

In this issue of Blood, convincingly demonstrated the contribution of clonal hematopoiesis of indeterminate potential (CHIP) to the risk of chronic obstructive pulmonary disease (COPD).1 Their epidemiological data are supported with a transgenic mouse study showing that mice with Tet2-deficient hematopoietic cells, used as a model for CHIP, have increased airway inflammation and an increased risk for emphysema after various stimuli.
Landmark studies, using pooled available cohorts, revealed that clonal hematopoiesis (CH), which is characterized by mutant peripheral blood cells carrying ≥1 acquired variant of a known leukemia driver gene (eg, mutated DNMT3A, TET2), emerges during aging.2,3 The term CHIP was introduced to distinguish CH in a nonmalignant setting from malignant clonal hematopoiesis.4 By definition, CHIP requires a variant allele fraction ≥2% (ie,≥2% of the sequenced alleles bear a specific mutation that corresponds to roughly 4% of cells for heterozygous mutations). The prospective data from the above-mentioned landmark studies revealed that subjects with CH had an ∼10-fold increased risk of developing hematological cancer. However, this is probably an overestimation because the estimate was based on only 20 cases of hematological cancers (12 myeloid, 8 lymphoid) in individuals with CH, and only relatively large clones were detected in the studies. If smaller clones had been included, the incidence of CH would increase, and the relative risk would decrease.
Several studies have demonstrated that mutations in common genes associated with CHIP (like DNMT3A, TET2 and JAK2) can be found in immune cells and alter their function (see Jaiswal5 for additional details). This triggered the hypothesis that CH, whose incidence increases with age, is an important mediator of the chronic inflammatory state observed during aging (“inflammaging”).5 In recent years, the association between CHIP and inflammation in cardiovascular disease has been established. CHIP is associated with a hazard ratio ∼2 for incident coronary heart disease and ischemic stroke, which is similar to that reported for previously established risk factors like smoking, diabetes, hypertension, and dyslipidemia (see figure).5 Now, as shown by Miller et al, the clinical repercussions of CHIP also include COPD, another aging-associated inflammatory disorder. Although previous studies suggested an association between COPD and CHIP,6-9 this has been convincingly proven in a study that was specifically designed to assess this association. Miller et al compared a large number of COPD cases from 4 distinct cohorts (N = 8444), defined by quantitative spirometric measurements, Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria, and available comprehensive cigarette smoking data (including pack years), with >40 000 controls. Based on data from 48 835 included individuals, they found that CHIP was significantly associated with an increased incidence of COPD (hazard ratio [HR], 1.4; 95% confidence interval [CI], 1.2-1.6 for GOLD 2-4 COPD and HR, 1.8; 95% CI, 1.5-2.3 for GOLD 3-4 COPD). Thus, the impact of CHIP on the odds of GOLD 2-4 and GOLD 3-4 COPD is equivalent to 12 to 21 and 25 to 41 pack-years of smoking, respectively. Because smoke exposure by itself was associated with a small, but significant, increased risk for CHIP (odds ratio [OR], 1.03 per 10 pack-years), it is important to note that CHIP increased the risk of COPD and disease severity independent of age, cigarette smoke exposure, or an inherited polygenic risk score. The dedicated and well-annotated cohorts used by Miller et al are an important strength of their epidemiological study.
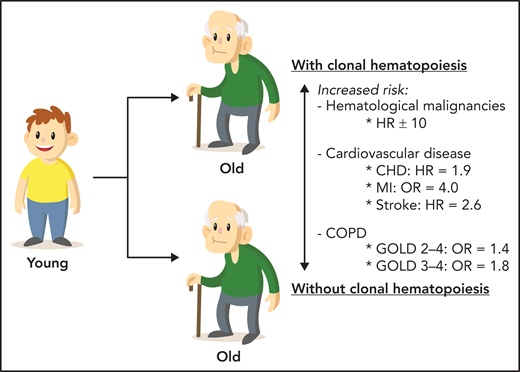
CHIP is a common age-related condition that represents a clonal prephase of hematologic neoplasms and is also an important risk factor for cardiovascular disease and COPD. CHD, coronary heart disease; MI, myocardial infarction.
CHIP is a common age-related condition that represents a clonal prephase of hematologic neoplasms and is also an important risk factor for cardiovascular disease and COPD. CHD, coronary heart disease; MI, myocardial infarction.
The presence of GOLD 3-4 COPD was significantly associated with mutations in DNMT3A (OR 1.7; adjusted 95% CI, 1.1-2.6), TET2 (OR, 2.4; adjusted 95% CI, 1.0-5.6) and, interestingly, TP53 (OR, 9.2; adjusted 95% CI, 1.5-57.7). Because TET2 was commonly mutated and independently associated with COPD in the human cohorts, the epidemiological data were probed using a Tet2-knockout mouse CHIP model. Exposing these mice to various inflammatory stimuli (cigarette smoke and polyinosinic-polycytidylic acid) showed that Tet2 loss in hematopoietic cells enhances pulmonary inflammation, increases interferon signaling, decreases tumor growth factor-β signaling, and accelerates emphysema. These findings provide additional confirmation for the hypothesis that aberrant immune cell function, caused by CHIP, can augment inflammatory stimuli and drive COPD.
In a broader context, it is tempting to speculate about a role for CHIP in other aging-associated inflammatory diseases. However, CHIP mutations are generally present in granulocytes and monocytes, occasionally in B cells, and rarely in T cells.5 Therefore, the impact of CHIP on inflammatory diseases in which T cells have a major role (eg, rheumatoid arthritis) may be limited. We also need to explore why only some individuals develop CHIP by assessing which extrinsic and intrinsic factors determine the growth of these clones. Clearly, future studies are needed to assess the role of CHIP in other inflammatory diseases and determine factors that trigger emergence and affect the clonal dynamics of CHIP over time.
Given the association between CHIP, especially in cases with mutated TET2, and inflammation, and more specifically with the secretion of interleukin-1β (IL-1β; the immediate upstream precursor to IL-6), it would be interesting to study clinically available IL-1β inhibitors (eg, canakinumab or anakinra) in patients with COPD who also have CHIP with mutated TET2. An exploratory analysis of the Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) revealed that canakinumab reduced the relative risk of major adverse cardiovascular events by 64% in patients with TET2 mutations and by 15% in all patients being treated.10 The potential benefits of personalization of IL-1β inhibitor therapy based on CHIP status is an intriguing, but untested, hypothesis in COPD.
In summary, the results of the study by Miller et al further support the possibility that age-associated chronic inflammation may be a key common factor in CH and aging-related inflammatory diseases (eg, cardiovascular disease and COPD) (see figure). Indeed, the potential of CHIP is gradually becoming less indeterminate.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal