

In this issue of Blood, 1 report on the germline screening of 391 patients from the BEAT acute myeloid leukemia (AML) study, finding that 13.6% of patients unselected by family history have pathogenic or likely pathogenic variants of known cancer-predisposition genes.
Germline predisposition to hematological malignancies (HMs) is a topic of increasing interest to the research community and increasing concern to clinicians, particularly with regard to the selection of hematopoietic stem cell donors from family members.2 Over the past 2 decades, family studies of the germline predisposition to myeloid neoplasms have increased our understanding of the genes known to underlie this predisposition. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia included a new category for myeloid neoplasms with germline predisposition.3-5 Despite these advances, genetic analysis of well-curated families who are predisposed to myeloid malignancy identifies known or potential predisposition candidates at ∼50%, suggesting that additional predisposition genes are yet to be identified.5 Likewise, the frequency of germline predisposition mutations in patients in a typical leukemia clinic is not known. Because most genomic sequence data in the latter context come from tumor sequencing at leukemia presentation, a range of recommendations have emerged for identification and prioritization of potential germline variants from tumor-only sequencing.1,6 It is also important to remember that tumor panels are not a replacement for dedicated germline screening.
In their study, Yang and colleagues tackled the frequency question by performing tumor/normal tissue sequencing of 391 patients from the BEAT AML cohort.1 Casting their net wide, they performed exome sequencing of skin fibroblasts and used a set of 291 genes associated with cancer predisposition, curated from guidelines and the literature. Variants were selected and prioritized based on parameters including variant allele frequency (>40%), population frequency (Minor Allele Frequency <1.0%), and predicted pathogenicity (eg, Combined Annotation–Dependent Depletion >15). This presented the investigators with the unenviable task of classifying 1547 unique variants from 228 genes, utilizing the American College of Medical Genetics and Genomics guidelines.7 They identified pathogenic or likely pathogenic (P/LP) germline variants in 13.6% of patients with AML (see figure). They then looked at this group in greater detail to identify clinical and phenotypic criteria (other than family history) with which to prioritize individuals for germline screening. There was no statistical association with AML subtype, sex, or age. Notably, no patients in this group fell into the favorable category based on European LeukemiaNet stratification.8 There were some exceptions to the above findings for patients with variants, in particular clinically actionable genes (found in 6.4% of patients). For example, DDX41 was the most recurrently mutated clinically actionable gene (7 patients; 1.8%); as seen in previous studies,9 P/LP variants were more prevalent in older individuals and expressed a marked male bias (6:1 males/females). Other familiar familial genes included singleton cases with germline P/LP variants in GATA2 and TERT and the RAS pathway genes PTPN11 and NF1.
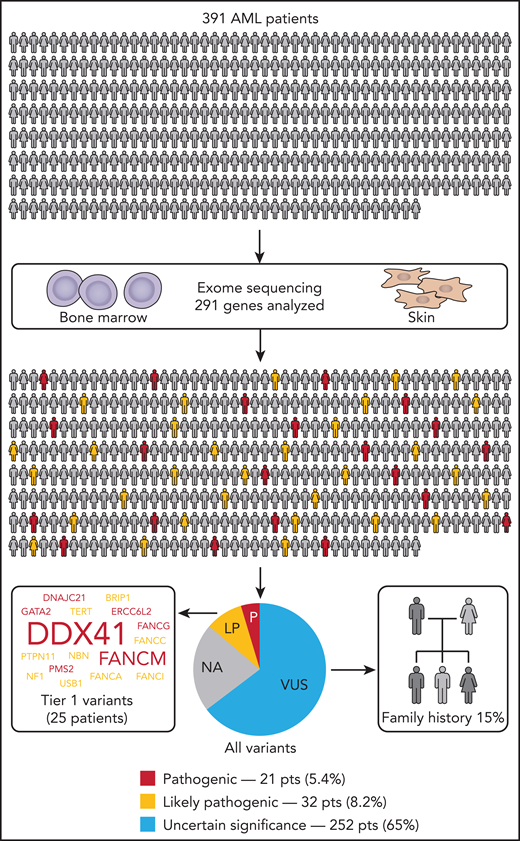
Germline screening for HM-predisposition variants in the Beat AML cohort identified 13.6% of patients with AML with P/LP variants classified according to American College of Medical Genetics and Genomics guidelines. Family history was also assessed independent of genomic findings, with 15% of assessed individuals positive for a family history of HMs. LP, likely pathogenic; NA, patients without variants in the other categories; P, pathogenic; pts, patients; VUS, variant of uncertain significance.
Germline screening for HM-predisposition variants in the Beat AML cohort identified 13.6% of patients with AML with P/LP variants classified according to American College of Medical Genetics and Genomics guidelines. Family history was also assessed independent of genomic findings, with 15% of assessed individuals positive for a family history of HMs. LP, likely pathogenic; NA, patients without variants in the other categories; P, pathogenic; pts, patients; VUS, variant of uncertain significance.
Moving beyond this small set of known myeloid-predisposition genes, an interesting pattern emerged within the remaining P/LP variants. Recognizing genes in a similar functional category, 22 patients were found to have variants in DNA damage response (DDR) genes, including 8 patients with P/LP variants in CHEK2 and other patients with variants in Fanconi anemia (FA) pathway genes (FANCM, FANCA, BRIP1, FANCC, FANCG, and FANCI) and other DDR genes. FA genes are well known for their increased risk of myeloid leukemia with an autosomal-recessive inheritance associated with FA.2 However, the potential for autosomal-dominant predisposition to myeloid malignancies due to heterozygous variants is an intriguing, but not well-understood, possibility. Segregation studies to look for high penetrance for myeloid malignancy predisposition for these genes may be difficult because variants in some of these genes also predispose to breast cancer and a range of solid tumor malignancies.2 These families may present with heterogeneous pan-cancer phenotypes, complicating segregation studies. Alternative evidence for risk of myeloid predisposition for some genes or alleles in the DDR pathway could be more amenable to careful case-control studies, because, collectively, they are identified more frequently than very rare myeloid predisposition gene mutations, such as in RUNX1, which was not identified as germline in any patients in this study. Understanding the role of DDR pathway mutations in HMs could lead to the extension of specific treatment approaches that are efficacious for this subgroup, such as PARP inhibitors, which, for unselected AML, have limited activity, at least in the single-agent trial setting.10
This study also highlights 1 of the principal challenges for clinical laboratories today. Of the 1547 variants curated, 252 patients (65%) had ≥1 variant classified as a variant of uncertain significance (VUS). Attempting to understand whether a VUS is vicious (truly pathogenic), vexing (unable to be resolved), or veiled (truly benign) often requires extensive follow-up, including de novo/segregation studies or functional studies.
Finally, this study made clear that routine germline genetic profiling of patients with leukemia could overcome some of the deficits of incomplete family history (due to biology [ie, incomplete penetrance] or missing information). Although, overall, their clinical information showed that 15% of individuals had a known family history of HMs, these overlapped, but did not intersect completely, with individuals who had P/LP germline variants in clinically actionable genes. As the field continues to move toward precision medicine in the diverse fields of diagnosis, prognosis, treatment, and monitoring for AML, this study adds to the accumulating evidence that including prospective assessment of the germline genetics in patients is an important consideration.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal