

In this issue of Blood, 1 describe a cohort of 24 children with Evans syndrome (pES) harboring a distinct signature of immune dysregulation: increased circulating follicular T-helper (cTfh) cells associated with chronic T-cell activation/exhaustion and decreased naïve CD4+ T cells and class switched memory B (CSMB) cells.
The seminal publication in 1951 by Robert Evans and colleagues titled “Primary thrombocytopenic purpura and acquired hemolytic anemia: evidence for a common etiology” led to the disorder being referred to as Evans syndrome (ES).2 Many astute and farsighted observations were made in 1940s and 1950s by Evans and others on the etiopathogenesis of immune-mediated peripheral destruction of red cells and platelets even before the structure of DNA and proteins like immunoglobulins were described.3,4
The first sentence of 1 publication about “acquired” hemolytic anemia read “It is now evident that the syndrome of acquired hemolytic anemia represents a distinct entity which is separate in pathogenesis and course from the commonly described familial hemolytic jaundice,” noted as distinct from what were then known as inherited red blood cell disorders such as thalassemia and sickle cell disease.5 Coombs et al6 described agglutination of red blood cells weakly sensitized to human Rh blood group antibodies observable under a microscope in the hemolytic disease of the newborn in 1945. In 1947, Evans and colleagues demonstrated antibodies (they called it a “fraction of the plasma protein, probably a globulin”) against red blood cells in “acquired” hemolytic anemia with antihuman globulin containing rabbit serum.7 However, Evans et al7 also lamented “Since there is no technic comparable to the reticulocyte count for estimating the rate of formation of thrombocytes or to the determination of the pyrrhole pigment excretion for measuring the rate of their destruction, it has not been possible to measure the rate of thrombocyte destruction or formation.” Alas, to this day, elucidating peripheral platelet destruction remains equally daunting for any student of hematology because there is no reticulocyte count or Coomb’s direct antiglobulin test for platelets analogous to the clinical tests readily available for the study of red cell fate in anemia. The paper dating back to 1951 from Evans et al reports their 29 patients with primary thrombocytopenic purpura and acquired hemolytic anemia divided into 5 groups with cross-relationships in a chart to surmise common etiology and familial inheritance patterns (see figure). In addition to the occurrence of 2 diseases in the same person, spontaneous remissions and exacerbations were also noted in many. The authors posited that the spleen is a site of antibody production, although not the sole organ at fault. Their cohort also included 1 instance of a father and daughter presenting with primary thrombocytopenic purpura, which led them to observe “the tendency to produce anti-thrombocyte antibody was inherited.”2 Incidentally the terms immune hemolytic anemia and immune thrombocytopenia currently in vogue were first coined by Evans, who eschewed use of the term “idiopathic” thrombocytopenic purpura as far back as in 1951. Known to his colleagues and friends as Bud Evans, he was a native Seattleite. Unfortunately, Evans was killed in a tragic car accident on Mercer Island in 1974 at the young age of 62.
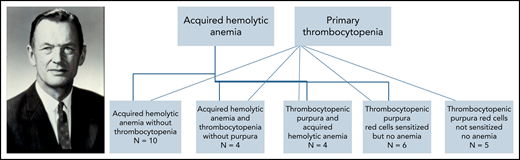
This figure was modified from Evans et al2 and shows their cohort of 29 patients with ITP and AIHA in 5 groups with spectrum-like cross-relationships in a chart to surmise common etiology and 1 example of familial inheritance pattern. Photograph of Robert Evans—credit: University of Washington School of Medicine (https://medicine.uw.edu/people/about-dr-evans).
This figure was modified from Evans et al2 and shows their cohort of 29 patients with ITP and AIHA in 5 groups with spectrum-like cross-relationships in a chart to surmise common etiology and 1 example of familial inheritance pattern. Photograph of Robert Evans—credit: University of Washington School of Medicine (https://medicine.uw.edu/people/about-dr-evans).
Today we have reached full circle identifying many inherited monogenic causes of what Evans believed to be “acquired” immune hemolytic anemia and immune thrombocytopenia.8 However, despite relatively easy and affordable access to panel-based next-generation genetic testing in 2021, we do not have a good genetic explanation in 50% to 60% of patients with ES to implicate their underlying pathology and use it for targeted treatments.9,10 In the cohort reported here by Kumar et al, only 12 of 24 patients had a pathogenic or likely pathogenic genetic variant. Hence, they propose a 4-point scoring system of biomarkers based on cTfh frequency, CD4 effector memory cell activation, frequency of naïve CD4+ T cells, and B-cell maturation defects seen as decreased CSMB cells noted in all 24 patients with pES, irrespective of their genetic etiology. However, this degree of obedient clustering of distinct immune profiles in patients with ES need to be reproducible in the hands of other investigators in larger cohorts. Most patients in their cohort of 24 had some degree of lymphoproliferation and pulmonary or gastrointestinal manifestations. These associations underscore the importance of looking for similar clinical clues in all patients presenting with suspected ES. These observations might help us better understand the pathobiology of pES and provide some rational and empirical basis for therapies guided by cellular phenotyping through immune profile-based scoring system, especially in the remainder of the patients with pES where no known genetic cause can yet be discerned. Today the eponymous syndrome named after Evans remains an omnibus placeholder clinical diagnosis just like another immune disorder known as common variable immune deficiency, where the main function is to help insurance providers code the patients for reimbursements. Complete and further diagnostic work-up of both adult and pediatric patients with ES can provide clues for treatments. This goal can be accomplished by multicenter collaborative clinical trials performing upfront genetic testing and cellular immunophenotyping of every patient presenting with multilineage autoimmune cytopenias as has been proposed by Kumar et al.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal