cells (circle), CD19+CD20− B cells (square), CD19+ ASCs (triangle), and CD19− ASCs (inverted triangle). (C) Relative gene expression of bcl2-family genes on BM cell populations. (D) Relative expression of cyclin genes on sorted BM cell populations. None of the scatter plots showed a significant difference between the CD19+ and CD19− ASCs.
CD19+and CD19−BM ASCs exhibit similar genetic mechanisms for survival. (A) A heat map showing expression of genes with differential expression between 1 of any 2 BM cell populations. Most gene expression appears similar between ASCs and distinct from BM B-cell populations. (B) Scatter plot of 4 caspase-family relative gene expression between BM CD19+CD20+ B cells (circle), CD19+CD20− B cells (square), CD19+ ASCs (triangle), and CD19− ASCs (inverted triangle). (C) Relative gene expression of bcl2-family genes on BM cell populations. (D) Relative expression of cyclin genes on sorted BM cell populations. None of the scatter plots showed a significant difference between the CD19+ and CD19− ASCs.
Discussion
Exploitation of humoral immunity through vaccination has undeniably been 1 of the single most important advancements to the practice of Medicine. Recent efforts have provided direct evidence for long-lived immunity found in human BM CD19− ASCs,17-19 but the role of the predominant CD19+ ASC subset remained unclear. Here we show that these cells exist in each tissue we studied: BM, spleen, tonsil, and PB, and were capable of providing long-lasting immunity in a similar fashion to the CD19− BM ASCs. CD19+ ASCs are targets of CD19-targeted CAR-T and antibody-drug conjugated therapies, but not CD20-directed therapy; therefore, they represent an important cell subset to consider to further improve cancer treatment strategies.
Our results revealed minor differences between ASC subsets based on surface antigen expression. The CD19+ ASCs could be further divided by HLADR, Ki67, and CD20 expression, whereas these antigens were not expressed on CD19− ASCs. The CD19+ ASCs in PB largely expressed the proliferation marker Ki67, indicating blasting cells or that a cycling or proliferating phenotype may be required for ASCs to enter or exist in the circulation. The tonsil CD19+ ASCs expressed low levels of CD20, but all other tissue ASCs were negative for CD20 (Figure 1). Although neither CD19+ or negative ASCs expressed HLA-DR in BM10,23,39 or spleen, CD19+ ASCs in the tonsil and PB could be further divided into either HLADR+ or negative subsets. Our results demonstrated that a CD38highCD27+ phenotype was a simple and robust measure of all BM and spleen IgG-secreting cells via ELISpot or cytoplasmic immunoglobulin detection. The absence of CD19− ASCs in PB and tonsil, combined with our finding that CD19+ ASCs are nearly threefold more frequent than CD19− ASCs in BM and spleen, demonstrated that they were the most abundant immunoglobulin-secreting cell subset in man.
In BM and spleen, the predominant immunoglobulin isotype produced by ASCs and secreted was IgG (Figure 2). Our results in BM agreed with Halliley et al17 but differ from Mei et al,18 which indicated a higher IgA secretion found in the CD19+ ASCs in BM. Significantly, the analysis of BM ASCs by cytoplasmic FCM revealed that the CD19+ ASCs contained a much greater number of IgG-secreting cells compared with CD19− ASCs. Therefore, the potential secreted immunoglobulin contribution to humoral immunity of CD19+ ASCs is greater than CD19− ASCs based on frequency and tissue distribution.
Long-lived immunity and the presence of high-affinity serum IgG antibodies specific for previously encountered antigens, like vaccine antigens, was described as existing for decades independent of memory B cells3,17,40,41 and was thought to be contributed only by CD19− BM ASCs. Our work shows both CD19+ and negative ASCs also exist in the spleen and have similar capability as the BM ASCs to recognize vaccine antigens (Figure 3). We found no significant difference in the frequency of BM vaccine-specific IgG-secreting cells for Daptacel between the CD19+ or negative fractions, but we did observe an increase in CD19− ASCs for Fluzone, as Mei et al18 reported. In addition, we identify CD19+ and negative ASCs-secreting IgG, which react to MMRII, Varivax, and IPOL vaccine antigens in the spleen. Interestingly, we show polio-specific reactivity from CD19+ ASCs from the tonsil. Our data indicate that long-lived serological memory in man is derived from CD19+ and negative ASCs, found in the BM, spleen, and CD19+ ASCs in the tonsil, signifying that protective serum IgG levels may be contributed from multiple ASC subsets, most of which are CD19+.
Principal component analysis of whole genome expression profiling allowed us to compare and contrast BM ASC subsets. The cluster of CD19+ and negative ASC genetic composition indicated that these cells had a similar genetic composition (Figure 4). Our finding of identical immunoglobulin sequences (Figure 4F) shared between CD19+ and negative ASCs from 6 separate BM donors indicated that ASCs share a similar developmental path as well as nearly identical messenger RNA expression. Comparative microarray analysis pointed to a nearly identical genetic composition where the difference between the 2 ASC subsets, out of 54 000 probes measured, was only 21 probes for 14 genes. CD19− genes: FAM13A, KIAA1244, OVOL1, FOXP2, MLF1, DST, MYOF, and RBMS1, were increased compared with CD19+ ASCs. These genes have largely unknown function and should be explored in greater detail for relevance to ASC function, survival, tissue restriction, or long life. The genes with increased expression found in CD19+ ASCs were HCST, CD19, RFTN1, SWAP70, EBF1, and C12orf75. CD19, HCST, and SWAP70 have been shown to signal directly, or aid in recruitment of PI3K signaling.42-44 CD19 is a major mediator of PI3K on B cells45 necessary for cell survival,46 function,47 chemotaxis, and homing,48 which we observed as a novel and unique characteristic of this ASC subset.
We examined the gene family regulation of caspases, bcl-2, and cyclins as a means of describing the genetic mechanisms for cell survival between BM CD19+ and negative ASC subsets. The caspase family member, CFLAR, was demonstrated as a key gene, which provided an antiapoptotic pathway49 where increased expression was identified in quiescent ASCs found in the spleen of rituximab-resistant idiopathic thrombocytopenic purpura patients.21 This antiapoptotic regulator had elevated relative expression on both BM ASC subsets, along with CASP10, which was previously shown to interact with CFLAR, important in blocking an autophagy-dependent cell death pathway in multiple myeloma.50 Both ASCs exhibited decreased expression of CASP6 and CASP8AP2, which may play a protective role from apoptosis and cell cycle inhibition51-53 (Figure 5). The ASC subsets show enhanced relative expression of MCL-1, found to be essential for ASC survival.54 Autophagy-inducing BH3-only genes: BCL2L11, BNIP3, and BIK, with roles in apoptosis and drug resistance,55 cancer pathogenesis,56 and DNA strand-break repair,57 had increased expression on both CD19+ and negative ASCs relative to B-cell subsets. Our observation of shared expression of bcl-2 family members may play an important role in ASC development and malignancy because autoimmune inflamed stromal cells could promote bcl-xl–dependent survival of B cells in vitro,58 and deregulation of bcl-xl and the cell cycle initiating gene c-myc have been implicated in the generation of ASC neoplasms in cotransgenic mice.59 We showed reduced ASC expression of the cyclin family genes: CCNA2, CCNB1, CCNB1IP1, and CCNE2, all of which play a role in cell cycle progression. Thus, both BM ASC subsets likely have equal capacity for cell cycle regulation. The only difference we found was between CD19+ and negative BM ASCs for CCND2. Although our results appeared to confirm Mei et al,18 we did not see a statistically significant difference between the CD19 ASC subsets. The similar messenger RNA expression of CD19+ and negative ASCs for caspase, and bcl-2, and cyclin family genes indicated both BM ASC subsets were long lived and thus provided novel insights to the genetic mechanisms determining humoral immunity.
The finding that CD19+ vaccine-specific ASCs outnumbered CD19− vaccine-specific ASCs could have great importance to CD19-directed cell therapy and vaccine development. The long-held belief that 80% to 90% of long-lived ASCs were found in the BM60 may need revision given the presence of both CD19 ASC subsets in the spleen and vaccine-specific ASCs found in the tonsil. As such, CD19+ ASC subsets add further complexity to our concept of humoral immunity. The CD19+ ASCs are essential partners in humoral immune memory, and their capability to signal through PI3K may allow these cellular effectors to migrate and concomitantly reside in multiple tissues. Our discovery of both CD19+ and negative ASC subsets in the spleen adds complexity to the theory of BM niche competition suggested by Amanna and Slifka60 and Radbruch et al61 as a requirement for long-lived serological immunity and extends it to the spleen and possibly other tissues. In addition, our findings challenged the presumption that CD19− ASCs own the BM niche as more abundant CD19+ ASCs exist in BM, spleen, and other tissues and point to a more complex makeup of immune protection in man.
A CD19-directed CAR-T study by Bhoj et al19 showed that depletion of CD19+ ASCs had a measurable effect on serum immunoglobulin levels following treatment, which, together with our data, led us to conclude the importance of CD19+ ASCs to humoral immunity, immune memory, and vaccine-derived immunity is much greater than previously thought. Our observations of BM and spleen CD19+ and negative ASCs, and Mei et al,18 who discussed the appearance of CD19− ASCs found at sites of inflammation in immune disorders, show the importance of ASC biology for safe and effective immunotherapy. Our discovery of shared immunoglobulin gene sequences between CD19+ and negative ASCs suggested these nearly genetically identical subsets were derived and then differentiated. As we increase our understanding of the complex mechanisms involved in humoral immunity, we may not only improve our vaccine development capabilities but also open a new era where these cells and their products, antibodies, can be identified, isolated, and manipulated in order to provide curative treatments to cancers and the plagues of our time.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE107683).
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Sepideh Farshadi, Ying (Kevin) Li, and Miguel Sanjuan.
Authorship
Contribution: C.J.G. designed, analyzed, and interpreted the majority of experiments; J.C. conducted, analyzed, and interpreted intracellular cytoplasmic FCM assays and cell sorting; R.G. conducted antigen-specific ELISpot assays; B.R. conducted, analyzed, and interpreted ELISpot assays and cell sorting; C.A.M. conducted all aspects of RNA purification and isolation, conducted microarray experiments, and interpreted results; R. Halpin and Y.S. prepared and processed samples for sequencing; J. Wang and J. Wu provided statistics and Informatics support and analyzed the gene expression and sequencing experiments, respectively; R.R. conducted cell sorting experiments and cell lysate preparation for array; C.J.G., under the guidance of Y.W., R.K., and R. Herbst, conceived, designed, and supervised the project; and C.J.G., J.C., Y.W., and R. Herbst wrote and revised the manuscript with input from all authors.
Conflict-of-interest disclosure: C.J.G., J.C., B.R., C.A.M., R.G., J. Wang, J. Wu, R. Halpin, Y.S., R.R., R.K., Y.W., and R. Herbst are/were full-time employees and shareholders of MedImmune/AstraZeneca.
Correspondence: C. J. Groves, MedImmune LLC, One MedImmune Way, Gaithersburg, MD 20878; e-mail: grovesc@medimmune.com.

