

Key Points
13.6% of adult AML patients harbor pathogenic/likely pathogenic germline variants, including 6.39% of patients with variants in clinically actionable genes, supporting the routine screening for germline variants in these patients.
Most commonly mutated genes are from DNA damage response pathway and variants were identified in new candidate genes including DNAH5, DNAH9, DNMT3A, and SUZ12.

Abstract
Inherited predisposition to myeloid malignancies is more common than previously appreciated. We analyzed the whole-exome sequencing data of paired leukemia and skin biopsy samples from 391 adult patients from the Beat AML 1.0 consortium. Using the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines for variant interpretation, we curated 1547 unique variants from 228 genes. The pathogenic/likely pathogenic (P/LP) germline variants were identified in 53 acute myeloid leukemia (AML) patients (13.6%) in 34 genes, including 6.39% (25/391) of patients harboring P/LP variants in genes considered clinically actionable (tier 1). 41.5% of the 53 patients with P/LP variants were in genes associated with the DNA damage response. The most frequently mutated genes were CHEK2 (8 patients) and DDX41 (7 patients). Pathogenic germline variants were also found in new candidate genes (DNAH5, DNAH9, DNMT3A, and SUZ12). No strong correlation was found between the germline mutational rate and age of AML onset. Among 49 patients who have a reported history of at least one family member affected with hematological malignancies, 6 patients harbored known P/LP germline variants and the remaining patients had at least one variant of uncertain significance, suggesting a need for further functional validation studies. Using CHEK2 as an example, we show that three-dimensional protein modeling can be one of the effective methodologies to prioritize variants of unknown significance for functional studies. Further, we evaluated an in silico approach that applies ACMG curation in an automated manner using the tool for assessment and (TAPES) prioritization in exome studies, which can minimize manual curation time for variants. Overall, our findings suggest a need to comprehensively understand the predisposition potential of many germline variants in order to enable closer monitoring for disease management and treatment interventions for affected patients and families.
Introduction
Acute myeloid leukemia (AML), a clonal disorder of hematopoietic progenitor cells, is one of the most common and fatal myeloid malignancies.1 Familial clustering of myeloid malignancies, including AML, has increasingly been recognized. Several constitutional syndromes have been associated with an increased susceptibility to AML, such as Fanconi anemia, dyskeratosis congenita, and Down syndrome.2,3 Such clinical associations have spurred interest in better defining the germline variants involved in increased susceptibility to myeloid malignancies. Since the first predisposing gene, RUNX1, was discovered twenty years ago,4 a growing number of genes have been found to be associated with germline predisposition to myeloid neoplasms, including ANKRD26, CEBPA, ETV6, GATA2, and DDX41.2,5,6,7
The World Health Organization recently introduced “myeloid neoplasms with germline predisposition” as a component of the myeloid neoplasms in its revised 2016 diagnostic classification scheme,8,9 which was subsequently adopted by other clinical guidelines.10,11 The diagnosis of a hereditary predisposition has significant clinical implications for the patient and his/her family, influencing therapeutic decisions such as donor selection for allogeneic hematopoietic stem cell transplant12,13 and the choice of transplant preparative regimens for the AML patients, proper genetic counseling and cancer surveillance for affected family members, and possible variant-informed therapeutic interventions. Early identification of AML predisposition is thus integral to providing optimal clinical care.
The goal of this study was to catalog the germline variants in cancer-susceptibility genes in a cohort of AML patients who were representative of those found in a typical adult leukemia clinic.
Methods
Patients and cohort
All patients involved in this study gave informed consent to participate, which was approved by the institutional review boards at 7 participating institutions.14 391 AML patients with tumor samples and matched skin biopsies were collected. Patient electronic medical records were accessed to obtain clinical, prognostic, pathologic, cytogenetic, and molecular genetic data (supplemental Table 1; supplemental Figure 1 available on the Blood Web site).14 The past medical and family histories of 324 AML patients were reviewed for evidence of familial clustering of hematological malignancies (HM) in first-, second-, or third-degree relatives.
Sequencing of tumor and constitutional material
Paired tumor and skin specimens were assessed using whole-exome sequencing as previously described14 and in the supplemental information.
Germline variants prioritization
High confidence germline variants, including single-nucleotide variants and small insertions and deletions, in 291 genes (supplemental Table 2) were called by VarScan2. The selection criteria for genes (5 tiers) and the parameters for variant calling are described in supplemental Methods. Variants were prioritized using these criteria: (1) variant allele frequency (VAF) at least 40% in both skin and tumor specimens, except for genes SAMD9 and SAMD9L in patients with aberrant chromosome 7, and (2) minor allele frequency <1% in normal populations (ExAC and gnomAD databases), except for the known founder variants in CHEK2, including c.1100delC, c.444 + 1G>A, p.Ile157Thr and p.Ser428Phe, were not subjected to the population frequency cutoff.15 The median range of VAF is within 40% to 60% for heterozygous status or 94% to 100% for homozygous (or hemizygous) status based on the assessment of common single nucleotide polymorphisms (supplemental Figure 2). Missense variants were prioritized by multiple in silico computational predictions of pathogenicity and functional impact (eg, SIFT, Polyphen, REVEL,16 DANN, CADD)17 with a REVEL score ≥0.5 or a CADD score ≥15.
Variant interpretation and classification
The prioritized germline variants were interpreted according to the 2015 American Society of Medical Genetics and Genomics (ACMG) recommendations18 by 2 geneticist/pathologist curators independently (supplemental Methods). To assess computational prediction, we developed a workflow using the tool for assessment and prioritization in exome studies (TAPES)19 (supplemental Methods).
Three-dimensional structural modeling of CHEK2 protein
A 3D structural model of CHEK2 was built as described in supplemental Methods.
Statistical analysis
The associations of categorical variables were examined using χ2 tests with the significance level set as P < .05. For testing the correlation between age of AML onset and the pathogenic/likely pathogenic (P/LP) germline variant detection rate, a Monte Carlo simulation was applied to the χ2 test. All the calculations were performed in R (v4.0.3).
Results
Patient characteristics and acquired genomic/genetic alterations
The clinical characteristics of this cohort of 391 patients are typical of adult AML patients seen in hematology/oncology clinics (Table 1; supplemental Table 1).9,20,21 The mean age of AML diagnosis was 59 years (interquartile range 51-71), with 59% of patients at least 60 years of age and 13% of patients younger than 40 years of age. Of the 321 patients whose cytogenetic report was available, 44% (142/321) had a normal karyotype for the available data. Within the 292 AML patients possessing molecular profiling, the most commonly somatically mutated genes were FLT3 (34%), NPM1 (32%), FLT3-ITD (25%), DNMT3A (16%), and IDH2 (15%) (supplemental Figure 3).20,21
Clinical characteristics of 391 patients in this analysis
| Characteristics at diagnosis | Value (%) |
|---|---|
| Age (years, mean and SD) | 59 ± 15 |
| Male | 221 (57) |
| Origin of AML | |
| de novo AML | 302 (77) |
| s-AML | 68 (17) |
| t-AML | 24 (6) |
| 2017 ELN risk | |
| Favorable | 99 (25) |
| Favorable or intermediate | 10 (3) |
| Intermediate | 78 (20) |
| Intermediate or adverse | 6 (2) |
| Adverse | 122 (31) |
| No information | 76 (19) |
| Cytogenetic aberrations | |
| Favorable | 33 (8) |
| Intermediate | 205 (52) |
| Normal karyotype | 142 (36) |
| Adverse | 84 (21) |
| Complex karyotype | 64 (16) |
| No information | 70 (18) |
| Characteristics at diagnosis | Value (%) |
|---|---|
| Age (years, mean and SD) | 59 ± 15 |
| Male | 221 (57) |
| Origin of AML | |
| de novo AML | 302 (77) |
| s-AML | 68 (17) |
| t-AML | 24 (6) |
| 2017 ELN risk | |
| Favorable | 99 (25) |
| Favorable or intermediate | 10 (3) |
| Intermediate | 78 (20) |
| Intermediate or adverse | 6 (2) |
| Adverse | 122 (31) |
| No information | 76 (19) |
| Cytogenetic aberrations | |
| Favorable | 33 (8) |
| Intermediate | 205 (52) |
| Normal karyotype | 142 (36) |
| Adverse | 84 (21) |
| Complex karyotype | 64 (16) |
| No information | 70 (18) |
Abbreviations: ELN, European LeukemiaNet; AML, acute myelogenous leukemia.
Percentages may not total 100 due to rounding.
Three patients are in both the s-AML and t-AML subgroups.
AML, acute myeloid leukemia; ELN, European LeukemiaNet; s-AML, secondary acute myeloid leukemia; t-AML, therapy-related acute myeloid leukemia.
Identification of pathogenic (P)/likely pathogenic (LP) germline variants
Using the ACMG criteria,18 4991 germline variants (1547 unique variants) from 228 genes that passed the prioritization filters were interpreted manually (supplemental Figure 1; supplemental Table 3). We found that 13.6% (53/391) of patients harbored pathogenic (P: 5.37%; 21 patients)/likely pathogenic (LP: 8.18%; 32 patients) variants and 64.45% (252/391) of patients had at least 1 variant of uncertain significance (VUS; Figure 1A).
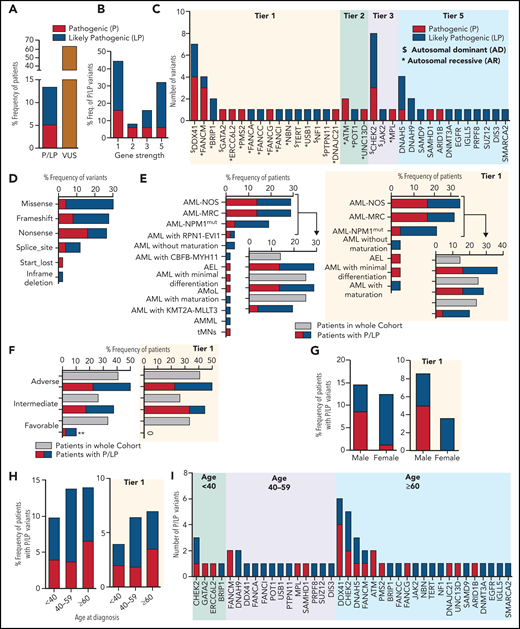
The pathogenic and likely pathogenic germline variants identified in 391 AML patients. (A) The percentage frequency of patients with pathogenic and likely pathogenic (P/LP) and variants of uncertain significance (VUS) in 391 patients. For patients 4039 and 2664, each has 2 germline variants, 1 in each P and LP category and thus counted once only in P category. (B) The percentage frequency of P and LP variants associated with gene strength for 50 unique variants. To define gene strength, 291 genes were selected for curation based on a comprehensive literature review. The strength of the genes is assigned from 1 to 5, depending on the evidence supporting their association with cancer predisposition to either hematological malignancies or other types of cancer, inherited hematological disorders that may predispose to hematological neoplasms, or reported as a somatic variant contributing to the pathogenesis of hematological malignancies. (C) Number of pathogenic and likely pathogenic germline variants in 34 genes for 56 variants in 53 patients, categorized by the gene strength. Variants in genes with “$” were associated with autosomal dominant conditions and those with asterisks were associated with AR syndromes. (D) Percentage frequency of variants classified by type of variants for 50 unique P/LP variants. (E-F) Association of P and LP variants with disease specific subgroups (E) or 2017 ELN risk categories (F) based on available information for 52 and 40 patients, respectively, compared with the whole cohort recruitment for those categories. P and LP variants identified in tier 1 genes only are shown on the right side. (G-H) Percentage frequency of P and LP variants classified by gender and age where denominator is the number of samples in that particular group within 391 patients. A subset of patients with germline P and LP variants identified in the tier 1 genes is presented in the graphs on the right. (I) Genes harboring number of P and LP variants in each of 3 age groups. ** indicates P value <.01. AEL, acute erythroid leukemia; AML NOS, acute myeloid leukemia, NOS; AML NPM1mut, AML with mutated NPM1; AML with CBFB-MYH11, AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11; AML with KMT2A-MLLT3, AML with t(9;11)(p22;q23); KMT2A-MLLT3; AML with RPN1-EVI1, AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1; AML-MRC, AML with myelodysplasia-related changes; AMML, acute myelomonocytic leukemia; AMoL, acute monoblastic and monocytic leukemia; tMNs, therapy-related myeloid neoplasms.
The pathogenic and likely pathogenic germline variants identified in 391 AML patients. (A) The percentage frequency of patients with pathogenic and likely pathogenic (P/LP) and variants of uncertain significance (VUS) in 391 patients. For patients 4039 and 2664, each has 2 germline variants, 1 in each P and LP category and thus counted once only in P category. (B) The percentage frequency of P and LP variants associated with gene strength for 50 unique variants. To define gene strength, 291 genes were selected for curation based on a comprehensive literature review. The strength of the genes is assigned from 1 to 5, depending on the evidence supporting their association with cancer predisposition to either hematological malignancies or other types of cancer, inherited hematological disorders that may predispose to hematological neoplasms, or reported as a somatic variant contributing to the pathogenesis of hematological malignancies. (C) Number of pathogenic and likely pathogenic germline variants in 34 genes for 56 variants in 53 patients, categorized by the gene strength. Variants in genes with “$” were associated with autosomal dominant conditions and those with asterisks were associated with AR syndromes. (D) Percentage frequency of variants classified by type of variants for 50 unique P/LP variants. (E-F) Association of P and LP variants with disease specific subgroups (E) or 2017 ELN risk categories (F) based on available information for 52 and 40 patients, respectively, compared with the whole cohort recruitment for those categories. P and LP variants identified in tier 1 genes only are shown on the right side. (G-H) Percentage frequency of P and LP variants classified by gender and age where denominator is the number of samples in that particular group within 391 patients. A subset of patients with germline P and LP variants identified in the tier 1 genes is presented in the graphs on the right. (I) Genes harboring number of P and LP variants in each of 3 age groups. ** indicates P value <.01. AEL, acute erythroid leukemia; AML NOS, acute myeloid leukemia, NOS; AML NPM1mut, AML with mutated NPM1; AML with CBFB-MYH11, AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11; AML with KMT2A-MLLT3, AML with t(9;11)(p22;q23); KMT2A-MLLT3; AML with RPN1-EVI1, AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1; AML-MRC, AML with myelodysplasia-related changes; AMML, acute myelomonocytic leukemia; AMoL, acute monoblastic and monocytic leukemia; tMNs, therapy-related myeloid neoplasms.
Of the 53 patients with P/LP germline variants, 56 variants (50 unique, including 17 P and 33 LP) in 34 genes were identified (supplemental Table 4). Forty-four percent (22/50) of the unique P/LP variants were in genes with established association with HM predisposition (tier 1, Figure 1B), 9 of which were in genes associated with autosomal dominant disorders, and the other 13 heterozygous variants were associated with autosomal recessive (AR) conditions (Figure 1C). A predicted loss of function variant (instead of gain of function) was identified in a tier 1 gene SAMD9. Thus, it is considered, in addition to the other 15 P/LP variants identified in the tier 5 genes, as a new candidate gene/allele for potential association with germline susceptibility to AML. The remaining 12 P/LP unique variants were identified in tier 2 and 3 genes. No P/LP germline variants were identified in tier 4 genes.
Among the tier 1 genes, the most frequently mutated gene was DDX41 (7 patients), followed by FANCM (4 patients) and BRIP1 (2 patients) (Figure 1C). Nineteen unique (23 total) P/LP variants from 22/53 (41.5%) patients were in 11 genes associated with the DNA damage response (DDR) pathway (supplemental Table 4; supplemental Figure 4), of which CHEK2 was the most frequently mutated gene, seen in 8 patients (Figure 1C). Overall, most of the P/LP variants were missense, nonsense, or frameshift variants (Figure 1D).
Correlation of specific diagnosis, monosomy 7, gender, or age of AML onset with P/LP germline variant detection rate
Seventy-seven percent (40/52) of patients with these P/LP variants were from 3 major disease subgroup categories, including AML with myelodysplasia-related changes, AML not otherwise specified, and AML with mutated NPM1, which is higher than the AML patients found within the whole cohort for these 3 categories (63.3%; 247/390) but is not statistically significant (P = .063) (Figure 1E). The proportion of patients with P/LP variants in the tier 1 genes is also predominantly from these 3 major disease subgroup categories (84%; 21/25) with a borderline statistical significance compared with the whole cohort (P = .05). Most of the P/LP variants were associated with AML cases that were categorized as adverse (21/40; 52.5%) and intermediate (15/40; 37.5%) risk according to 2017 European LeukemiaNet (ELN) criteria available for 40 patients (Figure 1F). However, AML patient recruitment for the whole cohort within these 2 categories was 40.8% (122/299) and 26.1% (78/299), respectively. The proportion of patients with P/LP germline variant in the favorable category was statistically lower compared with the AML patient recruitment for the whole cohort for this category (10% (4/40) vs 33% (99/299); P = .002). No AML patient harboring P/LP germline in the tier 1 genes was identified in the favorable category. Similarly, 5 of the 16 patients (31%) with monosomy 7 had at least 1 P/LP germline variant(s) identified, which is higher compared with the variant detection rate in patients without monosomy 7 (41/264; 15.5%) but is not statistically significant (P = .062) (supplemental Methods; supplemental Table 5).
We observed no significant difference in predominance of P/LP variants in males 14.5% (32/221) compared with females 12.4% (21/170) (Figure 1G). In the subset of patients with P/LP germline variants identified in the tier 1 genes, more male patients were observed (19/221; 8.6%), but this was not statistically significant (P = .056) compared with female patients (6/170; 3.5%). To determine the correlation of the P/LP germline variant detection rate with age at diagnosis of AML in our cohort, we stratified the patients into 3 age groups: <40 years, 40-59 years, and ≥60 years. The P/LP variant detection rates in these age groups were 9.8% (5/51), 13.8% (15/109), and 13.9% (32/230), respectively (Figure 1H-I), with no statistically significant difference (P = .783; 99% CI 0.768-0.798). The correlation with age was also not observed in the subset of patients with the P/LP germline variants identified in the tier 1 genes. However, DDX41 and CHEK2 variants are more prevalent in aging individuals.
Germline variants in genes associated with familial myeloid malignancies
The most frequently mutated gene in this category is DDX41, seen in 7 patients (Figure 2A). The median age of AML onset is 65 years, with a male-to-female ratio of 6:1 (Figure 2B). Among them, 2 patients (ID 1339 and 1374) had a family history of HM and/or solid tumors (Figure 2B-C), both harboring the known pathogenic variant M1I13,22,23; 3 patients (43%) had a personal history of solid tumor(s) (Figure 2B-C). Of the 5 patients who had an available metaphase cytogenetics at diagnosis, 4 (80%) were normal karyotype, similar to previously reported.24 One normal karyotype patient (ID 1339) had a low level cryptic del(5q) determined by fluorescence in situ hybridization (FISH). Interestingly, only 29% of the patients (2/7) have acquired mutations in DDX41 gene, lower than previously reported.24 Other somatically mutated genes included DNMT3A, TP53, SRSF2, FLT3, and STAG2 (Figure 2B).
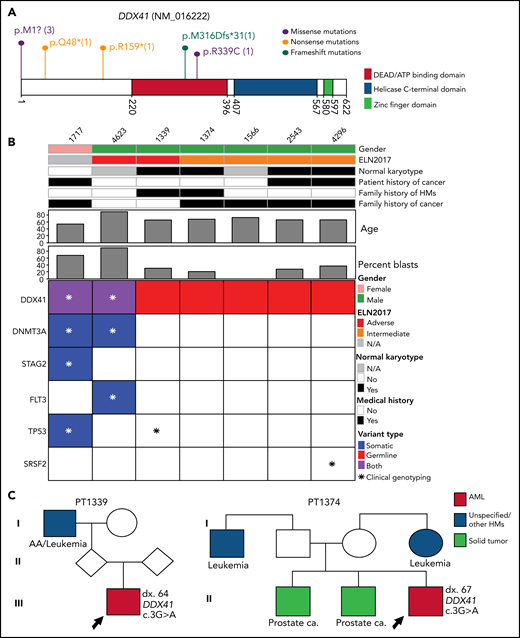
The pathogenic and likely pathogenic germline variants identified in DDX41. (A) Location of DDX41 P/LP variants, with the number of unique patients shown in parentheses. (B) Co-occurring somatic mutations as determined by clinical panel (*) and/or whole-exome sequencing (colored squares) for the patients with P/LP germline variants in DDX41. (C) Pedigree of 2 families with history of hematological malignancies and a P/LP germline variant identified in the DDX41 gene in the proband. The number designated by “dx.” indicates the age at diagnosis. The age at diagnosis is unknown for individuals if not indicated in the pedigree.
The pathogenic and likely pathogenic germline variants identified in DDX41. (A) Location of DDX41 P/LP variants, with the number of unique patients shown in parentheses. (B) Co-occurring somatic mutations as determined by clinical panel (*) and/or whole-exome sequencing (colored squares) for the patients with P/LP germline variants in DDX41. (C) Pedigree of 2 families with history of hematological malignancies and a P/LP germline variant identified in the DDX41 gene in the proband. The number designated by “dx.” indicates the age at diagnosis. The age at diagnosis is unknown for individuals if not indicated in the pedigree.
Patient 1378 (GATA2 T354M) presented with a subtype of AML with myelodysplasia-related changes and monosomy 7 at an early age of 21 years, consistent with previous reports.25,26 The somatic variants observed in this patient include RUNX1 and NRAS (Figure 3A). However, the patient’s clinical information is not available to assess for MonoMAC syndrome. This patient is also a carrier of a likely pathogenic variant in ERCC6L2. Patient 2664 (JAK2 E846D) was diagnosed with megakaryoblastic subtype (M7) AML, whose molecular profile of somatic mutations in MPL, TET2, and GATA1 (Figure 3A) is consistent with an underlying myeloproliferative neoplasm (MPN). This is the first report of the JAK2 E846D germline variant, previously described in a case of hereditary erythrocytosis with megakaryocytic atypia,27 in an AML patient. Of note, this patient is also a carrier of an ATM variant.

C-occurring somatic mutations in 53 patients with 50 unique P/LP variant(s). (A) Co-occurring somatic mutations for the patients with P/LP germline variants as determined by clinical panel and whole-exome sequencing. (B-E) Pedigree of 4 families with history of hematological malignancies and a P/LP germline variant identified in the genes TERT, MPL, DNAJC21, and CHEK2 in the proband, respectively. The number designated by “dx.” indicates the age at diagnosis, and that by “d.” indicates the age of death. The age at diagnosis or death is unknown for individuals if not indicated in the pedigree.
C-occurring somatic mutations in 53 patients with 50 unique P/LP variant(s). (A) Co-occurring somatic mutations for the patients with P/LP germline variants as determined by clinical panel and whole-exome sequencing. (B-E) Pedigree of 4 families with history of hematological malignancies and a P/LP germline variant identified in the genes TERT, MPL, DNAJC21, and CHEK2 in the proband, respectively. The number designated by “dx.” indicates the age at diagnosis, and that by “d.” indicates the age of death. The age at diagnosis or death is unknown for individuals if not indicated in the pedigree.
Germline variants in genes associated with HM-predisposing syndromes
A germline missense variant in TERT, R631W, was identified in a patient (ID 1552) who had a family history of HM in 4 first-degree relatives across 3 generations (Figure 3B; supplemental Table 6). This same variant has previously been reported in 1 three-generation family with dyskeratosis congenita, segregating with the disorder.28
Two patients were found carrying damaging germline variants in RAS regulatory genes with reported predisposition to juvenile myelomonocytic leukemia.29-31 Patient 1444 (PTPN11 G268S) had noticeable clinical features of short stature and prominent nasolabial folds that were consistent with Noonan syndrome. Patient 1224 (NF1 H781L) did not have an appreciable phenotype associated with neurofibromatosis. Another 2 patients (ID 1184 and 2010) were carriers of a germline loss-of-function variant in genes MPL and DNAJC21, respectively, which are responsible for AR disorders that cause inherited bone marrow failure and increased risk of transformation to myelodysplastic syndrome (MDS)/AML.32-34 The MPL variant carrier (ID 1184) did not have any blood count analysis prior to his diagnosis of AML but he had pancytopenia at the time of diagnosis. Unfortunately, both patients’ clinical phenotypes were limited or not ascertained for further evaluation of the syndromes; however, both of them have a family history of leukemia/lymphoma in their first-degree family members (Figure 3C-D; supplemental Table 6).
Germline variants in genes associated with DDR
Among the mutated DDR genes, CHEK2 was the most frequently mutated (8 patients; Figure 4A). The age of AML onset for these 8 patients ranged from 22 to 73 years. Sixty-three percent of these patients (5/8) had a reported history of other cancer, including prostate cancer, MDS, or MDS/MPN (supplemental Tables 4 and 6). One of the 2 AML patients in our cohort that harbored the most frequently reported pathogenic variant c.1229delC (aka. 1100delC in the literature) had HM reported in a first-degree relative (ID 4700; Figure 3E; supplemental Table 6). Patient 4039 (CHEK2 R160G) also had a heterozygous truncating variant identified in another DDR gene PMS2.
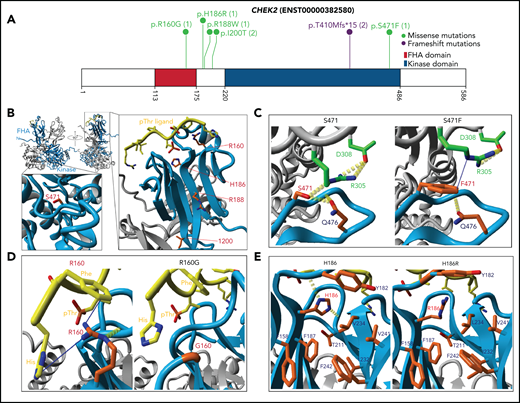
CHEK2 structural model for predicting impact of variants on protein functions. (A) Location of CHEK2 P/LP germline variants, with the number of unique patients shown in parentheses. (B) 3D CHEK2 dimer model with the functional domains (FHA and kinase domain), with respective monomers in light blue and gray, and a pThr ligand in yellow. Five residues affected by P/LP missense variants identified in this study cohort (red labels) are annotated in the extended windows. (C) The S471F in the kinase domain appears to abrogate interaction with residue R305 (intermolecular), and possibly residue Q476 (intramolecular), that destabilize the dimerization of CHEK2, reducing its efficacy of transphosphorylation and activation. (D) The R160G appears critical for stabilizing 2 N-terminal residues (H1 and F2) of the ligand by 2 Cation-Pi interactions. R160 engages in an inter-β-sheet H-bond which further stabilizes the FHA-ligand interface. Alteration to a glycine likely does 2 things to it: (1) abrogates interactions between the FHA domain and ligand and (2) entropically destabilizes the structure at position 160. (E) The H186R in the FHA domain disrupts the interactions with the polar cap and the hydrophobic core, destabilizing FHA domain structure. Additional stabilizing H-bonds (in yellow) are lost, which may be critical for ligand interface integrity and association.
CHEK2 structural model for predicting impact of variants on protein functions. (A) Location of CHEK2 P/LP germline variants, with the number of unique patients shown in parentheses. (B) 3D CHEK2 dimer model with the functional domains (FHA and kinase domain), with respective monomers in light blue and gray, and a pThr ligand in yellow. Five residues affected by P/LP missense variants identified in this study cohort (red labels) are annotated in the extended windows. (C) The S471F in the kinase domain appears to abrogate interaction with residue R305 (intermolecular), and possibly residue Q476 (intramolecular), that destabilize the dimerization of CHEK2, reducing its efficacy of transphosphorylation and activation. (D) The R160G appears critical for stabilizing 2 N-terminal residues (H1 and F2) of the ligand by 2 Cation-Pi interactions. R160 engages in an inter-β-sheet H-bond which further stabilizes the FHA-ligand interface. Alteration to a glycine likely does 2 things to it: (1) abrogates interactions between the FHA domain and ligand and (2) entropically destabilizes the structure at position 160. (E) The H186R in the FHA domain disrupts the interactions with the polar cap and the hydrophobic core, destabilizing FHA domain structure. Additional stabilizing H-bonds (in yellow) are lost, which may be critical for ligand interface integrity and association.
Fourteen patients in this study were carriers of other DDR genes, including those associated with Fanconi anemia35 (FANCM, FANCA, BRIP1, FANCC, FANCG, and FANCI, total 10 patients), ATM36 (2 patients), ERCC6L2 (1 patient), and NBN37 (1 patient). Unfortunately, for the majority of these patients, the relevant clinical information for the associated syndromes was not available, except patient 1919 (FANCG Q218*) who did not have noticeable congenital abnormalities or short stature or any personal history of other cancers. Heterozygous carriers of ATM and a subset of FA-related genes (such as FANCC and BRIP1) have been reported to have increased risk of breast cancer.38-41
Newly identified candidate genes/alleles
We identified novel P/LP germline variants based on the ACMG classification criteria; however, the contribution of these variants to the cancer susceptibility to AML needs to be established by further studies. These included variants in the genes that either are associated with predisposition to solid tumor (DNAH942 and EGFR43) or have somatic variants reported in HM (DNAH5, SAMHD1, ARID1B, DNMT3A, SUZ12, and PRPF8) (supplemental Table 4). The SUZ12 variant carrier acquired a somatic variant in the SUZ12 gene in the leukemic clone, consistent with the 2-hit model.
Gain-of-function germline alterations in SAMD9/SAMD9L, often missense variants, have been associated with the MIRAGE (myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy) syndrome and monosomy-7 MDS/AML in pediatric patients.44,45 No P/LP missense variants in SAMD9/SAMD9L have been identified in our cohort; however, we detected a SAMD9 nonsense variant in an elderly AML patient who had a history of prostate cancer. Recently, germline loss-of-function variants in SAMD9/SAMD9L have also been associated with adult onset MDS and BMF,45,46 and are implied as an underlying mechanism for adult onset MDS, as opposed to the gain-of-function variants for pediatric patients, a hypothesis that needs further investigation.
Using three-dimensional structural analysis for predicting functional impact of the proteins
Since CHEK2 variants were most prevalent in our cohort (Figure 4A), we used an in silico approach to predict protein functions of missense variants (Figure 4B; supplemental Figure 5A-B).47 The partial crystal structure of CHEK2 (PDB 3I6U) was used as a template, yielding a complete dimer including kinase and forkhead-associated (FHA) domains, unstructured loops, and a docked pThr ligand. S471 forms 2 stabilizing H-bonds with Q476 and R305 (intra- and intermolecular, respectively; Figure 4C). S471F results in a loss of H-bonding and hydrophobic repulsion, pushing R305 up and away from the interaction center of these residues. While F471 and R305 can theoretically engage in Cation-π interactions, the angle likely weakens the interaction compared with the H-bond lost by F471 variant. Ultimately, S471F destabilizes CHEK2 dimerization, reducing transphosphorylation and activation. An R160G variant reduces contacts between CHEK2 and pThr ligand, destabilizing the FHA-ligand interface (Figure 4D). H186 is found at the transition between the polar cap and hydrophobic core of the FHA domain. An H186R variant is likely disruptive due to the size and constitutive charge of arginine, perturbing the FHA core, deforming the ligand binding interface, and abrogating potential pH-dependent ligand interactions48 (Figure 4E). Similarly, I200T and R188W variants are predicted to confer a deleterious impact (supplemental Figure 5C-D; Table 2). The deleterious impact of these variants predicted by the protein modeling is consistent with the published results of the functional analysis (Table 2).47,49-59 These results serve as proof-of-concept and demonstrate that protein modeling helped in initial in silico predictions of protein function for CHEK2, and may be extended to other genes.
Comparison of CHEK2 variants characteristics as determined by protein modeling
| Mutation | Domain | Interaction(s) | Proposed impact | Functional data |
|---|---|---|---|---|
| S471F | Kinase/Dimer Interface | H-bonding | Abrogate interaction with residue R305 (intermolecular), and possibly residue Q476 (intramolecular) to destabilize the dimerization of CHEK2, reducing its efficacy of transphosphorylation and activation | Roeb et al, 2012 |
| R160G | FHA-ligand interface | Cation-Pi | The loss of the positively charged guanidinium group likely abrogates the Cation-Pi interactions with His and Phe | Wu et al, 2006; Delimitsou et al, 2019; Roeb et al, 2012; Chrisanthar et al, 2008; Sodha et al, 2006 |
| H186R | FHA domain | H-Bonding, Electrostatic | Introduction of bulky, constitutive charge within the core of the FHA domain, and removal of any potential for pH-dependent structural stability or ligand interaction. Similarly, H-bonding lost | Roeb et al, 2012; Walsh et al, 2011 |
| R188W | FHA domain | Electrostatic, Cation-Pi, Salt Bridge | Loss of positive charge, trans-β-sheet salt bridge and Cation-Pi interactions | Wu et al, 2001; Li et al 2002; Delimitasou et al, 2019 |
| I200T | FHA domain | Hydrophobic | Reduced hydrophobicity at a position proposed to be critical for hydrophobic driven dimerization and function | Falck et al, 2001a and 2001b; Kilpivaara et al, 2004; Li et al 2002; Roeb et al, 2012; Cai et al, 2009 |
| Mutation | Domain | Interaction(s) | Proposed impact | Functional data |
|---|---|---|---|---|
| S471F | Kinase/Dimer Interface | H-bonding | Abrogate interaction with residue R305 (intermolecular), and possibly residue Q476 (intramolecular) to destabilize the dimerization of CHEK2, reducing its efficacy of transphosphorylation and activation | Roeb et al, 2012 |
| R160G | FHA-ligand interface | Cation-Pi | The loss of the positively charged guanidinium group likely abrogates the Cation-Pi interactions with His and Phe | Wu et al, 2006; Delimitsou et al, 2019; Roeb et al, 2012; Chrisanthar et al, 2008; Sodha et al, 2006 |
| H186R | FHA domain | H-Bonding, Electrostatic | Introduction of bulky, constitutive charge within the core of the FHA domain, and removal of any potential for pH-dependent structural stability or ligand interaction. Similarly, H-bonding lost | Roeb et al, 2012; Walsh et al, 2011 |
| R188W | FHA domain | Electrostatic, Cation-Pi, Salt Bridge | Loss of positive charge, trans-β-sheet salt bridge and Cation-Pi interactions | Wu et al, 2001; Li et al 2002; Delimitasou et al, 2019 |
| I200T | FHA domain | Hydrophobic | Reduced hydrophobicity at a position proposed to be critical for hydrophobic driven dimerization and function | Falck et al, 2001a and 2001b; Kilpivaara et al, 2004; Li et al 2002; Roeb et al, 2012; Cai et al, 2009 |
Germline variants in patients with family history for HM
In our study, about 64.45% (252/391) of patients harbor at least 1 germline VUS without a P/LP variant. We used the available family history of prior cancer information from 324 AML patients to further prioritize VUSs. Among them, 15% (49/324) occurred in patients with reported history of at least 1 family member affected with HM, including AML, MDS, and lymphoma (Figure 5A; supplemental Table 6). P/LP germline variants were identified in 6/49 (12.2%) of patients (Figure 5B). Thirty-six out of 49 (73.5%) patients had at least 1 VUS. Mostly frequently occurring secondary variants are similar to typical AML cohort such as FLT3-ITD, NPM1, NRAS, RUNX1, and CEBPA (Figure 5C; supplemental Table 7). Among the VUS variants, FAT1 and FAT4 were frequently mutated (8 patients). Both genes belong to a family of FAT atypical cadherins and are known tumor suppressors with recurrent somatic variants in a variety of cancer types, including myeloid and lymphoid neoplasms. Six out of 8 (75%) of these patients had a first-degree relative(s) affected with HM. However, FAT genes appear tolerant to missense changes based on the constraint data in the gnomAD database. Additional studies are needed to evaluate the association of FAT genes to cancer susceptibility to AML.
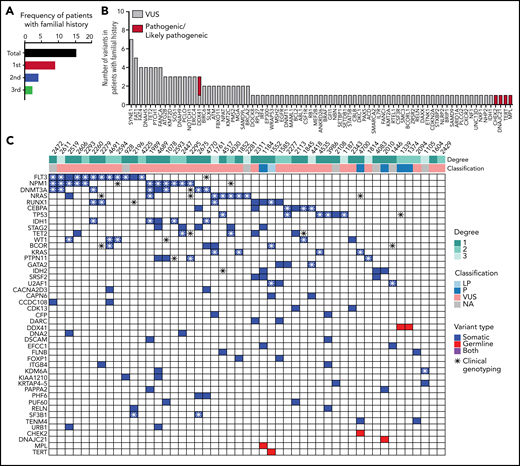
Forty-nine patients with familial clustering of hematological malignancies. (A) Familial history follow-up indicates that 15% of the patients have an affected first-degree, second-degree, or third-degree relative with a hematologic malignancy. (B) Number of pathogenic/likely pathogenic and VUS germline variants in patients with familial clustering. (C) Co-occurrence of somatic mutations in patients harboring germline variants and familial clustering. A full list of somatic and germline variants is included in supplemental Table 7.
Forty-nine patients with familial clustering of hematological malignancies. (A) Familial history follow-up indicates that 15% of the patients have an affected first-degree, second-degree, or third-degree relative with a hematologic malignancy. (B) Number of pathogenic/likely pathogenic and VUS germline variants in patients with familial clustering. (C) Co-occurrence of somatic mutations in patients harboring germline variants and familial clustering. A full list of somatic and germline variants is included in supplemental Table 7.
Automatic method in variant classification using the 2015 ACMG guidelines
The manual ACMG curation was performed only for the variants with high CADD and REVEL score. This manual curation is very cumbersome and time taking. Therefore, we developed an in silico approach that applies ACMG curation in an automated manner using TAPES19 to the 1532 unique germline variants manually interpreted (Table 3; supplemental Table 3; supplemental Methods). Our data show high specificity between manual and automated curation for assignment of non-P/LP classifications (99.1%, 1454/1466) as well as non-B/LB classifications (93.6%, 588/628) (supplemental Methods). However, ∼38.8% (19/49) P/LP variants were misclassified as VUS by the automated TAPES approach, mostly due to failure in applying the types of evidence that require human interpretation from the literature review, such as the functional studies, the mutational spectrum of the gene, and the segregation data. The lower sensitivity of this method (P/LP 61.2%, 30/49; B/LB 73.5%, 653/888) means its best use would be as a filter to deprioritize variants likely to be true negatives, thus saving time and streamlining tedious manual processes.
Comparison of manual and automated curation of variants using ACMG guidelines
| TAPES (automated curation) | |||||
|---|---|---|---|---|---|
| B/LB | P/LP | VUS | No call† | ||
| Manual curation | B/LB | 653 | 3 | 232 | 10 |
| P/LP | 0 | 30 | 19 | 0 | |
| VUS | 40 | 10 | 529 | 4 | |
| NA* | 71 | 0 | 498 | 2 | |
| TAPES (automated curation) | |||||
|---|---|---|---|---|---|
| B/LB | P/LP | VUS | No call† | ||
| Manual curation | B/LB | 653 | 3 | 232 | 10 |
| P/LP | 0 | 30 | 19 | 0 | |
| VUS | 40 | 10 | 529 | 4 | |
| NA* | 71 | 0 | 498 | 2 | |
TAPES (tool for assessment and prioritization in exome studies) was used for automated curation.
Predicted values where no review was carried out.
TAPES call where probability of pathogenicity disagreed with TAPES calls with Manual curation.
Discussion
Recent studies have demonstrated that germline cancer predisposition variants may not be uncommon, existing in 11% to 37% of families with hereditary MDS/AML.60-63 Most of these studies were based on small cohorts, a limited number of genes, pediatric patient population, or adults preselected by family history. The frequency of germline variants in unselected AML patients is therefore not well defined. Using a cohort of AML patients that is representative of those seen in a typical adult leukemia clinic, we found 13.6% of AML patients are carriers of 1 or more P/LP germline variants in genes associated with either cancer susceptibility or HM, similar to a recent report in elderly patients with de novo AML.64 To our knowledge, this is the largest AML cohort analysis to date that has prioritized germline variants from paired samples using gold standard curation for a large panel of genes. Among these 53 AML patients, 25 patients harbored P/LP germline variants in clinically actionable genes (tier 1) according to current guidelines, of which direct impact on clinical management would be considered in 11 patients with variants in genes associated with AD conditions (DDX41, GATA2, NF1, PTPN11, and TERT). For those carriers of genes associated with AR conditions, such as dyskeratosis congenital (POT1 and USB1) or Fanconi anemia, clinical evaluation for phenotype associated with the syndromes and/or further laboratory studies (copy number variation analysis, measurement of telomere length, or chromosomal breakage analysis) are warranted to establish the diagnosis. JAK2 germline variants have been associated with predisposition to MPN with incomplete penetrance.65 Thus the JAK2 carrier identified in our cohort may benefit from further segregation analysis. In addition, new candidate genes including DNAH962,66 and DNMT3A66,67 are identified, which were only recently reported in affected members of MDS/AML families. Germline nonsense variant in DHAH9 has been implied in cancer susceptibility to esophageal squamous cell carcinoma in one study, with loss of heterozygosity observed in one tumor.42 The constraint scores of DNAH5/9 based on the observed loss-of-function variants in the gnomAD population database support that these new candidate genes are not sensitive to haploinsufficiency, hence possibly require secondary alterations for the development of cancer. Further investigations including case-control studies and functional analysis are required to define the role for these new candidate genes or alleles in germline predisposition to AML.
The germline variants detection rate in our cohort was similar among patient groups with regard to age of leukemia manifestation or to gender. However, the 7 DDX41 carriers in our cohort are elderly patients with a median onset of AML ∼60 years of age and male-dominance, consistent with previous reports.22,24 In addition, only 15% (49) of patients in the study cohort had documented family history of HM, and only 12% of these 49 patients had a P/LP germline variant. Several factors could contribute to the low rates. Incomplete penetrance is noticeable in certain genes, such as CHEK2 or telomere disorder genes (TERT, TERC, and TINF2) where not every variant carrier develops clinical manifestations. Ascertainment bias could be another issue, as a comprehensive family history is frequently not taken, especially in adult clinics where there is low suspicion for inherited disorders. In addition, the late disease onset observed in certain genes (such as DDX41) may contribute to an unrevealing family history. Furthermore, our knowledge around germline predisposition to HM is still relatively limited, underscoring the importance of systemic screening for pathogenic germline variants in adult patients with myeloid disorders.
The most frequently mutated genes we observed were involved in the DNA damage response and repair pathways, particularly CHEK2 and genes associated with Fanconi anemia pathway, which cumulatively represented almost half of the P/LP germline variants. Pathogenic germline CHEK2 variants, in particular truncating variants, have been associated with a moderately increased risk of solid tumor (breast, prostate, and colorectal cancers) with reduced penetrance68-70 and recently with increased risk of HM including DLBCL, NHL, and MDS.71-73 Additionally, germline CHEK2 variants have been associated with the development of therapy-related myeloid malignancies.74,75 However, the 2 founder variants identified in our cohort, c.1229delC (aka 1100delC) and p.I200T (aka p.I157T), are relatively common in the general population and could be detected by chance (OR 1.21, P = .788, 95% CI [0.30, 4.86] for c.1229delC; OR 0.51, P = .336, 95% CI [0.12, 2.02] for p.Ile200Thr). Further studies are warranted to establish the clinical utility of these variants and the role of the CHEK2 gene in predisposition to AML. In addition, we identified novel genes that have roles in DNA repair, including ARID1B76 and SAMHB1.77 In general for pan-cancer, defects in DNA repair genes have long been recognized as the most commonly involved genetic event in hereditary cancers78-80 including HM.74,81 Therapeutic strategies to target DNA repair pathways have been actively explored as anticancer treatment, such as PARP inhibitor and compounds targeting transducers or effectors in DNA repair pathways.82 Furthermore, identifying these germline events can be very important in informing the choice of transplant preparative regimens for the AML proband. Cytotoxic hematopoietic stem cell transplant (HSCT) regimens may, for example, significantly increase the risk of solid tumors in FA patients.83
Many VUS were found in the majority of patients in our cohort who had reported family history of HM, underscoring the need for further studies to support their pathogenicity. Family segregation analysis can yield powerful data to reclassify a VUS; however, obtaining samples from family members for variant analysis has been a challenge for a retrospective study. In addition, using the CHEK2 as an example, we demonstrated that the structural analysis using a protein modeling approach may be useful in prioritizing VUSs for functional studies.84
Cultured skin fibroblast is considered the gold standard for constitutional tissue in the context of HM, with the caveat of being labor and time intensive, not routinely performed in all clinical diagnostic laboratories, and could occasionally be confounded by artifacts.85 In the current study, matched skin biopsy specimens were obtained at the incision site under anesthesia simultaneously during the bone marrow biopsy. There was a minimal contamination with blood in the washed skin tissues, as confirmed by the detection of low-level leukemia-associated somatic mutations (MeanVAF = 6.5%; supplemental Figure 6; supplemental Table 8) that could be distinguishable from the germline variants, suggesting this approach as a suitable alternative to skin fibroblasts.
Limitations of this study are that the intragenic insertions/deletions and copy number variations, alterations in the noncoding regions, as well as epigenetic events were not assessed. Some patients who had a heterozygous germline variant identified in an AR condition may harbor an alteration in the other allele that could not be detected, such as exon-level deletion in the Fanconi anemia genes or other genes (PMS2, NBN, DNAJC21, ATM, USB1, POT1, and MPL). In addition, certain mutational hotspots, such as that found in the 5'-UTR of the ANKRD26 gene or intron 4 of the GATA2 gene, were not evaluated. Therefore, the actual mutational rate could be higher. Potentially due to these reasons, some of the patients who have family member(s) affected with HMs may have genetic alterations that were not detectable with the current analysis. Furthermore, mosaic germline variants or somatic reversion of germline variants, such as in Fanconi anemia genes, could be missed in this study due to the filter criteria used. Lastly, being a retrospective study, there is incomplete information in the medical chart for assessing relevant clinical phenotypes associated with inherited disorders.
In summary, we observed 13.6% of adult AML patients harboring germline P/LP variants. Forty-four percent of these unique variants were in genes with established association with cancer predisposition to HMs. We also identified the variants in novel candidate genes for further investigation for their role in leukemia susceptibility. Overall, our findings highlight the importance of screening for germline variants even in elderly AML patients.
Acknowledgments
The authors thank all of our patients at all sites for donating precious time and tissue. DNA quality assessments, library creation, and short read sequencing assays were performed by the OHSU Massively Parallel Sequencing Shared Resource, as well as support from the Knight Diagnostic Laboratory and OHSU Clinical Pathology Core.
Funding for this project was provided by the Oregon Health & Science University Knight Cancer Research Institute (F.Y. and A.A.), School of Medicine Exploratory Research Seed Grants (F.Y.), and Cancer early detection advanced research center (A.A.) as pilot research projects. The project in part is supported by a Leukemia Lymphoma Therapy Acceleration Grant to B.D. and J.W.T. A.A. is supported by grants from the National Cancer Institute (R01 CA229875-01A1), National Heart, Lung, and Blood Institute (R01 HL155426-01), American Cancer Society (RSG-17-187-01-LIB), Alex Lemonade/Babich RUNX1 Foundation, EvansMDS Foundation, and V foundation Scholar award (A.A.).
Authorship
Contribution: A.A., S.M., F.Y., and N.L. provided project oversight for experimental design, data management, data analysis and interpretation, methods development; A.A., S.M., F.Y., N.L., T.A., D.B., T.L., J.S.-R., B.W., G.F., and R.D.P. helped with workflow development, data processing, management, and analysis; B.D., J.W.T., U.B., C.T., A.M.B., D.A.P., P.P., R.H.C., S.T., M.W.D., provided patient sample acquisition, clinical data structure, collection, and analysis; N.L., T.L., A.M.B., P.P., and S.T. provided curation and entry of patient clinical annotations; S.M. provided variant confirmation, and data analysis; D.B. assisted in workflow development for pre-processing and analysis of Exome-Seq and clinical data curation and integration, methods development; U.S. and J.C.S. performed protein modeling; and all of the authors wrote the manuscripts and provided feedback.
Conflict-of-interest disclosure: B.D. potential competing interests–SAB: Aileron Therapeutics, Therapy Architects (ALLCRON), Cepheid, Vivid Biosciences, Celgene, RUNX1 Research Program, Nemucore Medical Innovations, Novartis, Gilead Sciences (inactive), Monojul (inactive); SAB & Stock: Aptose Biosciences, Blueprint Medicines, EnLiven Therapeutics, Iterion Therapeutics, GRAIL, Recludix Pharma; Scientific Founder: MolecularMD (inactive, acquired by ICON); Board of Directors & Stock: Amgen, Vincerx Pharma; Board of Directors: Burroughs Wellcome Fund, CureOne; Joint Steering Committee: Beat AML LLS; Founder: VB Therapeutics; Sponsored Research Agreement: EnLiven Therapeutics; Clinical Trial Funding: Novartis, Bristol-Myers Squibb, Pfizer; Royalties from Patent 6958335 (Novartis exclusive license) and OHSU and Dana-Farber Cancer Institute (1 Merck exclusive license and 1 CytoImage, Inc. exclusive license).
Correspondence: Anupriya Agarwal, Knight Cancer Institute, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd, KR-HEM, Portland, OR 97239; e-mail: agarwala@ohsu.edu.
Beat AML Waves 1-2 are in the National Cancer Institute’s (NCI) Genomic Data Commons (GDC) (BEATAML1.0-COHORT; phs001657) and were published.14 We note the clinical annotation has been updated since the publication and original GDC submissions (supplemental Table 1). The genomic data for Wave 3 and clinical annotation for all waves is submitted to GDC (same accession) but its release is pending with the publication (Tyner et al in preparation).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal