

Key Points
LDHA regulates protein synthesis during MK maturation and proplatelet formation by directly binding to eEF2 in an NADH-dependent manner.
Targeting LDHA with stiripentol significantly promotes the production of platelets in the immune thrombocytopenia model.

Abstract
Translation is essential for megakaryocyte (MK) maturation and platelet production. However, how the translational pathways are regulated in this process remains unknown. In this study, we found that MK/platelet–specific lactate dehydrogenase A (LdhA) knockout mice exhibited an increased number of platelets with remarkably accelerated MK maturation and proplatelet formation. Interestingly, the role of LDHA in MK maturation and platelet formation did not depend on lactate content, which was the major product of LDHA. Mechanism studies revealed that LDHA interacted with eukaryotic elongation factor 2 (eEF2) in the cytoplasm, controlling the participation of eEF2 in translation at the ribosome. Furthermore, the interaction of LDHA and eEF2 was dependent on nicotinamide adenine dinucleotide (NADH), a coenzyme of LDHA. NADH-competitive inhibitors of LDHA could release eEF2 from the LDHA pool, upregulate translation, and enhance MK maturation in vitro. Among LDHA inhibitors, stiripentol significantly promoted the production of platelets in vivo under a physiological state and in the immune thrombocytopenia model. Moreover, stiripentol could promote platelet production from human cord blood mononuclear cell–derived MKs and also have a superposed effect with romiplostim. In short, this study shows a novel nonclassical function of LDHA in translation that may serve as a potential target for thrombocytopenia therapy.
Introduction
Platelets are essential components of peripheral blood and play key roles in hemostasis, thrombosis, inflammation, tumor growth, and metastasis.1,2 The normal number of human platelets remains stable at 150 to 450 × 109/L. Thrombocytopenia increases the risk of vascular, mucosal, and visceral bleeding, whereas thrombocythemia heightens the risk of thrombotic events, including stroke, peripheral ischemia, and myocardial infarction. A variety of factors can contribute to anomalous platelet counts, inappropriate platelet production being a major one. Platelets are formed and released into the bloodstream by precursor cells called megakaryocytes (MKs) that reside within the bone marrow.3 The production of platelets from MKs undergoes a series of biological processes, including MK maturation through endomitosis, membrane system invagination, proplatelet formation (PPF), and terminal platelet formation and release.4
Among all of the developmental processes, the translation of messenger RNAs (mRNAs), which results in de novo protein synthesis, is a critical step.5,6 Because the assembly of ribosomes consumes large amounts of energy, the metabolism in MKs is active to meet the high-energy requirement, particularly those of higher ploidy. Simultaneous deletion of glucose transporters 1 and 3 completely abolished glucose uptake and impaired PPF from MKs, illustrating the essential role of glucose metabolism in platelet production.7 However, the underlying mechanisms remain unclear.
Lactate dehydrogenase (LDH) is an important tetrameric enzyme that participates in anaerobic and aerobic glycolysis, catalyzing the interconversion of pyruvate to lactate with nicotinamide adenine dinucleotide (NADH)/NAD+ as a cofactor. LDH is a homo- or hetero-tetramer molecule of either muscle (M) or heart (H) subunit, encoded by LdhA (M) and LdhB (H). There are 5 isoenzymes of LDH: LDH-1 (4H), LDH-2 (3H1M), LDH-3 (2H2M), LDH-4 (1H3M), and LDH-5 (5M).8 Each isoenzyme has unique structures and is found with different combinations in different tissues.9
In the current study, mouse platelets only expressed LDH-5 (5M) encoded by LdhA. In addition, LdhA–/– mice developed an increase in platelet number caused by accelerated MK maturation and PPF. Mechanism studies revealed that lactate dehydrogenase A (LDHA) interacted with eEF2 and inhibited protein synthesis in an NADH-dependent manner. Inhibitors of LDHA targeting NADH-binding sites could upregulate translation and promote MK maturation in vitro. Stiripentol, an LDHA inhibitor, significantly enhanced the production of platelets in vivo under a physiological state and in the immune thrombocytopenia mouse model. Moreover, stiripentol could promote platelet production from human cord blood mononuclear cell–derived MKs, and it also has a superposed effect with thrombopoietin receptor agonist romiplostim. The present study reports an unrevealed role of LDHA in translation and MK maturation, which may serve as a new target for thrombocytopenia therapy.
Methods
Antibodies, reagents, mice, and additional methods
Detailed descriptions of antibodies, reagents, mice, and additional methods are presented in the supplemental Methods (available on the Blood Web site).
LDH isoenzyme analysis
LDH isoforms were separated by charge-to-mass ratio in native polyacrylamide gel electrophoresis. Electrophoresis was performed with pH 8.6 running buffer without sodium dodecyl sulfate and 40 to 60 V running voltage on ice. Gels were then stained with LDH staining buffer at room temperature for 5 to 10 minutes. LDH staining buffer included 2 mM phosphate buffer (pH 7.5), 0.2 mM 5-methylphenazinium methosulfate (Sigma), 3 mM nitro blue tetrazolium chloride (Sigma), 200 mM sodium lactate (Sigma), and 2.8 mM NAD (Sigma). Staining buffer was prepared just before use.
Platelet life span assay, platelet clearance and regeneration
Platelet survival in vivo was measured at different time points after mouse tail IV injection of sulfo-NHS-LC-biotin (30 μg/g weight; Pierce). Platelets were collected from mouse orbital blood and detected with allophycocyanin (APC)-streptavidin (2 μg/mL; BD) according to flow cytometry.
Basal platelet counts were determined before platelets were cleared by mice tail vein injection with anti-CD42b monoclonal antibody (2 μg/g weight; Emfret Analytics). The number of platelet regeneration was then measured by using a Hemavet automated hematologic analyzer.
MK differentiation, PPF, and polyploidy assay
Standard experimental methods for MK differentiation, PPF, and polyploidy assay have been described previously.10 Briefly, fetal liver cells were isolated from 13.5-day-old mouse embryos and cultured with 10 ng/mL interleukin 3 (Prospec Bio) and 15 IU/mL thrombopoietin (TPO) (3SBio). MKs were separated on a bovine serum albumin gradient (1.5% to 3%) at 37°C for 1 hour and were seeded onto slides coated with 50 μg/mL fibrinogen (Sigma) at 37°C. After 12 hours of culture, the MKs were stained with 10 μg/mL 4′,6-diamidino-2-phenylindole (DAPI; Yeasen) and anti–α-tubulin antibody (1:4000; Sigma). Statistical analyses were implemented with ImageJ (National Institutes of Health). For polyploidy assay, MKs were stained with 2 ng/μL anti–CD41-APC antibody (Invitrogen) and 50 μg/mL propidium iodide (Invitrogen), 100 μg/mL ribonuclease A (Yeasen), and 0.2% Triton X-100 (Sigma). MK polyploidy was then detected by using flow cytometry.
Cell-free protein expression assay
Cell-free protein expression was performed with TNT Coupled Rabbit Reticulocyte Lysate Systems (Promega) including 25 µL TNT Rabbit Reticulocyte Lysate, 2 µL TNT Reaction Buffer, 1 µL TNT SP6 RNA Polymerase, 1 µL Amino Acid Mixture (Minus Leucine, 1 mM), 1 µL Amino Acid Mixture (Minus Methionine,1 mM), 1 µL Ribonuclease Inhibitor (40 U/µL; Invitrogen), 1 µg luciferase DNA template plasmid, and nuclease-free water to a final volume of 50 µL. According to experimental requirements, different reagents were supplemented for 30 minutes before protein synthesis. Protein synthesis was then performed in a 30°C water bath for 90 minutes and was estimated by using a luciferase assay system (Promega); the program included a 2-second measurement delay followed by a 10-second measurement reading by NOVOstar (BMG LABTECH). MARS data analysis software (BMG LABTECH) was used to calculate average values.
Statistical analyses
Statistical analyses were implemented with GraphPad Prism 9 (GraphPad Software). Statistical significance was analyzed by using an unpaired 2-tailed t test, 1-way analysis of variance test, and 2-way analysis of variance test. Values are represented as mean ± standard error of the mean. P < .05 defined significance (*P < .05, **P < .01, ***P < .001, and ****P < .0001).
Results
LDHA deficiency caused an increase in platelet number
Glucose metabolism is necessary for platelet activation and production. To evaluate the metabolic pathway of glucose, metabolic flux analysis was performed by using the tracer [U-13C6]-glucose in platelets. Figure 1A shows that the labeling rates of glycolysis metabolites were >80%, whereas those of the tricarboxylic acid cycle metabolites were <10%, which confirmed that glucose metabolism was dominated by anaerobic glycolysis in platelets. LDH is a key enzyme in anaerobic glycolysis, and the expression levels of LDH were measured in platelets. We found that LDH-5 (5M) was the main form of LDH in mouse platelets (Figure 1B). In sharp contrast to the low expression of LDHB, mRNA and protein of LDHA were highly expressed in mouse platelets (Figure 1C-D).

LdhA−/− mice developed an increase in platelet number. (A) Glucose metabolic flux analysis in mouse platelets by liquid chromatography/tandem mass spectrometry (n = 3). (B) LDH isozyme assay in mouse platelet, myocardium (heart), and skeletal muscle (muscle). (C) mRNA relative expression assay of LdhA and LdhB gene in mice tissues by quantitative real-time polymerase chain reaction (n = 3). (D) The protein expression of LDHA and LDHB in mouse platelet, myocardium (heart), and skeletal muscle (muscle). (E) Knockout-first strategy for creating conditional LdhA knockout mice. Recombination steps with Flp or Pf4-cre recombinase are illustrated. (F) mRNA relative expression assay of LdhA in LdhAf/f and LdhA−/− mice platelets by quantitative real-time polymerase chain reaction (n = 3). (G) The protein expression of LDHA in LdhAf/f and LdhA−/− mice platelets. (H) LDH isozyme assay in LdhAf/f and LdhA−/− mice platelets. Mice myocardium (heart) and skeletal muscle (muscle) were used as positive control. (I) Red blood cell (n = 5) and white blood cell (n = 5) counts in LdhAf/f and LdhA−/− mice peripheral blood. (J) Platelet counts in LdhAf/f and LdhA−/− mice peripheral blood (n = 5). (K) Mean platelet volume in LdhAf/f and LdhA−/− mice (n = 8). (L) Representative transmission electron microscope images of LdhAf/f and LdhA−/− mice platelets (n = 15, left). Scale bar, 50 nm. Total platelet section area was measured, and dense granule, α granule, and mitochondria numbers were counted. The data are expressed as granule numbers per unit area of counted section (right). Male mice 8 to 10 weeks old were used for these animal experiments. **P < .01, ***P < .001, ****P < .0001. 3PG, 3‐phosphoglycerate; Cit, citrate; F6P, fructose-6-phosphate; Fum, fumarate; G6P, glucose-6-phosphate; GA3P, glyceraldehyde-3-phosphate; Glu, glucose; Lac, lactate; Mal, malate; ns, not significant; PA, poly A; PEP, phosphoenolpyruvate; SA, splice acceptor; Suc, succinate.
LdhA−/− mice developed an increase in platelet number. (A) Glucose metabolic flux analysis in mouse platelets by liquid chromatography/tandem mass spectrometry (n = 3). (B) LDH isozyme assay in mouse platelet, myocardium (heart), and skeletal muscle (muscle). (C) mRNA relative expression assay of LdhA and LdhB gene in mice tissues by quantitative real-time polymerase chain reaction (n = 3). (D) The protein expression of LDHA and LDHB in mouse platelet, myocardium (heart), and skeletal muscle (muscle). (E) Knockout-first strategy for creating conditional LdhA knockout mice. Recombination steps with Flp or Pf4-cre recombinase are illustrated. (F) mRNA relative expression assay of LdhA in LdhAf/f and LdhA−/− mice platelets by quantitative real-time polymerase chain reaction (n = 3). (G) The protein expression of LDHA in LdhAf/f and LdhA−/− mice platelets. (H) LDH isozyme assay in LdhAf/f and LdhA−/− mice platelets. Mice myocardium (heart) and skeletal muscle (muscle) were used as positive control. (I) Red blood cell (n = 5) and white blood cell (n = 5) counts in LdhAf/f and LdhA−/− mice peripheral blood. (J) Platelet counts in LdhAf/f and LdhA−/− mice peripheral blood (n = 5). (K) Mean platelet volume in LdhAf/f and LdhA−/− mice (n = 8). (L) Representative transmission electron microscope images of LdhAf/f and LdhA−/− mice platelets (n = 15, left). Scale bar, 50 nm. Total platelet section area was measured, and dense granule, α granule, and mitochondria numbers were counted. The data are expressed as granule numbers per unit area of counted section (right). Male mice 8 to 10 weeks old were used for these animal experiments. **P < .01, ***P < .001, ****P < .0001. 3PG, 3‐phosphoglycerate; Cit, citrate; F6P, fructose-6-phosphate; Fum, fumarate; G6P, glucose-6-phosphate; GA3P, glyceraldehyde-3-phosphate; Glu, glucose; Lac, lactate; Mal, malate; ns, not significant; PA, poly A; PEP, phosphoenolpyruvate; SA, splice acceptor; Suc, succinate.
To explore the function of LDH in platelets, MK/platelet–specific LdhA-deficient mice (LdhAf/fPf4-Cre+, LdhA−/−) were created by using a knockout-first strategy11–13 (Figure 1E). As expected, the mRNA and protein levels of LDHA in LdhA−/− platelets were markedly reduced compared with those in LdhAf/f platelets (Figure 1F-G). LDH isoenzyme analysis showed that LDHA deficiency led to a complete loss of LDH activity (Figure 1H). The whole blood cell counts of peripheral blood showed that LdhA−/− mice had 893 ± 23.38 × 109 circulating platelets/L compared with 672 ± 47.74 × 109 circulating platelets/L in LdhAf/f mice (Figure 1J), indicating that LDHA deficiency led to an increase in platelet numbers in mice. Meanwhile, there were no differences in the white blood cell counts and red blood cell counts between LdhA−/− mice and LdhAf/f mice (Figure 1I). In addition, the mean platelet volume of LdhAf/f mice and LdhA−/− mice was equivalent (7.35 ± 0.08 fL in LdhAf/f vs 7.30 ± 0.05 fL in LdhA−/−) (Figure 1K). The ultrastructure in platelets was detected by using a transmission electron microscope (Figure 1L). There was no significant difference in the number of particles such as dense granule, α granule, and mitochondria in platelets between LdhAf/f and LdhA−/− platelets. There were no alteration of platelet functions in LdhA−/− mice compared with LdhAf/f mice in vitro or in vivo (supplemental Figure 1A-E), although bleeding time slightly decreased in LdhA−/− mice (supplemental Figure 1F), which might be due to an increased platelet number. These results indicate that LDHA deficiency promotes the production of platelets with normal functions.
To explore the reason for the increased platelet numbers in LdhA−/− mice, platelet life span and platelet production were measured. The apoptosis ratio of LdhA−/− platelets had a similar level to LdhAf/f platelets in vitro (Figure 2A). The survival of biotin-labeled platelets in vivo was measured at different time points, with the data showing that LdhA−/− platelets had a normal life span (Figure 2B). Meanwhile, LdhA−/− mice had a percentage of 2.77% ± 0.18% reticulated platelets, and LdhAf/f mice had a percentage of 1.65% ± 0.06% reticulated platelets, suggesting a much higher number of newborn platelets in LdhA−/− mice (Figure 2C). Platelets were then cleared by tail vein injection of anti-CD42b monoclonal antibody, and the regeneration speed of LdhA−/− platelets was faster than that of LdhAf/f platelets (Figure 2D). These data indicate that the increased platelet number in LdhA−/− mice was caused by enhanced platelet production in vivo.
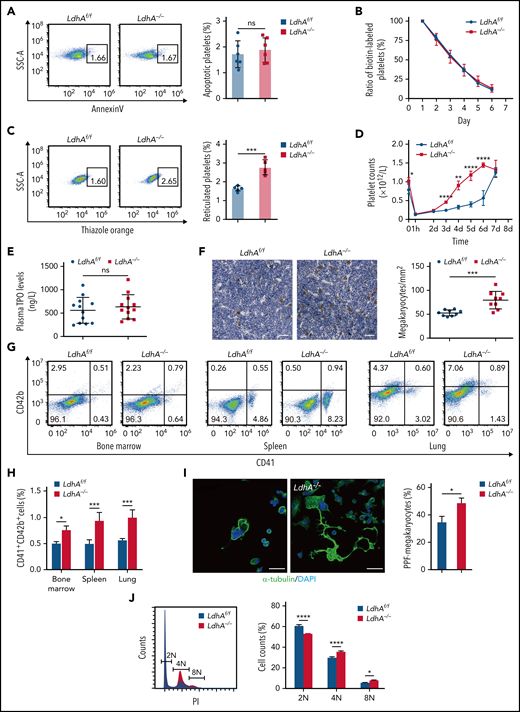
MK maturation and PPF were expedited in LdhA−/− mice. (A) Representative Annexin V+ gating strategy and percentage of apoptotic platelets in LdhAf/f and LdhA−/− mice (n = 6). (B) Biotin-labeled ratio of platelets at different time points after tail IV injection of Sulfo-NHS-LC-Biotin (30 μg/g weight) in LdhAf/f and LdhA−/− mice (n = 5; 8 weeks old, male). (C) Representative thiazole orange–positive gating strategy and percentage of reticulated platelets in LdhAf/f and LdhA−/− mice (n = 5). (D) Platelets were eliminated by tail IV injection of anti-CD42b antibody (2 μg/g weight) in LdhAf/f and LdhA−/− mice, and platelet counts were monitored at different time points (n = 5; 8 weeks old, male). (E) Detection of plasma TPO level by using enzyme-linked immunosorbent assay (n = 12). (F) Quantitative analysis of bone marrow MKs in LdhAf/f and LdhA−/− mice femur bone by immunohistochemistry with 5 μg/mL anti-CD42c antibody (n = 9; 8 weeks old, male). Scale bar, 50 μm. (G) Representative CD41+CD42b+ MK gating strategy. (H) Statistical analysis on the ratios of CD41+CD42b+ MKs in LdhAf/f and LdhA−/− mice bone marrow, spleen, and lung according to flow cytometry (n = 3; 8 weeks old, male). (I) Immunofluorescence images of PPF-MKs derived from the fetal liver in LdhAf/f and LdhA−/− pregnant mice; statistical analysis on PPF-MK/total MK ratio is shown (n = 3). Scale bar, 30 μm. (J) Detection of polyploidy (2N, 4N, and 8N) from fetal liver MKs of LdhAf/f and LdhA−/− pregnant mice by flow cytometry (n = 4). *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; ns, not significant; SSC-A, side scatter area.
MK maturation and PPF were expedited in LdhA−/− mice. (A) Representative Annexin V+ gating strategy and percentage of apoptotic platelets in LdhAf/f and LdhA−/− mice (n = 6). (B) Biotin-labeled ratio of platelets at different time points after tail IV injection of Sulfo-NHS-LC-Biotin (30 μg/g weight) in LdhAf/f and LdhA−/− mice (n = 5; 8 weeks old, male). (C) Representative thiazole orange–positive gating strategy and percentage of reticulated platelets in LdhAf/f and LdhA−/− mice (n = 5). (D) Platelets were eliminated by tail IV injection of anti-CD42b antibody (2 μg/g weight) in LdhAf/f and LdhA−/− mice, and platelet counts were monitored at different time points (n = 5; 8 weeks old, male). (E) Detection of plasma TPO level by using enzyme-linked immunosorbent assay (n = 12). (F) Quantitative analysis of bone marrow MKs in LdhAf/f and LdhA−/− mice femur bone by immunohistochemistry with 5 μg/mL anti-CD42c antibody (n = 9; 8 weeks old, male). Scale bar, 50 μm. (G) Representative CD41+CD42b+ MK gating strategy. (H) Statistical analysis on the ratios of CD41+CD42b+ MKs in LdhAf/f and LdhA−/− mice bone marrow, spleen, and lung according to flow cytometry (n = 3; 8 weeks old, male). (I) Immunofluorescence images of PPF-MKs derived from the fetal liver in LdhAf/f and LdhA−/− pregnant mice; statistical analysis on PPF-MK/total MK ratio is shown (n = 3). Scale bar, 30 μm. (J) Detection of polyploidy (2N, 4N, and 8N) from fetal liver MKs of LdhAf/f and LdhA−/− pregnant mice by flow cytometry (n = 4). *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; ns, not significant; SSC-A, side scatter area.
LDHA deficiency accelerated MK maturation and PPF
TPO is a key regulator of megakaryopoiesis and thrombopoiesis.14,15 However, we found that LDH deficiency did not affect the TPO levels in mice plasma (Figure 2E). To explore the cause of enhanced platelet production in LdhA−/− mice, the precursors of platelets (MKs) in the bone marrow of femurs were immunohistochemistry stained with anti-CD42c antibody. Figure 2F shows that the LdhA−/− mice had 51.05% ± 6.45% more MKs than LdhAf/f mice. At the same time, the CD41+CD42b+ cell ratios in the bone marrow, spleen, and lung, which are the major thrombopoiesis organs in mice,16,17 were measured by using flow cytometry. LdhA−/− mice had 51.88% ± 0.11%, 88.94% ± 0.10%, and 76.35% ± 0.09% more MKs than LdhAf/f mice in the bone marrow, spleen, and lung, respectively (Figure 2G-H; supplemental Figure 2D). MKs are derived from hematopoietic stem cells (HSCs) that undergo sequential differentiation through the MK–erythroid progenitor and terminal differentiation of MK progenitors (MKPs).18 There was no change in the hematopoietic populations upstream of the MKs, including long-term HSCs, short-term HSCs, multipotent progenitors, CD41+HSCs, common myeloid progenitors, megakaryocyte–erythrocyte progenitors, granulocyte–monocyte progenitors, and MKPs in the bone marrow of LdhA−/− mice compared with that in LdhAf/f mice (supplemental Figure 2A-B). However, colony-forming unit–MK assay results showed that MKPs from LdhA−/− mice formed 89.71% ± 3.42% more colony-forming unit–MKs than that from LdhAf/f mice (supplemental Figure 2C). These data indicate that LDHA could regulate terminal differentiation of MKs instead of early differentiation from HSCs.
Murine fetal liver is a suitable choice for high MK yield and is also a privileged organ to study MKP differentiation.19Figure 2I shows that LdhA−/− terminal differential MKs, which are also called PPF-MKs,20 obtained from murine fetal livers were morphologically more differentiated than those of LdhAf/f MKs in vitro (48.28% ± 0.02% in LdhA−/− vs 34.28% ± 0.03% in LdhAf/f). During the process of MK maturation and PPF, MK undergoes endomitosis and accumulates DNA content from diploid (2N) to tetraploid (4N), octoploid (8N), 16-ploid (16N), and even more.21 The proportions of the 4N and 8N cells were much higher, whereas the proportions of the 2N cells were much lower in primary LdhA−/− MKs in contrast to LdhAf/f MKs (Figure 2J). From these data, it was apparent that LDHA acted as a speed limit in MK maturation and PPF.
LDHA promotes glycolysis by catalyzing the conversion of pyruvate to lactate. LDHA deficiency led to a 69.49% ± 0.18% drop in lactate level in platelets, whereas there was no difference in lactate levels in peripheral blood plasma between LdhA−/− and LdhAf/f mice (Figure 3A). The extracellular acidification rate of LdhA−/− platelets was significantly reduced compared with that of LdhAf/f platelets (Figure 3B). These data manifested that LDHA deficiency indeed impaired glycolysis and the production of lactate. Interestingly, various concentrations of lactate had no effect on the differentiation of primary MKs from LdhA−/− or LdhAf/f fetal livers in vitro (Figure 3C), indicating that the role of LDHA in MK maturation may not depend on the production of lactate.
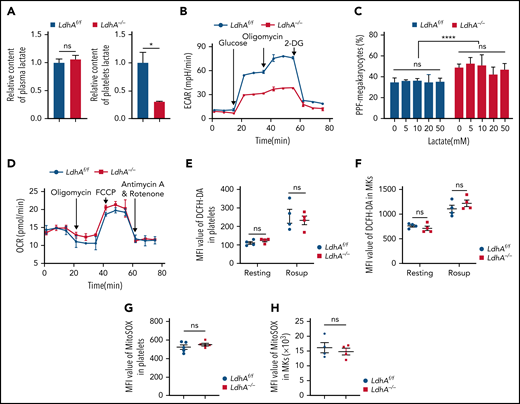
The role of LDHA in MK differentiation is independent of lactate. (A) Relative content of lactate in plasma (n = 9) and circulating platelet (n = 3). (B) Extracellular acidification rate (ECAR) in LdhAf/f and LdhA−/− mice platelets (n = 3). (C) Statistical analysis on the ratio of PPF-MKs/total MKs in LdhAf/f and LdhA−/− mice fetal liver MKs with different lactate concentrations (n = 3). (D) Oxygen consumption rate (OCR) in LdhAf/f and LdhA−/− mice platelets (n = 3). (E-F) Total reactive oxygen species (ROS) of LdhAf/f and LdhA−/− mice platelets and bone marrow MKs were detected by 2′,7′-dichlorofluorescin diacetate (DCFH-DA) probe in resting and Rosup-stimulated (50 μg/mL) group (n = 4). ROS-inducing reagent Rosup (Beyotime) was used as a positive control for ROS level. (G-H) Mitochondrial ROS of LdhAf/f and LdhA−/− mice platelets (n = 5) and bone marrow MKs (n = 4) were detected by MitoSOX Red (Thermo Fisher Scientific). *P < .05, ****P < .0001. MFI, mean fluorescence intensity; ns, not significant.
The role of LDHA in MK differentiation is independent of lactate. (A) Relative content of lactate in plasma (n = 9) and circulating platelet (n = 3). (B) Extracellular acidification rate (ECAR) in LdhAf/f and LdhA−/− mice platelets (n = 3). (C) Statistical analysis on the ratio of PPF-MKs/total MKs in LdhAf/f and LdhA−/− mice fetal liver MKs with different lactate concentrations (n = 3). (D) Oxygen consumption rate (OCR) in LdhAf/f and LdhA−/− mice platelets (n = 3). (E-F) Total reactive oxygen species (ROS) of LdhAf/f and LdhA−/− mice platelets and bone marrow MKs were detected by 2′,7′-dichlorofluorescin diacetate (DCFH-DA) probe in resting and Rosup-stimulated (50 μg/mL) group (n = 4). ROS-inducing reagent Rosup (Beyotime) was used as a positive control for ROS level. (G-H) Mitochondrial ROS of LdhAf/f and LdhA−/− mice platelets (n = 5) and bone marrow MKs (n = 4) were detected by MitoSOX Red (Thermo Fisher Scientific). *P < .05, ****P < .0001. MFI, mean fluorescence intensity; ns, not significant.
Previous studies reported that the reduction of LDHA activity resulted in stimulation of mitochondrial respiration and the decrease of mitochondrial membrane potential in cancer cells,22 indicating that there might be complementary roles of glycolysis and mitochondrial respiration. However, Figure 3D shows that the oxygen consumption rate is similar in LdhAf/f and LdhA−/− platelets, suggesting that LDH deficiency did not result in a shift in the metabolic patterns of mitochondrial oxidative respiration in platelets. In addition, total and mitochondrial reactive oxygen species levels in platelets and bone marrow MKs were measured. LdhAf/f and LdhA−/− MKs/platelets shared similar total and mitochondrial reactive oxygen species levels (Figure 3E-H).
LDHA interacted with eEF2 and inhibited protein synthesis in platelets
The possible associate protein of LDHA in MKs/platelets was then studied through the coimmunoprecipitation of human platelet lysate with anti-LDHA antibody. Coomassie brilliant blue staining revealed there was a specific band at 95 kD in the LDHA coimmunoprecipitation sample compared with immunoglobulin G control (Figure 4A). Then, unbiased proteomics of the LDHA coimmunoprecipitation sample and protein spectrum of the 95 kD band were both performed to identify the binding proteins of LDHA, and several proteins containing eEF2 were found in both analyses (Figure 4B). Furthermore, only the interaction between LDHA and eEF2 was confirmed in a human megakaryocytic cell line (DAMI), which was established from the blood of a patient with megakaryoblastic leukemia23 (Figure 4C). eEF2 is one of the important members of the guanosine triphosphate (GTP)-binding translation elongation factor family catalyzing the translocation of the peptidyl-transfer RNA from the A to P sites of the ribosome during the process of protein synthesis.24,25 eEF2 is found to be highly expressed in various cancers and is correlated with cancer cell progression and recurrence.26 However, the role of eEF2 in MK differentiation and platelet production remains unclear. By polyploidy assay, we found that enhancing eEF2 activity with A-484954 promoted the differentiation of DAMI cells (Figure 4D). On the contrary, inhibiting eEF2 activity with DDD107498 succinate restrained the differentiation of DAMI cells. These data indicate that eEF2 was involved in MK differentiation.
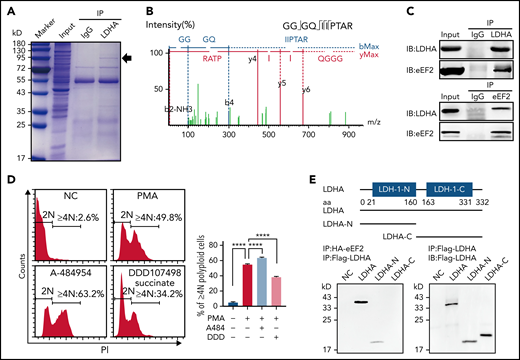
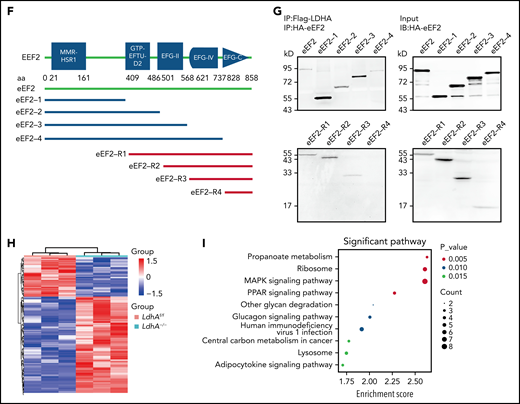
Changes in protein synthesis caused by LDHA deficiency are related to eEF2. (A) Coomassie brilliant blue–stained gel of human platelets coimmunoprecipitation (co-IP) with anti-LDHA or anti–immunoglobulin G (IgG) antibody. The black arrow indicates the location of differential protein. (B) Tandem mass spectrometry assay of eEF2-specific peptide (GGGQIIPTAR) from 95 kD protein gel. (C) Co-IP of DAMI whole-cell lysates with anti-LDHA and anti-eEF2 antibodies. Isotype-matched IgG as negative control (NC). (D) Detection of DNA content (≥4N) in DAMI cells treated with phorbol 12-myristate 13-acetate (PMA; 50 nM), A-484954 (A-484; 10 μM), and DDD107498 succinate (DDD; 1 μM) (n = 3). (E) Functional domains of LDHA and its truncations. Full-length LDHA and its truncated plasmids with Flag-tag (top). Co-IP of coexpression LDHA truncated plasmids and HA-eEF2 plasmid with anti-HA antibody/anti-Flag antibody in HEK-293T (bottom). NC transfected with empty HA/Flag fusion plasmid to control for nonspecific binding. (F) Functional domains of eEF2 and its truncations. Full-length eEF2 and its truncated plasmids with HA-tag. (G) Co-IP of coexpression eEF2-truncated plasmids and Flag-LDHA plasmid with anti-Flag antibody in HEK-293T. (H) Heatmap showing significant differential proteins in LdhAf/f and LdhA−/− mice platelets based on quantitative proteomic analysis. Three independent biological replicates were used for cluster analysis, and color represents quantitative ratio. (I) Kyoto Encyclopedia of Genes and Genomes assay of top 10 differential cell pathways in LdhAf/f and LdhA−/− mice platelets based on quantitative proteomic analysis. ****P < .0001. PPAR, peroxisome proliferator-activated receptor.
Changes in protein synthesis caused by LDHA deficiency are related to eEF2. (A) Coomassie brilliant blue–stained gel of human platelets coimmunoprecipitation (co-IP) with anti-LDHA or anti–immunoglobulin G (IgG) antibody. The black arrow indicates the location of differential protein. (B) Tandem mass spectrometry assay of eEF2-specific peptide (GGGQIIPTAR) from 95 kD protein gel. (C) Co-IP of DAMI whole-cell lysates with anti-LDHA and anti-eEF2 antibodies. Isotype-matched IgG as negative control (NC). (D) Detection of DNA content (≥4N) in DAMI cells treated with phorbol 12-myristate 13-acetate (PMA; 50 nM), A-484954 (A-484; 10 μM), and DDD107498 succinate (DDD; 1 μM) (n = 3). (E) Functional domains of LDHA and its truncations. Full-length LDHA and its truncated plasmids with Flag-tag (top). Co-IP of coexpression LDHA truncated plasmids and HA-eEF2 plasmid with anti-HA antibody/anti-Flag antibody in HEK-293T (bottom). NC transfected with empty HA/Flag fusion plasmid to control for nonspecific binding. (F) Functional domains of eEF2 and its truncations. Full-length eEF2 and its truncated plasmids with HA-tag. (G) Co-IP of coexpression eEF2-truncated plasmids and Flag-LDHA plasmid with anti-Flag antibody in HEK-293T. (H) Heatmap showing significant differential proteins in LdhAf/f and LdhA−/− mice platelets based on quantitative proteomic analysis. Three independent biological replicates were used for cluster analysis, and color represents quantitative ratio. (I) Kyoto Encyclopedia of Genes and Genomes assay of top 10 differential cell pathways in LdhAf/f and LdhA−/− mice platelets based on quantitative proteomic analysis. ****P < .0001. PPAR, peroxisome proliferator-activated receptor.
Furthermore, 5 functional domains of eEF2 (MMR-HSR1, GTP-EFTU-D2, EFG-II, EFG-IV, and EFG-C) (Figure 4F) and 2 functional domains of LDHA (LDH-1-N and LDH-1-C) (Figure 4E) were predicted by using SMART (http://smart.embl-heidelberg.de). The 9 plasmids expressing 8 truncated and a full-length eEF2 with HA fusion, and 3 plasmids expressing 2 truncated and a full-length LDHA with Flag fusion, were constructed. Coimmunoprecipitations manifested that eEF2 had multiple binding sites with LDHA, whereas only the LDH-1-N domain of LDHA could bind to eEF2 (Figure 4E-G).
Protein synthesis is essential for MK maturation and platelet production. Quantitative proteomic analysis was performed to explore the change of proteins in LdhA–/– platelets. More than 40% of proteins in LdhA–/– platelets were upregulated compared with LdhAf/f platelets (Figure 4H). Kyoto Encyclopedia of Genes and Genomes enrichment analysis revealed a variety of differential signaling pathways in LdhA–/– platelets. The ribosome pathway and MAPK signal pathway were upregulated, which were related to MK differentiation (Figure 4I).27,28
Because the data showed that LDHA was not directly located on the ribosome (Figure 5A), we speculated that LDHA may act as a “pool” located away from the ribosome that arrests eEF2 when binding to it and inhibits its participation in protein synthesis.

NADH restrained translation by enhancing the binding of LDHA and eEF2. (A) Coimmunoprecipitation (IP) of DAMI whole-cell lysates with anti-LDHA antibody and ribosomal protein antibodies. (B) Co-IP of Flag-LDHA and HA-eEF2 in HEK-293T treated with pyruvate, lactate, and NAD+. Negative control (NC) was transfected with empty HA/Flag-tag plasmid and used for nonspecific binding control. (C) Co-IP of Flag-LDHA and HA-eEF2 in HEK-293T treated with different concentrations of NADH; the gray values of eEF2-HA IP bands were counted (n = 3). (D) The protein expression of eEF2 and LDHA in mouse or rabbit reticulocyte lysate was detected by using western blot. β-Actin was used as a loading control. (E) The levels of protein synthesis were measured when different concentrations of LDHA proteins were added in the rabbit reticulocyte translation system (n = 3). (F) The levels of protein synthesis were measured when different concentrations of NADH were added in the rabbit reticulocyte translation system with or without 5 μg LDHA (n = 3). (G) The levels of protein synthesis were measured when different concentrations of NAD+ were added in the rabbit reticulocyte translation system with 5 μg LDHA (n = 3). (H) The levels of protein synthesis were measured when glucose (5 mM), pyruvate (1 mM), and lactate (2 mM) were added in the rabbit reticulocyte translation system with 5 μg LDHA (n = 3). (I) Megakaryocyte PPF assay in LdhAf/f and LdhA−/− mice liver fetal MKs incubated with 1 mM NADH. Statistical analysis of the PPF-MK/total MK ratio is shown (n = 4). Scale bar, 100 μm. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; ns, not significant; RPL28, ribosomal protein L28; RPL35, ribosomal protein L35; RPS3A, ribosomal protein S3A.
NADH restrained translation by enhancing the binding of LDHA and eEF2. (A) Coimmunoprecipitation (IP) of DAMI whole-cell lysates with anti-LDHA antibody and ribosomal protein antibodies. (B) Co-IP of Flag-LDHA and HA-eEF2 in HEK-293T treated with pyruvate, lactate, and NAD+. Negative control (NC) was transfected with empty HA/Flag-tag plasmid and used for nonspecific binding control. (C) Co-IP of Flag-LDHA and HA-eEF2 in HEK-293T treated with different concentrations of NADH; the gray values of eEF2-HA IP bands were counted (n = 3). (D) The protein expression of eEF2 and LDHA in mouse or rabbit reticulocyte lysate was detected by using western blot. β-Actin was used as a loading control. (E) The levels of protein synthesis were measured when different concentrations of LDHA proteins were added in the rabbit reticulocyte translation system (n = 3). (F) The levels of protein synthesis were measured when different concentrations of NADH were added in the rabbit reticulocyte translation system with or without 5 μg LDHA (n = 3). (G) The levels of protein synthesis were measured when different concentrations of NAD+ were added in the rabbit reticulocyte translation system with 5 μg LDHA (n = 3). (H) The levels of protein synthesis were measured when glucose (5 mM), pyruvate (1 mM), and lactate (2 mM) were added in the rabbit reticulocyte translation system with 5 μg LDHA (n = 3). (I) Megakaryocyte PPF assay in LdhAf/f and LdhA−/− mice liver fetal MKs incubated with 1 mM NADH. Statistical analysis of the PPF-MK/total MK ratio is shown (n = 4). Scale bar, 100 μm. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; ns, not significant; RPL28, ribosomal protein L28; RPL35, ribosomal protein L35; RPS3A, ribosomal protein S3A.
NADH enhanced the association of LDHA with eEF2 and inhibited MK differentiation
LDHA has 2 major active sites: a substrate-binding site and a cofactor-binding site.29–31 LDHA interacted with eEF2 through its N-terminal domain, which was reported to be the major NADH-binding site29 (Figure 4G). To elucidate the regulative function of LDHA in protein synthesis, the effects of pyruvate, lactate, NADH, and NAD+ on the binding strength between LDHA and eEF2 were measured. Pyruvate, lactate, and NAD+ had no effect on the binding between LDHA and eEF2 (Figure 5B). On the other hand, the binding strength of LDHA and eEF2 were significantly enhanced by NADH (Figure 5C).
Cell-free protein expression is a simplified and accelerated avenue for the translation of a specific protein in a quasi-cell environment. The system is an important tool for studying protein synthesis in vitro and has a significant advantage over the whole-cell in vivo systems.32,33 The Eukaryotic system-TNT coupled rabbit reticulocyte lysate system was chosen due to its lack of LDHA and richness in eEF2 (Figure 5D). Figure 5E shows that LDHA protein significantly hindered the process of protein synthesis in vitro. Protein synthesis was further inhibited by NADH in an LDHA-dependent manner (Figure 5F). NAD+, glucose, pyruvate, and lactate, conversely, had no effect on protein synthesis compared with the LDHA protein-alone group (Figure 5G-H). These results indicate that NADH could enhance the interaction of LDHA with eEF2 and suppress protein synthesis.
Furthermore, NADH was added to the primary MKs isolated from LdhAf/f and LdhA−/− fetal livers during the differentiation process induced by TPO in vitro. The PPF ratio of LdhAf/f MKs was significantly reduced by NADH (11.76% ± 0.90% in the LdhAf/f NADH group vs 18.31% ± 1.18% in the LdhAf/f control group). Conversely, NADH did not inhibit the differentiation process of LdhA−/− MKs (Figure 5I). The reduced expression of CD41 suggests that NADH also inhibited the differentiation of human DAMI cells induced by phorbol 12-myristate 13-acetate (supplemental Figure 3A). These data show that NADH inhibits MK differentiation through LDHA.
LDHA can be a potential target for thrombocytopenia therapy
Clinically speaking, 40% to 50% of patients in medical and surgical intensive care units are at risk of thrombocytopenia, which often results in bleeding.34 The inhibition of MK maturation and platelet release are key factors causing thrombocytopenia.35 Based on the role of LDHA in MK differentiation, we speculated that LDHA could be a potential target for thrombocytopenia therapy. First, a variety of small-molecule inhibitors that competitively occupied the NADH-binding pocket or pyruvate-binding pocket of LDHA were screened through coimmunoprecipitation of LDHA and eEF2. The binding strength of LDHA and eEF2 was reduced by NADH-competing inhibitors such as FX11, GNE-140, GSK 2837808A, and AZ-3336–39 (Figure 6A). Meanwhile, the binding strength of LDHA and eEF2 was hardly affected by substrate-competing inhibitors such as sodium oxamate.40 In addition, protein synthesis was significantly upregulated by FX11 and stiripentol (Figure 6B). These data showed that LDHA inhibitors, especially NADH-competitive inhibitors, could promote protein synthesis by releasing eEF2 from LDHA. Furthermore, the location of eEF2 on ribosomes was detected in DAMI cells. The content of eEF2 bound to ribosomes was obviously increased after the treatment with stiripentol and FX11, which showed that LDHA interacted with eEF2 and reduced its location to the ribosome (Figure 6C). Interestingly, stiripentol, an antiepileptic drug regarded as a novel inhibitor of LDH,41,42 has an apparent translational regulatory ability and long-time clinical use experience.43 The functions of stiripentol in MK differentiation and platelet production were further determined. Figure 6D shows that PPF was significantly increased by stiripentol in LdhAf/f MKs, whereas PPF was not affected in LdhA−/− MKs in vitro. As is shown in Figure 6E, the number of mice platelets was significantly increased by day 4 (0.72 ± 0.06 × 109/L in 10% dimethyl sulfoxide–treated mice vs 0.93 ± 0.02 × 109/L in stiripentol-treated mice). The newborn platelet ratios were significantly increased after stiripentol administration, while platelet apoptosis and other blood cell counts remained unchanged (supplemental Figure 3B-E). Furthermore, stiripentol regulated the production of platelets in vivo through LDHA (Figure 6F). To investigate the therapeutic effect of stiripentol in thrombocytopenia, an immune thrombocytopenic purpura (ITP)44 mouse model was constructed through tail vein injection of anti-CD42b monoclonal antibody (Figure 6G). The rate of platelet regeneration in stiripentol-treated wild-type mice was faster than that in the dimethyl sulfoxide group (Figure 6H), suggesting that stiripentol had a significant therapeutic effect on ITP.
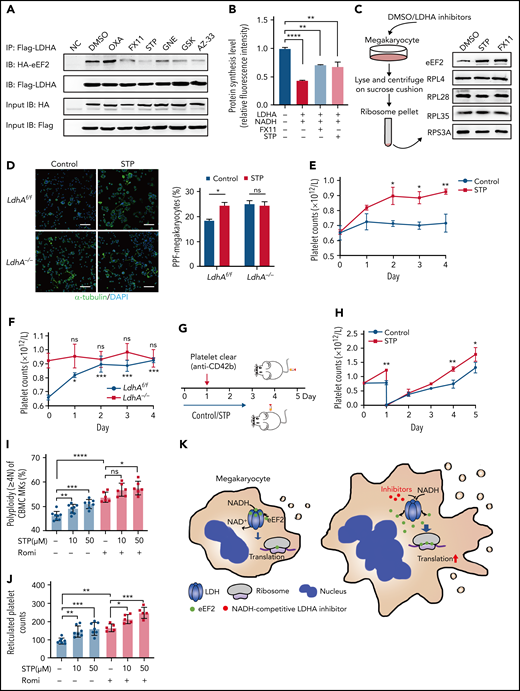
Screening and applying of LDHA inhibitors in thrombocytopenia. (A) Coimmunoprecipitation (Co-IP) of Flag-LDHA and HA-eEF2 in HEK-293T treated with LDHA inhibitors. OXA = sodium oxamate 1 mM; FX11 = FX11 20 μM; STP = stiripentol 100 μM; GNE = (R)-GNE-140 20 μM; GSK = GSK 2837808A 20 nM; AZ-33 = AZ-33 20 μM. (B) The levels of protein synthesis were measured when FX11 (1 μM) and STP (10 μM) were added in the rabbit reticulocyte translation system with 2.5 μg LDHA and 2 mM NADH (n = 3). (C) Isolation of human MK (DAMI) ribosomes and detection of eEF2 content on the ribosomes under dimethyl sulfoxide (DMSO)/LDHA inhibitors treated for 24 hours. Ribosomal protein L4 (RPL4), ribosomal protein L28 (RPL28), ribosomal protein L35 (RPL35), and ribosomal protein S3A (RPS3A) were used to indicate the equal loading. (D) Megakaryocyte PPF assay in mice fetal liver MKs incubated with STP (100 μM). DMSO was used as control. Statistical analysis of the PPF-MK/total MK ratio is shown (n = 4). Scale bar, 100 μm. (E) Platelet counts of wild-type mice that were administered STP (200 mg/kg per 12 hours) or 10% DMSO through intraperitoneal injection (n = 3; 8 weeks old, male). (F) Platelet counts of LdhAf/f and LdhA−/− mice that were administered STP (200 mg/kg per 12 hours) through intraperitoneal injection (n = 4; 8 weeks old, male). (G-H) STP (200 mg/kg per 12 hours) or 10% DMSO was intraperitoneally injected in wild-type mice, and platelets were cleared by tail IV injection with anti-CD42b antibody (2 μg/g weight) at hour 24; platelet counts were monitored at different time points (n = 3; 8 weeks old, male). (I) Polyploidy (≥4N) assay of cord blood mononuclear cell (CBMC)-derived MKs treated with STP (10 μM; n =8 or 50 μM; n = 7), romiplostim (Romi; 100 ng/mL) (n = 6), and the combination of STP with Romi (n = 6) by using flow cytometry. (J) Detection of reticulated platelet counts from CBMC-derived MK culture supernatant that were incubated with STP (10 μM or 50 μM; n = 7), Romi (100 ng/mL; n = 5), and the combination of STP with Romi (n = 5) according to flow cytometry. (K) Schematic diagram of eEF2 release from LDH with NADH or LDHA inhibitors to regulate translation in maturation and differentiation of MKs. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; NC, negative control; ns, not significant.
Screening and applying of LDHA inhibitors in thrombocytopenia. (A) Coimmunoprecipitation (Co-IP) of Flag-LDHA and HA-eEF2 in HEK-293T treated with LDHA inhibitors. OXA = sodium oxamate 1 mM; FX11 = FX11 20 μM; STP = stiripentol 100 μM; GNE = (R)-GNE-140 20 μM; GSK = GSK 2837808A 20 nM; AZ-33 = AZ-33 20 μM. (B) The levels of protein synthesis were measured when FX11 (1 μM) and STP (10 μM) were added in the rabbit reticulocyte translation system with 2.5 μg LDHA and 2 mM NADH (n = 3). (C) Isolation of human MK (DAMI) ribosomes and detection of eEF2 content on the ribosomes under dimethyl sulfoxide (DMSO)/LDHA inhibitors treated for 24 hours. Ribosomal protein L4 (RPL4), ribosomal protein L28 (RPL28), ribosomal protein L35 (RPL35), and ribosomal protein S3A (RPS3A) were used to indicate the equal loading. (D) Megakaryocyte PPF assay in mice fetal liver MKs incubated with STP (100 μM). DMSO was used as control. Statistical analysis of the PPF-MK/total MK ratio is shown (n = 4). Scale bar, 100 μm. (E) Platelet counts of wild-type mice that were administered STP (200 mg/kg per 12 hours) or 10% DMSO through intraperitoneal injection (n = 3; 8 weeks old, male). (F) Platelet counts of LdhAf/f and LdhA−/− mice that were administered STP (200 mg/kg per 12 hours) through intraperitoneal injection (n = 4; 8 weeks old, male). (G-H) STP (200 mg/kg per 12 hours) or 10% DMSO was intraperitoneally injected in wild-type mice, and platelets were cleared by tail IV injection with anti-CD42b antibody (2 μg/g weight) at hour 24; platelet counts were monitored at different time points (n = 3; 8 weeks old, male). (I) Polyploidy (≥4N) assay of cord blood mononuclear cell (CBMC)-derived MKs treated with STP (10 μM; n =8 or 50 μM; n = 7), romiplostim (Romi; 100 ng/mL) (n = 6), and the combination of STP with Romi (n = 6) by using flow cytometry. (J) Detection of reticulated platelet counts from CBMC-derived MK culture supernatant that were incubated with STP (10 μM or 50 μM; n = 7), Romi (100 ng/mL; n = 5), and the combination of STP with Romi (n = 5) according to flow cytometry. (K) Schematic diagram of eEF2 release from LDH with NADH or LDHA inhibitors to regulate translation in maturation and differentiation of MKs. *P < .05, **P < .01, ***P < .001, ****P < .0001. DAPI, 4′,6-diamidino-2-phenylindole; NC, negative control; ns, not significant.
Romiplostim has been shown to be successful in ITP treatment after standard treatment failure and is recommended as a second-line treatment.45 We found that stiripentol could promote MK ploidy and reticulated platelet production from human cord blood mononuclear cell–derived MKs alone, and also had a superposed effect with romiplostim (Figure 6I-J). These data indicate that LDHA inhibition might serve as a promising strategy in thrombocytopenia therapy (Figure 6K).
Discussion
During MK development, the active process of protein synthesis supports size increases, cytoskeletal protein content expansion, membrane system development, and so on.46 Various means by which interactions between RNA-binding proteins, translation initiation factors, and the ribosome can be regulated in cellular and developmental processes have been reported.47 However, the mechanism of how protein synthesis is regulated and whether targeting this process can be applied to promote or inhibit MK development and platelet production remains unclear. LDHA is a cytosolic enzyme, predominantly involved in anaerobic and aerobic glycolysis (Warburg effect). However, its multiple additional functions in non-neoplastic and neoplastic tissues have been reported in recent years, including as a transcription factor in the nucleus, an integral part of the sarcolemmal ATP-sensitive K+ channel in the heart, affecting the cell cycle, and so on.8 In this study, we revealed a novel function of LDHA in regulating protein synthesis by direct interaction with eEF2. eEF2 is a key translation elongation factor that is highly expressed in tumor cells with vigorous protein synthesis.48,49 Targeting eEF2 is considered to be an attractive strategy to inhibit tumor growth. It is widely believed that the function of eEF2 is mainly regulated through its phosphorylation by eEF2 kinase (eEF2K). Upon phosphorylation by eEF2K at Thr56, eEF2 is unable to locate on the ribosome and is thus inactivated.50 In this study, we found that eEF2K inhibitor A-484954 promoted MK differentiation, suggesting that eEF2 phosphorylation is involved in MK differentiation. However, the absence of LDHA did not affect the phosphorylation level of eEF2 (data not shown), indicating that the inhibitory role of LDHA in protein synthesis is eEF2K independent. Both ways of regulation of eEF2 by LDHA and eEF2K play important roles in protein synthesis and MK differentiation.
eEF2 binds to the N-terminal of LDHA, which is the main NADH-binding site. Moreover, by screening the LDHA inhibitors, we found that the binding of LDHA to eEF2 was inhibited by NADH-competitive inhibitors rather than substrate-competitive inhibitors, and the addition of NADH, but not substrates, enhanced the binding strength. These data showed that the binding of LDHA and eEF2 required NADH. As a cofactor, the formation of the LDH/NADH binary complex precedes the binding of pyruvate to the substrate-binding site, and the binding of NADH induces small conformational changes in the active-site loop and an adjacent helix.30 Therefore, we speculate that NADH binding causes a conformational shift of LDHA that increases its affinity for eEF2, although further research is needed to obtain concrete evidence. Stiripentol is an orphan drug that effectively reduces seizure frequency in infantile epilepsy as an adjunct therapy and also exhibits therapeutic advantages in improving the efficacy of other antiepileptic drugs.51 Mechanism studies have shown that stiripentol inhibits the activity of LDH through a nonsubstrate competitive pathway.41 However, the binding site and mechanism of stiripentol remain unclear. Here, we found that stiripentol could inhibit the binding of LDHA to eEF2 and promote protein synthesis, which is similar to the function of NADH-competitive inhibitors.
NAD(H) is an intracellular coenzyme involved in multiple biological processes.52 Two NADH molecules are formed when one glucose enters the glycolytic pathway. The energy-rich intermediates are then shuttled into the mitochondria to fuel the electron transport chain. Alternatively, the cytoplasmic NADH can also be oxidized during lactate fermentation.53 In normal cells, lactate fermentation is not preferred due to its ATP output (2 ATP/glucose) being much lower than that of the oxidative phosphorylation pathway (30-34 ATP/glucose). However, when cells face adverse conditions such as limited oxygen availability or mitochondrial capacity, NADH can be accumulated in both the mitochondria and the cytoplasm. In our findings, NADH has an obvious role in inhibiting protein synthesis by enhancing the detainment of eEF2 by LDHA. This may be a regulatory mechanism that helps the cell avoid excess energy consumption and adapt to environmental changes. In tumor cells, aerobic glycolysis is vigorous, and targeting LDHA is considered to be a promising strategy for antitumor therapy.54,55 However, some studies have pointed out that silencing LDHA does not inhibit the growth of normal cells and even promotes their proliferation.56 Our data here show that targeting LDHA with NADH-competitive inhibitors promotes protein synthesis, which may be a positive factor for cell growth. Therefore, the application of LDHA inhibitors in antitumor therapy should be carefully evaluated. In short, this study reports an unrevealed role of LDHA in protein synthesis and MK maturation, which may serve as a new target for thrombocytopenia therapy.
Acknowledgments
The authors thank the Core Facility of Basic Medical Sciences, Shanghai Jiao Tong University School of Medicine for support. They also thank Kevin Liu from the College of Arts and Sciences, Vanderbilt University, for proofreading the article.
This work was supported by the National Key R&D Program of China (2021YFA0804900, J.L.), National Natural Science Foundation of China (31830050, 82030004, and 81721004, J.L.; 81970123, X.F.; 81800129, L.Z.; 81900138, Y.X.; and 81900121, Y.H.), and China National Postdoctoral Program for Innovative Talents (BX2021186, H.J.). The authors thank members of Cambridge-Suda Genomic Resource Center for generating knockout mice supported by grant from the National Key R&D program of China (2018YFA0801100).
Authorship
Contribution: X.F., J.L., and Q.C. designed the experiments, analyzed data, and wrote the paper; Q.C., M.X., and L.W. performed the experiments; and L.L., Y.S., H.J., Y.G., Y.W., L.Z., Y.X., and Y.H. helped with the experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xuemei Fan, Department of Biochemistry and Molecular Cell Biology, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Rd, Shanghai 200025, China; e-mail: fanxuemei@sjtu.edu.cn; or Junling Liu, Department of Biochemistry and Molecular Cell Biology, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Collaborative Innovation Center of Hematology, Shanghai Jiao Tong University School of Medicine; Shanghai Synvida Biotechnology Co, Ltd, 280 South Chongqing Rd, Shanghai 200025, China; e-mail: liujl@shsmu.edu.cn; or Yu Hou, Department of Hematology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, 107 Wenhuaxi Rd, Jinan 250012, China; e-mail: houyu2009@sina.com.
Requests for data sharing may be submitted to Xuemei Fan (fanxuemei@sjtu.edu.cn). Mass spectrometry data are available via ProteomeXchange with identifier PXD030794. Quantitative proteomics analysis data are available via ProteomeXchange with identifier PXD030795.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal