

Abstract
Factor VIII C domains contain key binding sites for von Willebrand factor (vWF) and phospholipid membranes. Hemophilic patients were screened for factor VIII C-domain mutations to provide a well-characterized series. Mutated residues were localized to the high-resolution C2 structure and to a homology model of C1. Of 30 families found with mutations in the C domains, there were 14 missense changes, and 9 of these were novel. Of the missense mutations, 10 were associated with reduced vWF binding and 8 were at residues with surface-exposed side chains. Six of the 10 mutants had nearly equivalent factor VIII clotting activity and antigen level, suggesting that reduced vWF binding could cause hemophilia by reducing factor VIII stability in circulation. When the present series was combined with previously described mutations from an online international database, 11 C1 and C2 mutations in patients with mild or moderately severe hemophilia A were associated with antibody-inhibitor development in at least one affected individual. Of these substitutions, 6 occurred at surface-exposed residues. As further details of the C1 structure and its interface with C2 become available, and as binding studies are performed on the plasma of more patients with hemophilic C-domain mutations, prediction of surface binding sites should improve, allowing confirmation by site-specific mutagenesis of surface-exposed residues.
Factor VIII circulates as a precursor to factor VIIIa, an essential cofactor for intrinsic factor X activation that is a critical early step in coagulation. The factor VIII gene is 186 kilobases (kb) and contains 26 exons.1,2 The transcribed protein contains a signal peptide and a mature sequence of 2332 amino acid residues. Domain structures from the amino terminus are A1-A2-B-A3-C1-C2. The carboxy-terminal 313 amino acids form 2 highly homologous C domains. The C2 domain, and possibly C1, contribute to von Willebrand factor (vWF) binding,3,4 which is essential for the stabilization of factor VIII in circulation. When thrombin activates factor VIII, factor VIIIa dissociates from vWF and is concentrated by binding to a phospholipid surface, where it interacts with factor IXa. Factor VIIIa enhances the rate of activation of factor X by factor IXa by more than 105-fold.5 Lipid binding involves the C2 domain6-8 and possibly other sites in the A3-C1-C2 light chain of factor VIIIa.9 The factor VIII C domains contain surface epitopes both for clinically significant alloimmune and autoimmune inhibitors of factor VIII and for monoclonal antibodies.10-13 C2 may also bind to factor Xa.14
A deficient factor VIII clotting activity leads to hemophilia A, a congenital bleeding tendency of variable severity that is due to distinct factor VIII gene mutations. Some hemophilic mutations lead to circulating dysfunctional proteins whereas others affect expression, secretion, or stability in circulation. Comparison of the baseline clotting activity with the antigen level in the plasma of hemophilic patients provides an estimate of the specific activity of their factor VIII and helps identify those mutations associated with a dysfunctional protein. An international database of hemophilic point mutations lists 48 missense mutations in the 2 C domains.15 Unfortunately, only a quarter of these C-domain mutations have had plasma factor VIII antigen levels reported; of these, only 2 circulate dysfunctional antigen.
Each of the factor VIII C domains has a single disulfide bond connecting the cysteine residues that are near the amino- and carboxy-terminal ends.16 Comparisons of sequences of human factors VIII and V show that the C domains share about a 45% sequence identity17; human factor VIII C1 is 7 amino acids shorter than C2 but still shares 42% identity.1 A comparison of factor VIII complementary DNA sequences from different species shows that predicted amino acid residues in the C1 domains are more than 90% identical and the C2 domains are around 80% identical.18-20 The C2 domain of factor VIII also shares 20% sequence identity with galactose oxidase, and a molecular model based on the crystal structure of galactose oxidase correctly predicted an 8-stranded β-sandwich core.21 Pratt et al22 reported the 1.5 Å crystal structure of a recombinant human C2 domain. This structure provided a basis for predictions of surface binding sites and generation of models to examine homologous portions of the C1 domain. Furthermore, the crystal structure allowed modeling of hemophilic missense mutations in both the C1 and the C2 domains, which can indicate residues that are important for function or stability of the protein.
In this study, unrelated hemophilia A families were screened for heteroduplex formation in the 7 exonic sequences coding for the 2 C domains of the factor VIII gene: exons 20 through 26. Thirty families had a heteroduplex band in 1 exon, and DNA sequencing identified a mutation in each. Twenty-two families had 14 distinct missense mutations, including 9 that have not been reported previously.15 Missense mutations were localized within the crystallographic structure of C2 and a homology model of the C1 domain; preliminary results were presented.23
Patients and methods
Patients and family members
DNA samples were from 76 unrelated families including patients or obligate carriers with hemophilia A not previously described. Those with severe hemophilia A were negative for gene inversions, recurrent arginine to stop codons, and gross deletions. This series did not include 82 families previously diagnosed with factor VIII gene inversions, 2 families with partial gene deletions, or 96 small mutations within the coding sequences for the A domains in 121 families, the majority of which have been published.24-26Samples were submitted for clinical carrier testing or obtained following informed consent under a protocol approved by the University of Washington Human Subjects Review Committee.
Factor VIII determinations
Factor VIII clotting activities were performed by one-stage assays.27 Antigen levels were measured by modification of an enzyme-linked immunosorbent assay (ELISA)26,28 with the use of a commercial factor VIII antigen kit with monoclonal antibodies (Immuno AG, Vienna, Austria).29 The commercial ELISA kit is sensitive to 0.2% of the pooled normal plasma level, and antigen was undetectable in plasmas from the 2 patients with frameshift mutations when they had no residual transfused factor VIII.
vWF binding ELISA
A factor VIII–vWF binding assay using a polyclonal antibody against vWF and monoclonal antibody against the factor VIII light chain was carried out to detect the vWF binding abilities of hemophilic plasmas.30-32 Briefly, microtiter wells were coated with rabbit anti-vWF (Sigma, St Louis, MO, diluted 1 to 8000 in 0.1 mol/L NaHCO3, pH 9.5, coating buffer) and incubated in 2% bovine serum albumin in phosphate-buffered saline (PBS). After washing with PBS and incubation with a 1:100-fold dilution of normal plasma as a source of vWF, wells were washed with 0.4 mol/L CaCl2 to remove bound plasma factor VIII and were reequilibrated in PBS. Dilutions of patients' plasmas, normal plasma, or a purified recombinant factor VIII standard were then added, and the bound factor VIII was detected with the same anti–factor VIII light chain monoclonal antibody (conjugated with horseradish peroxidase) that was used in the standard factor VIII ELISA, described above.
Polymerase chain reaction amplification conditions
Fifty to 100 ng of genomic DNA from different patients' leukocytes were used for polymerase chain reaction (PCR) amplification. DNA amplification was performed in 50 mmol/L KCl, 10 mmol/L Tris-HCl, pH 8.3, 5 mmol/L MgCl2, 200 μmol/L dNTPs (ATP, GTP, CTP, and TTP), and 0.5 μmol/L for each primer set (Table1). AmpliTaq DNA polymerase (1 to 2 units, Perkin Elmer, Foster City, CA) was added for each 100 μL reaction. PCRs were performed in a TwinBlock (Ericomp, San Diego, CA) or PTC-100 (MJ Research, Incline Village, NV) programmable thermal controller. Cycling parameters for the reaction were optimized for each exon. Amplified PCR products included both 5′ and 3′ intronic splice junctions and from 19 to 114 base pairs of intronic sequence (excluding the primer sequences), depending on the amount of published sequence and size of the exon. Intragenic polymorphic alleles were determined as before.25 26
Primers and PCR amplification conditions for factor VIII mutation screening
| Exon | 5′ Primer | 3′ Primer | Size (base pairs) | Annealing time (sec), °C | Extension,* sec at 72° |
|---|---|---|---|---|---|
| (primers listed from 5′ to 3′ sequence) | |||||
| 20 | ACG TTG AGT ACA GTT CTT GG | ACT AAT AGA AGC ATG GAG ATG | 320 | 30, 52° | 50 |
| 21 | GAA TTT AAT CTC TGA TTT CT | GAG TGA ATG TGA TAC ATT TC | 279 | 30, 52° | 50 |
| 22 | AAA TAG GTT AAA ATA AAG TGT TAT | TTA ATG GTA TGT AAT TAG TCA TTT A | 218 | 35, 43° | 50 |
| 23 | ACT CTG TAT TCA CTT TCC AT | AAC TAG AAC AGT TAG TCA CC | 258 | 40, 55° | 50 |
| 24 | ATA ACT GAG GCT GAA GCA TG | CTC TGA GTC AGT TAA ACA GT | 270 | 30, 54° | 60 |
| 25 | GAG TGA GAA GTG CTG TGG | TTG CTC TGA AAA TTT GGT CAT A | 381 | 30, 58° | 50 |
| 26 | GCT TTG CAG TGA CCA TTG TC | AGC TGA GGA GGG AGA GGT GA | 265 | 40, 60° | 50 |
| Exon | 5′ Primer | 3′ Primer | Size (base pairs) | Annealing time (sec), °C | Extension,* sec at 72° |
|---|---|---|---|---|---|
| (primers listed from 5′ to 3′ sequence) | |||||
| 20 | ACG TTG AGT ACA GTT CTT GG | ACT AAT AGA AGC ATG GAG ATG | 320 | 30, 52° | 50 |
| 21 | GAA TTT AAT CTC TGA TTT CT | GAG TGA ATG TGA TAC ATT TC | 279 | 30, 52° | 50 |
| 22 | AAA TAG GTT AAA ATA AAG TGT TAT | TTA ATG GTA TGT AAT TAG TCA TTT A | 218 | 35, 43° | 50 |
| 23 | ACT CTG TAT TCA CTT TCC AT | AAC TAG AAC AGT TAG TCA CC | 258 | 40, 55° | 50 |
| 24 | ATA ACT GAG GCT GAA GCA TG | CTC TGA GTC AGT TAA ACA GT | 270 | 30, 54° | 60 |
| 25 | GAG TGA GAA GTG CTG TGG | TTG CTC TGA AAA TTT GGT CAT A | 381 | 30, 58° | 50 |
| 26 | GCT TTG CAG TGA CCA TTG TC | AGC TGA GGA GGG AGA GGT GA | 265 | 40, 60° | 50 |
The first step is 93°C for 2 min for each exonic fragment followed by 35 cycles each of 1 min at 93°, annealing, and then extension.
Heteroduplex analysis
For heteroduplex formation, 5 to 7 μmol/L of amplified patient and normal fragments were mixed and then cycled through a mutation-detection enhancer (MDE) denature-annealing program: 94°C for 3 minutes, 80°C for 2 minutes, 70°C for 2 minutes, 65°C for 15 minutes, 50°C for 5 minutes, 40°C 2 minutes, and 37°C for 15 minutes. Standard loading dye (3 to 4 μL) was added to each mixture before loading. Electrophoresis was performed on 43 × 26-cm gels consisting of 1 × MDE gel solution (FMC Bioproducts, Rockland, ME) with 15% (wt/vol) urea in 0.6 × TBE buffer (1 × is 8.9 mmol/L Tris base, 8.9 mmol/L boric acid, and 0.2 mmol/L EDTA, pH 8.0) for 16 to 24 hours at 800 V at room temperature. The gels were stained with ethidium bromide to visualize heteroduplex bands.26 33
DNA sequencing
Amplified fragments were sequenced in both directions in an Applied Biosystems (Foster City, CA) automated sequencer, model 373, with the use of amplification primers. Separate amplified fragments were used to verify positive findings with either predicted restriction recognition site changes (in 16) or repeat sequencing (in 14) of the distinct mutations. For 10 of the 30 families in which a mutation was determined, verification was also obtained on a sample from an obligate carrier or another affected family member.
Structural analyses of the C1 domain and missense mutation sites
A model of factor VIII C1 domain was built and energy minimized with the use of the program Modeller (Molecular Simulations, San Diego, CA) based on a 1.5 Å resolution x-ray structure of the highly homologous C2 domain of the same molecule. The sequences of the C1 and C2 domains share 42% identity (85% homology) as shown in Figure 1. Sequence alignment by Bestfit program of Genebank and Align of Modeller showed that most hydrophobic residues in the β-sheet sandwich core of the C2 domain were conserved (Figure 1). Additional gaps in the sequences of both C1 and C2 were introduced in the above alignment to eliminate large distances (greater than 8 Å) between the Cα atoms of the C1 model and C2 template. The structures of loops corresponding to the gaps in Figure 1 were refined separately starting from the initial model generated by Modeller. Models with the lowest energy and best stereogeometry were selected for further refinement. The final model displayed no outliers in the Ramachandran plot produced by Procheck.37 Solvent accessibility of the side chains in the molecular models was analyzed with the use of the program Naccess,38 where values range from 0% (completely buried) to 100% (completely exposed) as compared with the same residue in a fully extended tripeptide sequence, “A-X-A” (A is single-letter amino acid code and X, the residue being considered).

Alignment of C domain sequences.
Amino acids are aligned by CLUSTAL W 1.7, GenomeNet. Factor VIII (VIII) sequences are as indicated for different species: human (h),1 porcine (p),19 murine (m),18canine (c)20; factor V(V) sequences are: human (h),17,34 murine (m),35 bovine (b).36 Bold residues indicate differences from human factor VIII, underlined are C2 residues that may bind to phospholipids, and boxes indicate a potential Asn-linked carbohydrate. HphA (above each human factor VIII sequence row) indicates hemophilic mutations; an asterisk is for those associated with an inhibitor (see Table 3). Shading is for β-strands in the human C2 crystal structure.22 In panel A, C1 homologous sequences are shown and β-strands are distinguished from those in C2 with a prime, with no β3′ nor β4′ (see Figure 2). In panel B, C2 homologous sequences are shown including a C2 inhibitor epitope as defined by Healey et al.13 Note that exact residues at the ends of β-strands are subject to interpretation as they often end within a residue (coordinates are available; see Acknowledgments).
Alignment of C domain sequences.
Amino acids are aligned by CLUSTAL W 1.7, GenomeNet. Factor VIII (VIII) sequences are as indicated for different species: human (h),1 porcine (p),19 murine (m),18canine (c)20; factor V(V) sequences are: human (h),17,34 murine (m),35 bovine (b).36 Bold residues indicate differences from human factor VIII, underlined are C2 residues that may bind to phospholipids, and boxes indicate a potential Asn-linked carbohydrate. HphA (above each human factor VIII sequence row) indicates hemophilic mutations; an asterisk is for those associated with an inhibitor (see Table 3). Shading is for β-strands in the human C2 crystal structure.22 In panel A, C1 homologous sequences are shown and β-strands are distinguished from those in C2 with a prime, with no β3′ nor β4′ (see Figure 2). In panel B, C2 homologous sequences are shown including a C2 inhibitor epitope as defined by Healey et al.13 Note that exact residues at the ends of β-strands are subject to interpretation as they often end within a residue (coordinates are available; see Acknowledgments).
Results and discussion
Mutation identification
Exon fragments coding for the C domains, exons 20 through 26 and flanking regions of the factor VIII gene, were examined in 76 families with hemophilia A. A single fragment with a heteroduplex band was found in 30 families. Direct sequencing revealed a single point mutation in each of these positive fragments. Among the 20 distinct mutations, 13 were novel, having not been reported to an international database of hemophilic factor VIII mutations.15 We found 6 non-missense mutations in 8 families with severe hemophilia A. Of these families, 2 had a single base microdeletion (A in codons 2184 to 2185 and T in codons 2139 to 2140) predicting frameshifts and premature termination. We found 4 nonsense mutations in the other 6 families (in codons W2111 [single-letter amino acid code], R2116 in 3 families each with a different haplotype, W2203; and R2209). Of these nonsense mutations, R2209 and one of the families with R2116 were previously reported.39 The remaining 22 families had 14 distinct missense mutations, and 9 of these were novel (Table2). Two of these mutations in C1 were associated with normal levels of dysfunctional factor VIII antigen, and an additional 3 in C2 had significant excess antigen over clotting activity, indicating at least partial dysfunction. vWF-bound antigen level was less than the total factor VIII antigen in all mutations except for 3: R2164, C2304, and T2320. In T2320, it was actually enhanced.
Newly reported hemophilic C-domain missense mutations
| Exon | Mutations (no. families) | Factor VIII (% normal) | Phenotype (severity) | ||
|---|---|---|---|---|---|
| VIII:C | VIII:Ag | VIII-vWF | |||
| C1 domain | |||||
| 22 | Q2087E† | 11 | 11 | 1 | Mild |
| 22 | R2090C† | 14 | 107 | 2 | Mild |
| 23 | R2150C† (3) | 33 | 30 | <1 | Mild |
| 23 | R2159C (2) | 20 | 104 | <1 | Mild |
| 23 | R2163C | 1 | 1 | <1 | Moderate |
| 23 | M2164R† | 2 | 2 | 3 | Moderate |
| C2 domain | |||||
| 24 | S2173I† (3) | 16 | 41 | 14 | Mild |
| 24 | A2201P† | 7 | 36 | 1 | Mild |
| 25 | V2232A† | 9 | 8 | 2 | Mild + Inh |
| 26 | P2300L (2) | 18 | 18 | 5 | Mild + Inh |
| 26 | R2304C (3) | 2 | 3 | 2 | Mod + Inh |
| 26 | R2304G† | 8 | 3 | <1 | Moderate |
| 26 | R2307Q | 10 | 10 | <1 | Mild |
| 26 | R2320T† | 5 | 20 | 100 | Moderate |
| Exon | Mutations (no. families) | Factor VIII (% normal) | Phenotype (severity) | ||
|---|---|---|---|---|---|
| VIII:C | VIII:Ag | VIII-vWF | |||
| C1 domain | |||||
| 22 | Q2087E† | 11 | 11 | 1 | Mild |
| 22 | R2090C† | 14 | 107 | 2 | Mild |
| 23 | R2150C† (3) | 33 | 30 | <1 | Mild |
| 23 | R2159C (2) | 20 | 104 | <1 | Mild |
| 23 | R2163C | 1 | 1 | <1 | Moderate |
| 23 | M2164R† | 2 | 2 | 3 | Moderate |
| C2 domain | |||||
| 24 | S2173I† (3) | 16 | 41 | 14 | Mild |
| 24 | A2201P† | 7 | 36 | 1 | Mild |
| 25 | V2232A† | 9 | 8 | 2 | Mild + Inh |
| 26 | P2300L (2) | 18 | 18 | 5 | Mild + Inh |
| 26 | R2304C (3) | 2 | 3 | 2 | Mod + Inh |
| 26 | R2304G† | 8 | 3 | <1 | Moderate |
| 26 | R2307Q | 10 | 10 | <1 | Mild |
| 26 | R2320T† | 5 | 20 | 100 | Moderate |
Bold Ag levels indicate dysfunctional protein; bold vWF levels, bound Ag, where significantly different from total Ag; Inh, high titer inhibitor in at least one of the reported patients with this mutation.
Factor VIII determined in patient plasma as clotting activity (VIII:C), total factor VIII antigen (VIII:Ag), or vWF-bound factor VIII antigen (VIII-vWF) and reported as percentage of clotting reactivity in a pool of normal donor plasmas.
Novel mutation, not previously reported in database.15
Homology model of the C1 domain
The final C1 model is similar to the C2 template with a root mean squared deviation (rmsd) of 0.9 Å between equivalent Cα atoms in the superposition of the 2 domains (Figure 2). Loop structures connecting the β-strands are highly similar except for 3 fewer carboxy-terminal residues in C1 than in C2, and 3 loops with length differences relative to C2, where the model may be less predictive of C1 structure. The loops connecting β5 to β6 and β13 to β14 are 2 residues shorter and longer in C1 than C2, respectively. A third loop where the C1 and C2 domain structures differ is at the first of 2 β-strand hairpins (β3-turn-β4) in C2 that has 4 residues fewer and only a β-turn in C1 (Figure 2).

Homology model of the factor VIII C1 domain.
The homology model of the C1 domain (red) was created as described in the text and is superimposed onto the crystal structure of the factor VIII C2 domain (blue) and is shown in stereo views. The locations of the 19 β-strand structures in C2 are indicated,22referred to as β′ strands in C1. The major differences are in the loop lengths (Figure 1), which are indicated by gray arrows. The loop corresponding to the β3-β4 hairpin structure within the C2 domain, a putative membrane-binding surface, is shortened to a turn in C1 (thus, there is no β3′ or β4′ strand in C1; homologous numbering of strands 5′ through 19′ are preserved). The β5-to-β6 loop is 2 residues shorter in C1. The loop between β-strands 13 and 14 is 2 residues longer in C1.
Homology model of the factor VIII C1 domain.
The homology model of the C1 domain (red) was created as described in the text and is superimposed onto the crystal structure of the factor VIII C2 domain (blue) and is shown in stereo views. The locations of the 19 β-strand structures in C2 are indicated,22referred to as β′ strands in C1. The major differences are in the loop lengths (Figure 1), which are indicated by gray arrows. The loop corresponding to the β3-β4 hairpin structure within the C2 domain, a putative membrane-binding surface, is shortened to a turn in C1 (thus, there is no β3′ or β4′ strand in C1; homologous numbering of strands 5′ through 19′ are preserved). The β5-to-β6 loop is 2 residues shorter in C1. The loop between β-strands 13 and 14 is 2 residues longer in C1.
For the C2 domain, a membrane-binding surface has been proposed that consists of the 2 tight β-hairpin turns that present M2199, F2200, L2251, and L2252 to solvent and a loop that presents V2223 on the same surface.22 These protruding hydrophobic residues are surrounded by an underlying cleft lined with basic side chains, consistent with anionic phospholipid membrane binding. In the C1-domain model, the loops corresponding to this surface are truncated and are less hydrophobic overall. F2093 is solvent-exposed at the homologous surface and is surrounded by basic side chains (R2090, R2159, R2163, and K2092). It remains to be established whether the C1 domain contacts the membrane surface when native factor VIIIa is bound. Modeling studies based on electron microscopy and 2-D electron diffraction of factors Va and VIIIa have been interpreted in favor of a bound orientation in which only the C2 domain contacts the membrane surface.40 41 However, a sterically reasonable model of a C1-C2 construct can be generated that would form a surface with the β-loops of C1 described above in the same plane as their counterpart hairpins in C2; therefore, C1 might also contact the membrane (B.L.S., unpublished observations, November 1999). Resolution of this issue awaits structural analyses of larger factor VIIIa constructs in the presence and absence of an associated membrane bilayer and structural data on the C1-C2 interface.
The C1-domain protein core is predominantly hydrophobic: out of 44 residues with less than 10% surface exposure, 34 (77%) are nonpolar, aromatic, or glycine residues. The remaining 10 residues are more polar, such as T2086, which is conserved in the structure of C2 (T2245). Other buried polar residues in C1 include 3 sites of hemophilic missense mutations: S2069, Q2087, and N2129. In comparison, the C2 domain core contains 47 residues with less than 10% surface exposure; of these, 36 (77%) are hydrophobic, aromatic, or glycine residues. Three of the remaining 11 buried polar residues (R2209, Q2246, and R2320) are sites of hemophilic missense mutations.
Hemophilic mutation localization
Clinical data and the factor VIII antigen levels (where available) for hemophilic missense mutations are listed in Table3, combining the current series (Table 2) with previously reported C-domain missense mutations.15 As shown, 57 missense mutations have been reported in the factor VIII C domains, corresponding to substitutions at 43 different residues. These sites were localized to the factor VIII C2 domain crystal structure and the C1 homology model (Figure 3). Missense mutation sites are evenly divided between the 2 C domains; together they occur at 14% of the C-domain residues. Figure 3 also distinguishes the severity of the clinical bleeding tendencies; only 20% are associated with clinically severe hemophilia A.
Molecular consequences of hemophilic C-domain missense mutations
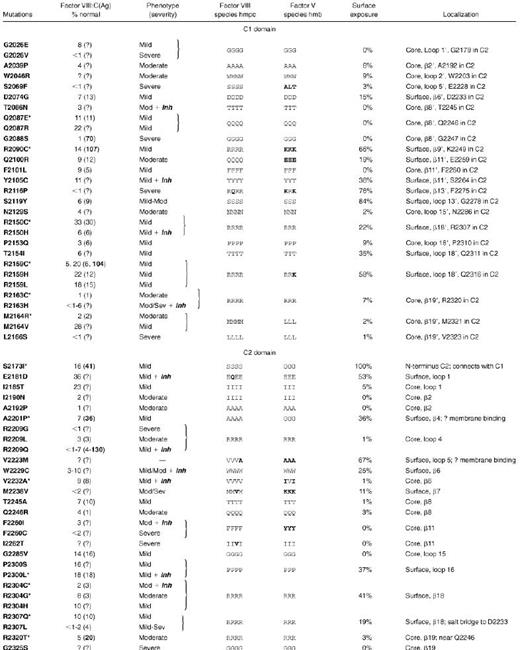 |
|
Mutations are from database15 and if marked by an asterisk, from Table 2. Factor VIII was determined in patient plasma as clotting activity (VIII:C) or total antigen (Ag) with bold for dysfunctional (excess) antigen; “?” indicates value not reported.15Inh, high titer inhibitor in at least one patient; β, beta strand (numbered) with loops numbered following strands. Factor VIII, homologous residues in h, human; m, murine; p, porcine; c, canine. Factor V, homologous residues in h, human; m, murine; b, bovine (see Figure 1 for sequence references).

Ribbon diagrams of the C domains: localization of hemophilic mutations.
Sites of hemophilic mutations (Table 3) are shown as spheres at the positions of their Cα atoms in the protein structure in stereo views. Spheres are color-coded according to the reported severity; lighter lavender represents mild to moderately severe, and darker violet represents more severe phenotypes (as reported for at least one affected individual). Core β-strands are green (foreground) and red (opposite side) ribbons with additional strands blue; each pair is oriented as in Figure 2. (A) The C1 model is based on homology modeling against the C2-domain crystal structure as described in Figure 2 and the text. (B) C2 is from the previously described crystal structure.22
Ribbon diagrams of the C domains: localization of hemophilic mutations.
Sites of hemophilic mutations (Table 3) are shown as spheres at the positions of their Cα atoms in the protein structure in stereo views. Spheres are color-coded according to the reported severity; lighter lavender represents mild to moderately severe, and darker violet represents more severe phenotypes (as reported for at least one affected individual). Core β-strands are green (foreground) and red (opposite side) ribbons with additional strands blue; each pair is oriented as in Figure 2. (A) The C1 model is based on homology modeling against the C2-domain crystal structure as described in Figure 2 and the text. (B) C2 is from the previously described crystal structure.22
Of the 12 missense mutations in which at least one reported individual was clinically severe, the native side chain has over 20% surface exposure in only one residue, R2116. Therefore, C-domain missense mutations associated with severe hemophilia A most often appear to destabilize protein folding and/or alter the trafficking and secretion of factor VIII by disrupting the protein core. One would predict that these mutations should be associated with very low factor VIII antigen levels; unfortunately, most of these cases have not had antigen levels reported. The possible effects of hemophilia A missense mutations are discussed below.The mutations were grouped according to the positions of the native residues in the protein core, in a putative membrane-associated surface, or at other positions on the protein surface.
Missense mutations in the protein core
At 25 of the 43 residues in C1 and C2 that are sites of missense mutations (Table 3), surface exposure is less than 10%, and these are classified as core positions. Of these 25 residues, 12 reported mutations at 11 sites replace the native side chains with bulkier groups, and only 3 of these are associated with mild hemophilic phenotypes. Factor VIII antigen levels were reported for 5 of these 12 mutations, and significant dysfunctional protein was found in 1: G2088S. Nine substitutions at 8 residues incorporate smaller side chains and may disrupt the core structure by altering internal hydrogen bonds or by causing cavities that decrease van der Waals contact stabilization of the protein core. Curiously, substitution of G2026 to E in C1 has a mild effect on protein function, while substitution of the same residue to V is associated with a severe bleeding tendency. Clearly, the effect of mutations in the protein core are generally more disruptive than surface mutations. Moreover, the effect of any one particular substitution is a function of the inherent flexibility and chemistry of the side chain's environment, as well as the backbone and side-chain torsion angles supported by the native and mutant residues.
Missense mutations at a possible membrane association surface
It is interesting to note that a relatively small percentage of the residues thought to be involved in membrane binding have been identified as mutation sites in hemophilia A patients. In the C2 domain, which is likely to be the primary anchor for membrane binding, only one mutation of a hydrophobic residue on this surface (V2223M) has been found in a hemophilic patient. Details of the phenotype for this mutation were not reported15; this same residue is alanine in canine factor VIII and in factor V (Table 3). In the C1 model, a hemophilic mutation site (R2159) is found on the same relative surface of the protein as V2223 and is significantly surface-exposed. Mutation of this residue to C, H, or L is associated with a clinically mild bleeding tendency. If this surface of the C1 domain within factor VIIIa is located near the membrane, this site may represent a basic residue that also makes electrostatic contact with phospholipid head groups.
Another mutation predicted to affect the membrane association surface of C2 is A2201P, at the beginning of the β4 strand. This mutation may alter the orientation of the preceding M-F2200 β-hairpin, a central feature of the proposed binding site.22 The fact that this patient also displayed a mild phenotype, and the general underrepresentation of hemophilic mutations in residues thought to interact with a phospholipid membrane, suggest that factor VIIIa generates binding energy to anionic lipid bilayers through a large number of relatively nonspecific interactions. In addition, unlike packing interactions in protein cores or at protein-protein interfaces, a change in the size or dimension of a single side chain is unlikely to cause an energetically costly steric clash against the lipid bilayer. Supporting evidence for this suggestion comes from the observations that in porcine factor VIII M2199 is I and in murine factor VIII L2252 is F. Furthermore, the residues homologous to M2199–F2200 in human factor VIII are W-W in the 3 factor V sequences reported (Figure 1). The structure of a human factor V C2 fragment also places these residues at a potential membrane binding site.42
Missense mutations across the protein surface
The remaining hemophilic missense mutation sites correspond to residues with side chains that are partially to extensively surface-exposed across regions separate from the putative membrane-binding surface. Amino-acid side chains having lower solvent accessibility will usually have significant core interactions. Thus, their substitutions could affect either core stability or a surface binding site. The interface between C1 and C2 remains to be defined and will undoubtedly involve burial of some sites listed here as surface-exposed. In light of these limitations, residues with greater than 10% surface exposure that are sites of mutations are identified in yellow in Figure 4, a van der Waals surface representation of each C domain. In the C1 model, 9 hemophilic mutation sites have 15% to 84% solvent accessibility (Table 3). Similarly, 9 exposed residues in C2 that are also sites of hemophilic mutations exhibit 11% to 100% accessibility (Table 3). The latter include A2201 and V2223 discussed under possible membrane association, above.

Van der Waals surface space–filled model of factor VIII C domains.
C1 (upper panels) and C2 (lower panels) are viewed in similar orientations to Figure 3A-B, with the left series being the “front” and the right, the “back” surface and where the “back” surface corresponds to a 180° rotation about the vertical axis. The domains are color-coded according to element type—carbon atoms are light green; oxygen, red; and nitrogen, blue—except that side chains of residues associated with hemophilia A point mutations have been colored yellow and labeled where visible. Residues that are sites of other hemophilic mutations that are either not shown or not labeled exhibit less than 10% relative solvent accessibility and are defined as buried in the hydrophobic core (Table3).
Van der Waals surface space–filled model of factor VIII C domains.
C1 (upper panels) and C2 (lower panels) are viewed in similar orientations to Figure 3A-B, with the left series being the “front” and the right, the “back” surface and where the “back” surface corresponds to a 180° rotation about the vertical axis. The domains are color-coded according to element type—carbon atoms are light green; oxygen, red; and nitrogen, blue—except that side chains of residues associated with hemophilia A point mutations have been colored yellow and labeled where visible. Residues that are sites of other hemophilic mutations that are either not shown or not labeled exhibit less than 10% relative solvent accessibility and are defined as buried in the hydrophobic core (Table3).
Regions of the factor VIII C-domain surfaces that are sites of hemophilic missense mutations, other than the proposed membrane-associated region, are likely to include surfaces that interact with other proteins such as vWF. Interaction surfaces between domains of native factor VIIIa, including A3-C1 and C1-C2 interfaces, might also be represented by clusters of such sites. Mutations of at least some of the residues that constitute protein-protein interaction surfaces should produce defects in protein-protein binding or affect factor VIII stability and function. In particular, disruption of vWF binding should lead to increased degradation and clearance of factor VIII from the circulation. Clustering of surface-exposed hemophilic mutations might be more likely to represent potential areas for these interactions.
In the surface representations of the C1 and C2 domains, 3 regions are particularly apparent as clusters of hemophilic mutation sites (Figure4, left panels). The first is localized to the C2 domain and is composed of R2307 and W2229. These residues are in immediate contact with one another, where the guanidino group of the R side chain makes a polar contact with the imidazole ring face of the W residue. These residues display approximately 10 nm2 of exposed surface area. Mutations of R2307 are associated with reduced vWF binding (Table2). R2304 is found within 1 nm of this cluster. A second cluster of hemophilic mutation sites occurs on C1 at surface-exposed residues: T2154, Q2100, and R2150. The mutation R2150H was associated with reduced vWF binding,43 as was R2150C (Table 2). Although this suggests that R2150 is involved in vWF binding, it is also possible that the native R side chain is buried in the C1-C2 interface. In the latter scenario, the substitutions at R2150 could have profound effects on the protein structure.
As striking as the surfaces described above are with respect to their exposed hemophilic mutation sites, the opposite sides of the C1 and C2 domains are notable for the relative paucity of such sites (Figure 4, right panels). Across these opposite surfaces, only the mutation E2181D in C2 occurs at a nonstructural, exposed residue that might be expected to interact with other macromolecules. This residue is a glutamine in murine factor VIII. Thus, the sides of C1 and C2 shown in the left panels of Figure 4 are primary candidates for sites of other interactions. These surfaces consist of the exterior of β-strands 6 and 18 in C2 and the corresponding region in C1.
A third cluster of polar surface-exposed residues is found near the N-terminus of the C1 domain. This cluster is composed of S2119, R2116, and Y2105. On the basis of the proximity of these residues to the amino terminus of the C1 domain, it seems plausible that this surface associates with the A3 domain of factor VIII. An A3-domain model, based on the structure of ceruloplasmin (see website15), predicts only 5 amino acid residues between the last β-strand of A3 and C2121 in C1. Further analysis and verification of this and other proposed binding surfaces awaits continued structural and biochemical studies.
Inhibitory antibodies
The development of inhibitory antibodies is a serious complication of factor VIII concentrate therapy that occurs in 10% to 30% of patients with severe hemophilia A and, as autoantibodies, in acquired hemophilia. Although uncommon, several cases of high titer inhibitors in mild or moderately severe hemophilia A patients have also occurred.44 The inhibitor antibodies bind predominantly to epitopes in the A2 and/or C2 domains of factor VIII11,13,28,44,45 and often interfere with the binding of factor VIII to vWF or to phospholipid.3,10,45-47 Phage display experiments indicated that significant inhibitor binding required 157 amino acids of C2, including its disulfide bond.48
In families with 11 hemophilic C-domain mutations, at least one affected individual developed an inhibitor. Of these mutations, 6 were at surface-exposed residues (Table 3, Figure 4). Thus, a bulky core residue substitution is not a prerequisite to alloimmune inhibitor formation. The substitution of W2229 to C was associated with development of an inhibitor in 7 of 18 affected individuals.44 W2229 is partially exposed on the surface of the β6 strand in C2 (Table 3) and, as discussed above, may be part of a binding surface. The antibodies of patients with less severe phenotypes usually neutralize the activity of native factor VIII as well as the mutant factor VIII that previously circulated at low but readily detectable levels. This converts the patient's hemophilia to a severe bleeding tendency. Over time, the inhibitors may predominantly neutralize native as opposed to the mutant factor VIII. One exception to this pattern of response was an inhibitor in a patient with a R2150H mutation. This inhibitor was first associated with allogeneic but not autologous inhibitory factor VIII antibodies that interfered with factor VIII–vWF binding, and was predicted to reduce factor VIII stability in circulation.42 In support of this prediction, the patient had comparable levels of factor VIII clotting activity and antigen. Similarly, a patient with a mutation to C2150 (Table 2) had an antigen level that was comparably reduced to his clotting activity.
An analysis of the effect of hemophilia A missense mutations in the factor VIII C domains has been recently appeared49and is based on previously published homology models against discoidin domains.21 These models, while correctly predicting the general fold of the β-sandwich cores of the C domains, exhibit substantial differences from the recently determined crystal structures of the factors V43 and VIII22 C2 domains (rmsd approximately 2 Å for backbone atoms in the protein core; rmsd greater than 5 Å in the peripheral loop regions). The published assignment of hemophilic missense sites as buried or exposed, based on the discoidin-based homology models,49 was compared with the same assignments based on the factor VIII C2-crystal structure (Table3). There was significant disagreement in the calculated surface area for approximately one third of the residues that constitute hemophilic mutation sites.
Structural definition of the factor VIII C2 domain22 has allowed identification of residues that contribute to exposed surfaces. By modeling the homologous C1 domain, one can tentatively localize similar areas, pending future structural studies on C1 and C1-C2. Surface areas suggest potential sites of interactions with phospholipid membranes and vWF. Furthermore, they should include epitopes for common immune responses to factor VIII. By localizing hemophilic mutations and comparing the phenotypes and, where available, properties of the hemophilic proteins, one can identify specific surface areas as candidates for future studies using site-specific mutagenesis. These may indicate potential therapeutic strategies, such as creation of a recombinant factor VIII that is relatively resistant to antibody inhibitors,50 and new approaches to antithrombotic therapy that target intrinsic factor X activation.
Acknowledgments
Dr John J. Peutz, St. Louis University, St. Louis, MO; Drs Frederick R. Rickles and Sidney Stein, Emory University, Atlanta, GA, for providing samples on a patient with an inhibitor; and members of the Fred Hutchinson Cancer Research Center's Structural Biology Program for their support. The coordinates for the homology model of the factor VIII C1 domain are available for download athttp://www.fhcrc.org/science/basic/labs/stoddard/coords.html. Requests for structural information should be directed to B.L.S. (bstoddar@fhcrc.org).
Supported in part by the American Heart Association Grants-in-Aid 9750412N (A.R.T.) and 0050336N (B.L.S.); by Public Health Service grants GM49857 and HL62470 (B.L.S.) and HL16919 (E.W.D.) from the National Institutes of Health; and by Baxter, Novo-Nordisk and Speywood for mutation screening (A.R.T.).
M.-L.L. and B.W.S. contributed equally to this study.
Reprints:Arthur R. Thompson, Puget Sound Blood Center, 921 Terry Ave, Seattle WA 98104-1256; e-mail: arthomps@u.washington.edu.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal