

In this issue of Blood, have for the first time developed a preclinical model of T/myeloid mixed phenotype acute leukemia (MPAL) that faithfully recapitulates the human disease by using a fusion oncoprotein that they had previously identified in this leukemia subtype.1 This work provides important insights into the cellular origin and molecular pathogenesis of this disease and provides, for the first time, a preclinical model that can be used to test new therapeutics.
MPAL is a rare leukemia subtype with poor prognosis that is characterized by immunophenotypic features of both myeloid and lymphoid lineages. The majority of patients with MPAL have features of either B-cell and myeloid lineages or T-cell and myeloid lineages (T/myeloid MPAL).2 MPAL is particularly difficult to treat, in part because it is unclear whether MPAL should be treated as acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), or a combination of both.3 The cell of origin and molecular basis of this disease, in particular the causes of its biphenotypic nature, are unclear. This in turn makes optimal clinical care equally unclear.
Recent sequencing studies have defined the molecular landscape of T/myeloid MPAL and identified alterations in transcriptional regulators as a defining feature of this disease, including recurrent mutually exclusive alterations or deletions in WT1, ETV6, RUNX1, and CEBPA, which are complemented by mutations in epigenetic regulators and activating mutations in signaling pathways.4,5 This has also revealed that this disease is distinct from T-cell ALL (T-ALL) and AML but shares significant molecular and genomic similarity to early T-cell precursor-like ALL (ETP-ALL), another subtype of immature leukemia with poor prognosis.4,6
The ETS variant 6 (ETV6) gene is a member of the ETS family of transcription factors that encodes a transcriptional repressor. In addition to ETV6 loss-of-function mutations in T/myeloid MPAL, several ETV6 fusion oncogenes have been identified.4,7 One of these, ETV6-NCOA2, which fuses ETV6 with the coactivator NCOA2, was first identified in 6 patients with T/myeloid MPAL and is associated with activating mutations in the key T-ALL oncogene NOTCH1.7
To determine the function of ETV6-NCOA2, Fishman et al ectopically expressed this fusion oncoprotein in murine hematopoietic stem and progenitor cells (HSPCs). Strikingly, this induced immature lymphoid genes in these cells, even when they were cultured in myeloid conditions, which implies an instructive role of this oncogene in driving lymphoid differentiation. Notably, this lymphoid program was not induced in cells transduced with KAT6A-NCOA2 (also known as MOZ-TIF2), an NCOA2 fusion oncogene associated with AML. This implies that the differential targeting of NCOA2 by ETV6 vs KAT6A DNA-binding domains is important in driving the lymphoid vs myeloid phenotype of leukemias containing these fusion oncoproteins. Indeed, when transferred in vivo, ETV6-NCOA2–transduced HSPCs induced T/myeloid MPAL that was highly similar to the human disease and was accompanied by secondary mutations in Notch1 (see figure). Experiments performed in human HSPCs showed similar results, with ETV6-NCOA2 driving a lymphoid gene expression program. In this setting, ETV6-NCOA2 could cooperate with nontransforming NOTCH1 mutations to drive T/myeloid MPAL in human cells, which showed remarkable similarity to human T/myeloid MPAL xenografts that carried ETV6-NCOA2 mutations. This confirms that ETV6-NCOA2 cooperates with NOTCH1 mutations in human T/myeloid MAL and that NOTCH1 is important in driving the leukemia phenotype.
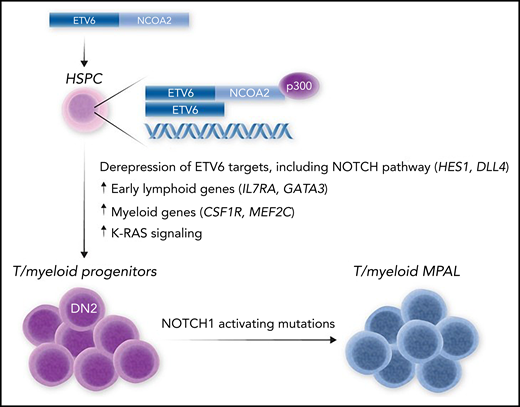
Model for T/myeloid MPAL induced by ETV6-NCOA2. Expression of the EVT6-NCOA2 fusion oncoprotein in HSPCs leads to formation of an aberrant transcriptional complex via binding to wild-type ETV6 and the transcriptional coactivator p300. This causes de-repression of ETV6 target genes, including NOTCH targets (HES1 and DLL4) and early lymphoid genes (IL7RA and GATA3) that promote lymphoid commitment while maintaining the expression of myeloid regulatory genes (CSF1 and MEF2C) that block further lymphoid differentiation. This causes differentiation arrest of multipotent T/myeloid progenitors, which accumulate further mutations, such as activating NOTCH1 mutations, that result in overt T/myeloid MPAL.
Model for T/myeloid MPAL induced by ETV6-NCOA2. Expression of the EVT6-NCOA2 fusion oncoprotein in HSPCs leads to formation of an aberrant transcriptional complex via binding to wild-type ETV6 and the transcriptional coactivator p300. This causes de-repression of ETV6 target genes, including NOTCH targets (HES1 and DLL4) and early lymphoid genes (IL7RA and GATA3) that promote lymphoid commitment while maintaining the expression of myeloid regulatory genes (CSF1 and MEF2C) that block further lymphoid differentiation. This causes differentiation arrest of multipotent T/myeloid progenitors, which accumulate further mutations, such as activating NOTCH1 mutations, that result in overt T/myeloid MPAL.
Although ETV6 encodes a transcriptional repressor, ETV6-NCOA2 fuses the ETV6 N-terminal pointed domain that mediates ETV6 dimerization with the activation domains of NCOA2, implying that this fusion functions to de-repress ETV6 target genes (see figure). Indeed, Fishman et al show that this oncoprotein binds to both wild-type ETV6 and the coactivator p300, forming an aberrant transcriptional complex that binds and de-represses key ETV6 targets, including the NOTCH1 pathway activators HES1 and DLL4. This also leads to upregulation of early lymphoid genes such as IL7RA and GATA3 that may be important in driving lymphoid specification of HSPCs. However, expression of key myeloid regulators is maintained, which results in a block in T-cell development at the DN2 stage, when these genes are normally downregulated to facilitate T-cell commitment and further T-cell development8 (see figure). These genes included CSF1R and MEF2C, which is known to be an important driver of ETP-ALL.9
Together, these results invoke a model whereby mutations that affect transcriptional regulators initiate T/myeloid MPAL in early hematopoietic progenitors. This is consistent with recent genomic analyses of MPAL, which showed that initiating mutations are acquired in early hematopoietic progenitors that retain myeloid and lymphoid potential.4 In the case of ETV6-NCOA2 fusion, the result is to invoke a transcriptional program that causes lymphoid specification, whilst maintaining the expression of key myeloid genes that sustain myeloid potential and block further T-cell differentiation (see figure). This developmental arrest sets the stage for further mutations, such as in NOTCH1, that result in overt leukemogenesis. It remains to be shown whether this model applies to other ETV6 fusion oncogenes that have been found in T/myeloid MPAL,4 to ETV6-inactivating mutations that have been found in both T/myeloid MPAL and ETP-ALL,4,10 and to other transcription factors such as WT1, RUNX1, and CEBPA that are recurrently mutated or deleted in T/myeloid MPAL. Nevertheless, these findings provide valuable insight into the pathogenesis of this unique leukemia subtype, as well as preclinical models that will help to identify new therapeutic vulnerabilities in this poor-prognosis disease.
Conflict-of-interest disclosure: M.P.M. has served as a consultant for AstraZeneca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal