

Key Points
Results support our previously published model and reveal the surprising role of the a1-loop in presenting Arg320 for initial cleavage.
Using pseutarin C as model prothrombinase, the interaction site for prothrombin was probed by site-directed PEGylation and other mutations.

Abstract
The prothrombinase complex processes prothrombin to thrombin through sequential cleavage at Arg320 followed by Arg271 when cofactor, factor (f) Va, protease, fXa, and substrate, prothrombin, are all bound to the same membrane surface. In the absence of the membrane or cofactor, cleavage occurs in the opposite order. For the less favorable cleavage site at Arg320 to be cleaved first, it is thought that prothrombin docks on fVa in a way that presents Arg320 and hides Arg271 from the active site of fXa. Based on the crystal structure of the prothrombinase complex from the venom of the Australian eastern brown snake, pseutarin C, we modeled an initial prothrombin docking mode, which involved an interaction with discrete portions of the A1 and A2 domains of fV and the loop connecting the 2 domains, known as the a1-loop. We interrogated the proposed interface by site-directed PEGylation and by swapping the a1-loop in pseutarin C with that of human fV and fVIII and measuring the effect on rate and pathway of thrombin generation. PEGylation of residues within our proposed binding site greatly reduced the rate of thrombin generation, without affecting the pathway, whereas those outside the proposed interface had no effect. PEGylation of residues within the a1-loop also reduced the rate of thrombin generation. The sequence of the a1-loop was found to play a critical role in prothrombin binding and in the presentation of Arg320 for initial cleavage.
Introduction
Thrombin is a serine protease with many functions in the process of blood coagulation (hemostasis), including the activation of platelets and the formation of fibrin, the 2 principal components of a hemostatic clot, and it must be generated rapidly at the site of vascular damage to prevent blood loss.1,2 The enzyme that converts the zymogen prothrombin to the active protease thrombin is the prothrombinase complex, composed of a cofactor, factor (f) Va, and a serine protease, fXa, assembled on the surface of a negatively charged phospholipid membrane in the presence of calcium ions.1,3,4 The molecular basis of prothrombinase assembly and function is of central importance to understanding normal hemostasis and its dysregulation, which can result in bleeding or thrombosis.5,6
fV circulates in blood as a large (330 kDa) single-chain glycoprotein composed of 3 A domains, 2 membrane-anchoring C domains, and an unstructured highly glycosylated B domain that maintains fV in an inactive procofactor state.7 Thrombin activates fV to fVa by cleavage at 3 sites, Arg709, Arg1018, and Arg1545, excising the B domain and unveiling the binding site of fXa.8 The heavy (A1‐A2) and light (A3‐C1‐C2) chains of fVa remain noncovalently associated in the presence of divalent copper and calcium ions. fXa is activated from its zymogen precursor fX by the extrinsic or intrinsic Xase complexes and is composed of a light chain containing the membrane-anchoring γ-carboxyglutamic acid (Gla) domain and 2 epidermal growth factor-like (EGF) domains, linked via disulfide bond to the heavy chain comprising the serine protease domain.9,10 The affinity of fXa for fVa is sufficiently low that prothrombinase can form only when both components are bound to the same membrane surface.11
Prothrombin is a single-chain molecule containing a Gla domain, 2 kringle domains (K1 and K2), and the protease domain.12 Prothrombin activation requires cleavage at 2 sites, Arg271 and Arg320 (Figure 1A).13 When prothrombinase is assembled on a membrane surface, the first cleavage event takes place at Arg320, resulting in formation of the active intermediate, meizothrombin, followed by the second cleavage at Arg271 to release the active protease thrombin from the Gla and kringle domains (F1.2).14 fXa alone or in the absence of both fVa and membranes cleaves prothrombin by a different route: first at Arg271 and then more slowly at Arg320.14 The most compelling explanation for this observation is that fXa inherently favors the sequence and/or accessibility of the 271 site, but that prothrombin interacts with fVa of prothrombinase in a manner that presents the less favored 320 site for cleavage.15-17 Once cleavage at Arg320 occurs, the protease domain of prothrombin undergoes a conformational change on conversion to the active intermediate meizothrombin, altering its interaction with prothrombinase to present the 271 site for cleavage. Although there have been reports suggesting that 2 separate conformations of prothrombinase account for the 2 cleavage events (ie, 2-enzyme hypothesis), the balance of data supports a single enzyme conformation and 2 distinct substrate binding modes.15-19 The 2-substrate mechanism is known as “ratcheting” and is predicated on the existence of an extensive interaction interface between prothrombin/meizothrombin and fVa.16,20,21
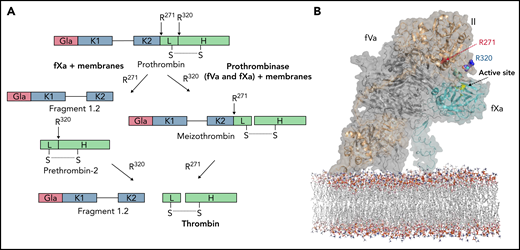
Prothrombin activation pathways and proposed model for prothrombin docking. (A) Schematic illustrates the 2 pathways for prothrombin processing by fXa and prothrombinase. fXa, with or without membranes, cleaves first at R271, releasing prethrombin-2 from fragment 1.2 (F1.2), followed by slow cleavage at R320 to form thrombin. Fully assembled prothrombinase first cleaves R320, leading to formation of the active intermediate, meizothrombin, followed by cleavage at R271, releasing thrombin from F1.2. (B) Representation of the proposed mode of prothrombin docking onto fully assembled prothrombinase. Prothrombin (gold semitransparent surface over cartoon) binds from the membrane (sticks) along the side of fVa (gray) to present the R320 cleavage site (indicated) to the active site of fXa (cyan; active site yellow). This initial docking mode keeps the otherwise favored R271 site remote from the active site of fXa.
Prothrombin activation pathways and proposed model for prothrombin docking. (A) Schematic illustrates the 2 pathways for prothrombin processing by fXa and prothrombinase. fXa, with or without membranes, cleaves first at R271, releasing prethrombin-2 from fragment 1.2 (F1.2), followed by slow cleavage at R320 to form thrombin. Fully assembled prothrombinase first cleaves R320, leading to formation of the active intermediate, meizothrombin, followed by cleavage at R271, releasing thrombin from F1.2. (B) Representation of the proposed mode of prothrombin docking onto fully assembled prothrombinase. Prothrombin (gold semitransparent surface over cartoon) binds from the membrane (sticks) along the side of fVa (gray) to present the R320 cleavage site (indicated) to the active site of fXa (cyan; active site yellow). This initial docking mode keeps the otherwise favored R271 site remote from the active site of fXa.
Several studies have investigated this interface, mostly by assessing the effect of mutations in human fV (hfV) on the rate and pathway of thrombin formation.22,23 In 2013, we published the crystal structure of a prothrombinase complex from the venom of the Australian eastern brown snake (Pseudonaja textilis).24 The complex, known as pseutarin C, consists of a catalytic subunit designated ptfXa (∼43 kDa) and a nonenzymatic subunit designated ptfV (∼163 kDa) that are orthologs of hfXa and hfV, with sequence identity of ∼50% (fX is 47.2% identical and B-domainless fV is 57.8% identical).24 It processes human prothrombin in the same way as fully assembled human prothrombinase (mostly through the meizothrombin intermediate), even in the absence of a phospholipid membrane. This suggests a conserved binding site for prothrombin on the fVa component of pseutarin C and human prothrombinase. Analysis of conserved N-linked glycosylation sites on fVa across 37 species revealed a glycosylation-free channel from the membrane surface to the active site of fXa into which we were able to dock prothrombin in a manner that presented Arg320 for cleavage (Figure 1B).24,25 Because prothrombin is organized into 2 structured regions (Gla/K1, known as F1, and K2/protease, known as prethrombin-1 [Pre-1]) separated by a long unstructured linker, the interaction interface relevant for understanding its sequential cleavage involves only Pre-1 and fVa. The proposed interface is a ridge formed by the A1 and A2 domains of fVa and the loop linking these domains, known as the a1-loop.
In this study, we investigated the interaction between fVa and prothrombin by mutating residues within and outside the proposed binding site on fV from pseutarin C (ptfV) and assessing the effects on the processing of human prothrombin. This system is ideal for such a study for several reasons: (1) the fXa component of pseutarin C (ptfXa) has no prothrombin cleavage activity in the absence of fV(a), so no background processing by free ptfXa takes place; (2) ptfXa is not allosterically activated by binding to ptfV, so prothrombin cleavage can occur only if prothrombin interacts with the fV component of pseutarin C; (3) the complex forms in the absence of membranes but still processes prothrombin predominantly through the meizothrombin intermediate, rendering phospholipid vesicles unnecessary; (4) the components of pseutarin C are much easier to produce in quantities required for biochemical and functional analyses than those of human prothrombinase; and (5) ptfV is fully active as a single-chain molecule and does not require activation by thrombin.
To probe the interface between prothrombin and pseutarin C, we mutated selected residues on the A1, A2, and A3 domains of ptfV to Cys for modification with a large polyethylene glycol (PEG) moiety to sterically interfere with docking. Residues in the a1-loop were also swapped with those of hfV and the unrelated fVIII to assess the role of a1-loop composition and length in prothrombin binding and processing. Our findings are consistent with the proposed prothrombin binding interface on fVa24 and reveal the surprisingly important role of the a1-loop in the initial interaction with prothrombin and presentation of the Arg320 site for cleavage.
Methods
Recombinant protein expression and purification
Full-length venom ptfX DNA (accession #AY631238.1) was synthesized (Gene Art), and the region encoding the full-length protein, including the prothrombin signal peptide for γ-carboxylation, was cloned into the pCEP4 vector and stably transfected into HEK-EBNA cells. To facilitate the removal of the activation peptide, the activation cleavage site was modified to that of hfX. Protein expression and purification are detailed in the data supplement. The gene coding the venom form of ptfV (#AY168281.1) with the human signal sequence was purchased from Gene Art and cloned into the pED vector for expression in BHK-M cells, as previously described.24,26 The free cysteine residue at position 540 was mutated to alanine (C540A) as the background before introducing subsequent cysteine mutations using the QuickChange Lightning (Agilent) site-directed mutagenesis kit. Plasmids carrying the correct mutations were stably transfected into BHK-M cells and expressed as described previously.24 fV cysteine variants were purified by a 2-step purification using Q FF and S FF ion exchange columns as described previously.24,26 Purified variants were concentrated and dialyzed into 50 mM of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) and 150 mM of sodium chloride (NaCl; pH 7.0) for labeling experiments. Because ptfV has a small B domain and is constitutively active, it does not require activation before use in functional assays. The gene coding the human prothrombin sequence (M17262.1) was purchased from Gene Art and cloned into the pCEP4 vector for stable expression in HEK-EBNA cells (Supplemental data, available on the Blood Web site).
Recombinant prothrombin was purified by a multistep purification process as described in the Supplemental data.
Site-specific labeling of fV cysteine variants and separation of labeled proteins on streptavidin mutein resin
Purified ptfV cysteine variants were reduced with Pierce Immobilized TCEP disulfide reducing gel (Thermo Fisher Scientific) at room temperature for 45 minutes, as per the manufacturer’s instructions. Reduced samples were separated from the TCEP gel by centrifugation using a spin cup paper filter and then labeled with 20-fold molar excess of maleimide-PEG40K (Merck) or EZ-Link maleimide-PEG11-biotin (922.09 Da; Thermo Fisher Scientific) at 4°C for 2 hours. Maleimide-PEG40K–labeled samples were used to estimate labeling efficiency by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE; Table 1; supplemental Figure 1). Maleimide-PEG11-biotin–labeled samples were applied to a Q Sepharose column (Cytiva; 1 mL) followed by dialysis to remove excess labeling reagent. Peak fractions containing both the mixture of labeled and unlabeled ptfV were pooled and dialyzed into a buffer of 100 mM of potassium phosphate and 150 mM of NaCl (pH 7.2) overnight at 4°C. Dialyzed samples were applied to a column packed with streptavidin mutein resin (Roche) to separate biotinylated from unbiotinylated protein, as per the manufacturer’s instructions (supplemental Figure 2). Peak fractions containing biotinylated ptfV were concentrated and dialyzed overnight into a buffer of 20 mM of Tris (pH 7.4), 150 mM of NaCl, and 2 mM of calcium chloride at 4°C.
Expression levels and labeling efficiency of ptfV Cys variants
| Mutation position | fV domain | BHK-M stable expression, mg/L media | PEGylation efficiency, %* |
|---|---|---|---|
| K134C | A1 | 1.95 | 49 |
| E168C | A1 | 1.65 | 72 |
| T276C | A1 | 2.27 | 76 |
| C221A (C302) | A1 | 0.38 | 39 |
| R310C | a1 | 1.26 | 65 |
| K311C | a1 | 0.90 | 71 |
| L312C | a1 | 2.89 | 75 |
| F314C | a1 | 2.01 | 85 |
| R315C | a1 | 4.38 | 85 |
| E316C | a1 | 4.83 | 50 |
| K321C† | a1 | NA | NA |
| T378C† | A2 | NA | NA |
| T403C | A2 | 4.63 | <6 |
| V420C | A2 | 3.55 | <15 |
| N441C | A2 | 1.66 | 83 |
| T455C | A2 | 2.93 | 27 |
| C540‡ | A2 | 3.98 | 59 |
| C540A§ | A2 | 2.09 | ND |
| S578C | a2 | 2.76 | 36 |
| Mutation position | fV domain | BHK-M stable expression, mg/L media | PEGylation efficiency, %* |
|---|---|---|---|
| K134C | A1 | 1.95 | 49 |
| E168C | A1 | 1.65 | 72 |
| T276C | A1 | 2.27 | 76 |
| C221A (C302) | A1 | 0.38 | 39 |
| R310C | a1 | 1.26 | 65 |
| K311C | a1 | 0.90 | 71 |
| L312C | a1 | 2.89 | 75 |
| F314C | a1 | 2.01 | 85 |
| R315C | a1 | 4.38 | 85 |
| E316C | a1 | 4.83 | 50 |
| K321C† | a1 | NA | NA |
| T378C† | A2 | NA | NA |
| T403C | A2 | 4.63 | <6 |
| V420C | A2 | 3.55 | <15 |
| N441C | A2 | 1.66 | 83 |
| T455C | A2 | 2.93 | 27 |
| C540‡ | A2 | 3.98 | 59 |
| C540A§ | A2 | 2.09 | ND |
| S578C | a2 | 2.76 | 36 |
NA, not applicable; ND, none detected.
Small-scale labeling was conducted with maleimide-PEG40K. All variants were run on 3% to 8% Tris-acetate gel postlabeling with maleimide-PEG40K (supplemental Figure 1). Percentage of PEGylation was estimated by comparing the PEGylated band with unPEGylated band on SDS-PAGE by densitometry.
BHK-M cells were transfected 3 times and stable cells selected; however, no expression was observed.
P textilis fV has 15 cysteine residues in total, 14 of which form disulfide bonds. C540 is a solvent-exposed free cysteine at the back of the A2 domain.
C540A mutation was generated to see if labeling is specific for cysteine residues. All mutations to introduce single cysteine residues were made on this background.
Swapping the ptfV a1-loop with that of hfV and hfVIII
Chimeric variants of ptfV carrying either the hfV a1-loop (ptfV_hfVa1) or the hfVIII a1-loop (ptfV_hfVIIIa1) were created by a Klenow-based ligation independent technique.27 Primers were designed to have 24-bp overlaps, and the pED-ptfV plasmid generated in the previous study used as a template in the reaction (supplemental Figure 3).24 All plasmids used in this study were purified and sequenced before transfection, and protein was purified as described.
Thrombin generation assays
The order of prothrombin cleavage was visualized by SDS-PAGE (Supplemental data). Thrombin generation was also monitored by chromogenic assay. In this assay, ptfV variants (2.5 μL and 50 nM final concentration) and ptfXa (2.5 μL and 1 nM final concentration) were added to a 96-well plate in an assay buffer (40 μL) containing 20 mM of HEPES, 0.15 M of NaCl, and 5 mM of calcium chloride (pH 7.4). Prothrombin (5 μL and 20 nM final concentration) was added, and time points were collected. Thrombin activity was assessed by addition of S-2238 (50 μL and 200 μM final concentration) and monitoring absorbance at 405 nm using a Versamax plate reader (Molecular Devices). The chromogenic assay does not distinguish between thrombin and meizothrombin, which also cleaves S-2238. Data were obtained from 2 independent experiments, performed at least in duplicate, and plotted with GraphPad Prism.
Results
Site-directed PEGylation method validation and controls
To investigate the prothrombin binding site on ptfV, we established a method to quantitatively PEGylate residues that had been mutated to cysteine. Similar to site-directed glycosylation, the addition of a large PEG moiety to a region involved in an interaction with another protein should be inhibitory.28 However, because even a small amount of residual unlabeled material would give a signal of prothrombin processing, it was necessary to develop a method to purify the PEGylated ptfV away from any unlabeled ptfV.
First, to test the efficiency of the conjugation method, we labeled wild-type ptfV, which has a free cysteine residue (Cys540), with maleimide-PEG40K and monitored change in molecular weight by SDS-PAGE (supplemental Figure 1). Labeling efficiency was good (∼60%) but underscored the need to purify labeled material from unlabeled for use in activity assays. To achieve this, maleimide-PEG11-biotin was used for labeling, and separation was achieved using a streptavidin mutein resin, as detailed in the Methods section (supplemental Figure 3). Cys540 is located on the back of the A2 domain in a region that is unlikely to be involved in prothrombin binding (Figure 2A), so the addition of PEG here was expected to have no effect on thrombin generation. Indeed, this is what was observed, with the PEGylated C540 exhibiting identical kinetics to unlabeled wild-type ptfV (Figure 2B).
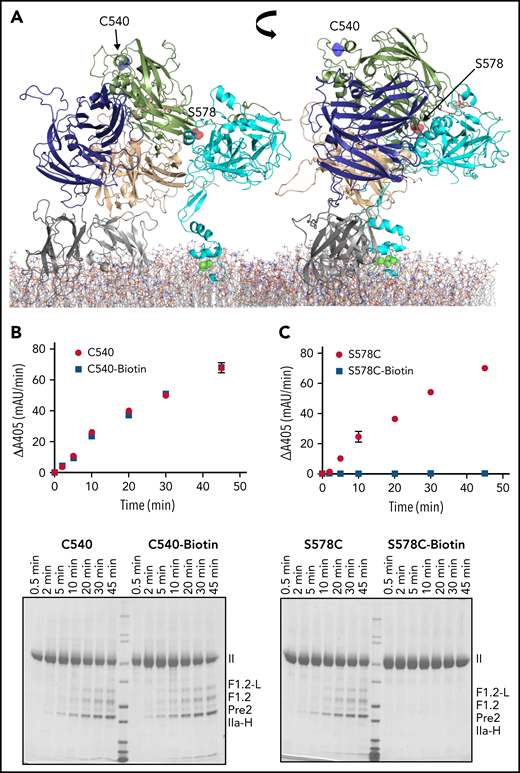
Validation of site-directed PEGylation method. (A) Cartoon representation of full-length pseutarin C (A1, purple; A2, green; A3, beige; C domains, gray; and fXa, cyan), indicating the position of Cys540 (blue balls) and Ser578 (red balls) that were selected as controls to validate the PEGylation method. ptfV has a free cysteine residue (Cys540) at the back of the molecule (A2 domain, green). This residue was selected as a negative control, because PEGylation was not expected to affect prothrombin binding. Ser578 is located in the binding interface between ptfV and ptfXa (cyan) in the pseutarin C structure and was therefore selected as a positive control to test the effect of PEGylation on pseutarin C activity. Both positions were labeled with maleimide-PEG11-biotin as described in the Methods section. The effects of both labeled (blue squares) and unlabeled (red circles) Cys540 (B) and S578C (C) were tested for prothrombin processing using chromogenic (top) and SDS-PAGE (bottom) assays. As expected, PEGylation of Cys540 had no effect on thrombin generation, whereas PEGylation of S578C completely prevented processing of prothrombin by pseutarin C. II, prothrombin; F1.2-L, F1.2+light chain; IIa-H, thrombin heavy chain; mAu, milli–absorbance unit.
Validation of site-directed PEGylation method. (A) Cartoon representation of full-length pseutarin C (A1, purple; A2, green; A3, beige; C domains, gray; and fXa, cyan), indicating the position of Cys540 (blue balls) and Ser578 (red balls) that were selected as controls to validate the PEGylation method. ptfV has a free cysteine residue (Cys540) at the back of the molecule (A2 domain, green). This residue was selected as a negative control, because PEGylation was not expected to affect prothrombin binding. Ser578 is located in the binding interface between ptfV and ptfXa (cyan) in the pseutarin C structure and was therefore selected as a positive control to test the effect of PEGylation on pseutarin C activity. Both positions were labeled with maleimide-PEG11-biotin as described in the Methods section. The effects of both labeled (blue squares) and unlabeled (red circles) Cys540 (B) and S578C (C) were tested for prothrombin processing using chromogenic (top) and SDS-PAGE (bottom) assays. As expected, PEGylation of Cys540 had no effect on thrombin generation, whereas PEGylation of S578C completely prevented processing of prothrombin by pseutarin C. II, prothrombin; F1.2-L, F1.2+light chain; IIa-H, thrombin heavy chain; mAu, milli–absorbance unit.
The free cysteine at position 540 was mutated to alanine (C540A) to create a template for introducing cysteines at other positions. This variant was also used to assess the specificity of maleimide-PEG40K for cysteine, because it is also able to modify primary amines under certain conditions. Under the conditions used, no labeling of ptfV C540A was detected with maleimide-PEG40K by SDS-PAGE (supplemental Figure 1).
To assess the ability of PEGylation to disrupt protein-protein interactions in this system, we mutated Ser578, which is in the binding interface between ptfXa and ptfV in the pseutarin C structure, to cysteine (Figure 2A). The S578C variant was labeled with maleimide-PEG11-biotin, purified, and used in thrombin generation assays (Figure 2C). The unlabeled S578C variant was able to process prothrombin in the presence of ptfXa in a manner indistinguishable from wild type. However, labeling this site with maleimide-PEG11-biotin completely abrogated thrombin generation (Figure 2C), confirming the ability of PEGylation to disrupt/inhibit protein-protein interactions.
Effect of site-directed PEGylation of ptfV on thrombin generation
The proposed binding site for the globular portion of prothrombin, composed of the second kringle domain and the protease domain (Pre-1), on fVa comprises parts of the A1 and A2 domains and the a1-loop linking them. Eighteen positions within and outside the proposed binding site for prothrombin were selected for mutation to cysteine, and we successfully expressed and labeled 14 in sufficient quantities for functional studies (Table 1).
In the A1 domain, Lys134, Glu168, and Thr276 were mutated to cysteine and labeled with maleimide-PEG11-biotin. In addition, Cys221 was mutated to alanine to disrupt the disulfide bridge with Cys302, therefore making Cys302 available for PEGylation (Figure 3A). Based on the model of Pre-1 docked onto ptfV, we predicted that 134 and 168 would be outside of the binding interface and 276 and 302 would be within the interface. Thrombin generation was monitored by chromogenic assay, and reaction products were visualized by SDS-PAGE (Figure 3B). Consistent with the modeled interface, PEGylation at residues 134 and 168 had no effect on thrombin generation, whereas PEGylation at 276 and 302 slowed thrombin generation by seven- and 12.6-fold, respectively (Figure 3B).
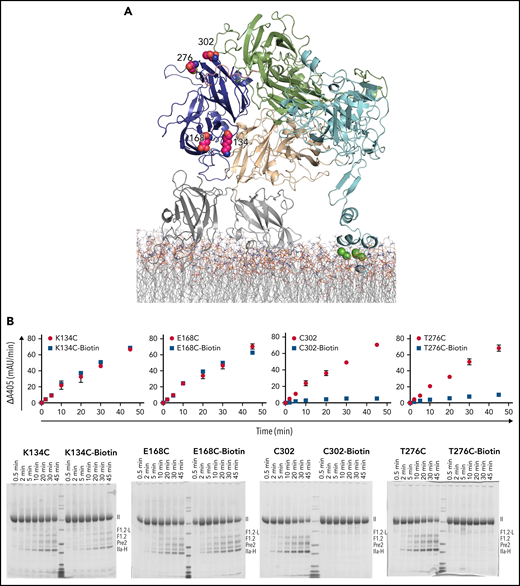
Site-directed PEGylation of A1 domain residues and effect on prothrombin processing. (A) Cartoon representation of full-length pseutarin C, colored as in Figure 2, indicating the positions of mutated and PEGylated residues on the A1 domain, shown as magenta balls. (B) Effects of both Cys mutation and PEGylation on thrombin generation were tested by chromogenic (top) and SDS-PAGE (bottom) assays (labeled as in Figure 2). II, prothrombin; F1.2-L, F1.2+light chain; IIa-H, thrombin heavy chain; mAu, milli–absorbance unit.
Site-directed PEGylation of A1 domain residues and effect on prothrombin processing. (A) Cartoon representation of full-length pseutarin C, colored as in Figure 2, indicating the positions of mutated and PEGylated residues on the A1 domain, shown as magenta balls. (B) Effects of both Cys mutation and PEGylation on thrombin generation were tested by chromogenic (top) and SDS-PAGE (bottom) assays (labeled as in Figure 2). II, prothrombin; F1.2-L, F1.2+light chain; IIa-H, thrombin heavy chain; mAu, milli–absorbance unit.
Similarly, PEGylation of selected residues within the A2 domain was assessed for effect on thrombin generation. Thr378, Thr403, Val420, Asn441, and Thr455 were mutated to cysteine and transfected to BHK-M cells for protein expression. Despite several transfection attempts, no expression was detected for T378C, and although stable clones were successfully selected for the remaining mutants, T403C and V420C had very low labeling efficiency (<6% and <15%, respectively; Table 1). Therefore, on the A2 domain, only N441C and T455C were labeled successfully with maleimide-PEG11-biotin, purified, and tested in thrombin generation assays (Figure 4A). PEGylation of N441C and T455C slowed thrombin generation by 408- and 12.4-fold, respectively (Figure 4B).
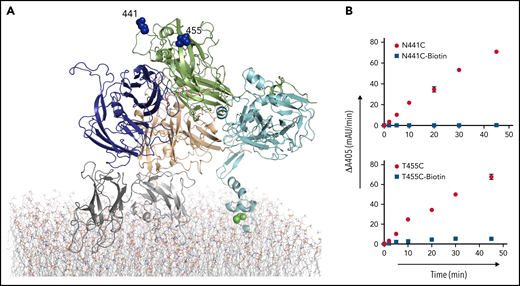
Site-directed PEGylation of A2 domain residues and effect on prothrombin processing. (A) Cartoon representation of full-length pseutarin C, colored as in Figure 2, indicating the positions of mutated and PEGylated residues on the A2 domain, shown as blue balls. (B) Effects of both Cys mutation (red circle) and PEGylation (blue square) on thrombin generation were tested by the chromogenic assay. mAu, milli–absorbance unit.
Site-directed PEGylation of A2 domain residues and effect on prothrombin processing. (A) Cartoon representation of full-length pseutarin C, colored as in Figure 2, indicating the positions of mutated and PEGylated residues on the A2 domain, shown as blue balls. (B) Effects of both Cys mutation (red circle) and PEGylation (blue square) on thrombin generation were tested by the chromogenic assay. mAu, milli–absorbance unit.
Individual residues on the a1-loop, Arg310, Lys311, Leu312, Phe314, Arg315, Glu316, and Lys321, were selected for mutation to cysteine and PEGylation (Table 1). K321C did not express, but all other a1-loop variants were successfully expressed, purified, and labeled with maleimide-PEG11-biotin (Figure 5A). PEGylation at residues 310, 311, 314, and 315 had similar effects on thrombin generation (9.4-, 5.4-, 7.4-, and 12.5-fold reduction, respectively). However, PEGylation at 312 and 316 reduced the rate of thrombin generation by only 2.5- and 1.7-fold, respectively (Figure 5B).
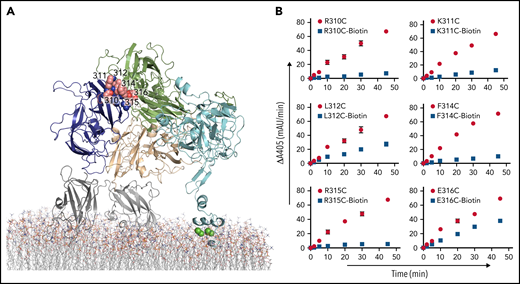
Site-directed PEGylation of a1-loop residues and their effect on thrombin generation. (A) Position of a1-loop mutations (balls) indicated in a cartoon representation of pseutarin C. (B) Effects of both Cys mutation (red circle) and PEGylation (blue square) on thrombin generation were tested by the chromogenic assay. mAu, milli–absorbance unit.
Site-directed PEGylation of a1-loop residues and their effect on thrombin generation. (A) Position of a1-loop mutations (balls) indicated in a cartoon representation of pseutarin C. (B) Effects of both Cys mutation (red circle) and PEGylation (blue square) on thrombin generation were tested by the chromogenic assay. mAu, milli–absorbance unit.
Effect of a1-loop composition on prothrombin processing
The effect of PEGylation of individual residues on the a1-loop confirms its involvement in prothrombin binding. The a1-loop is likely to have evolved to recognize, with some specificity, prothrombin from its target species; for pseutarin C, it will be primarily rodent prothrombin, and for hfV, it will be human prothrombin. To investigate the effect of a1-loop composition on human prothrombin processing by pseutarin C, we swapped the a1-loop on ptfV with those of hfV and hfVIII. The fVIII a1-loop is much longer than that of hfV and is cleaved by thrombin as a necessary part of its activation to fVIIIa, a key event enabling recognition of the substrate fX.29 The a1-loops of ptfV and hfV are both 20 amino acids in length; however, they differ greatly in composition: the hfV a1-loop has 11 basic and 1 acidic residues, and that of ptfV has 5 basic and 2 acidic residues (Figure 6A). The a1-loop of hfVIII is highly acidic, with 16 negatively charged residues, including a sulfated tyrosine (Figure 6A).
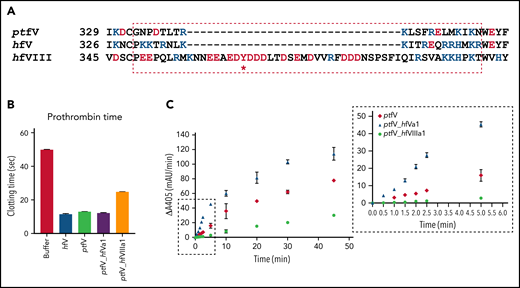
Effect of ptfV chimeric variants on prothrombin time and thrombin generation in fV-depleted human plasma. (A) Sequence alignment of the a1-loop regions of ptfV, hfV, and hfVIII with 4 flanking residues to either side is shown (* indicates sulfation). (B) Prothrombin times were determined in fV-depleted human plasma before and after spiking in the ptfV chimeric variants (ptfV_hfVa1 and ptfV_hfVIIIa1) and ptfV and B-domainless hfV as controls. (C) Thrombin generation was measured in a purified system using human prothrombin, ptfXa, and ptfV or the 2 chimeric variants by the chromogenic assay. Inset shows early time points between 0 and 5 minutes used in calculating initial rates. mAu, milli–absorbance unit.
Effect of ptfV chimeric variants on prothrombin time and thrombin generation in fV-depleted human plasma. (A) Sequence alignment of the a1-loop regions of ptfV, hfV, and hfVIII with 4 flanking residues to either side is shown (* indicates sulfation). (B) Prothrombin times were determined in fV-depleted human plasma before and after spiking in the ptfV chimeric variants (ptfV_hfVa1 and ptfV_hfVIIIa1) and ptfV and B-domainless hfV as controls. (C) Thrombin generation was measured in a purified system using human prothrombin, ptfXa, and ptfV or the 2 chimeric variants by the chromogenic assay. Inset shows early time points between 0 and 5 minutes used in calculating initial rates. mAu, milli–absorbance unit.
Wild-type ptfV and the resulting chimeras, ptfV_hfVa1 and ptfV_hfVIIIa1, were produced and tested for ability to clot fV-depleted human plasma, using B-domainless hfV as control. Clotting was initiated with vesicles, Ca2+, and tissue factor in a classic prothrombin time determination. Under these conditions, the mean clotting times were 11.5 seconds for hfV, 13.1 seconds for ptfV, 12.2 seconds for ptfV_hfVa1, and 24.8 seconds for ptfV_hfVIIIa1 (Figure 6B). These results confirm the importance of the a1-loop in recognizing prothrombin, with substitution with that of hfVIII doubling the clotting time and swapping in the a1-loop with that of hfV significantly reducing the time to clot human plasma (unpaired t test P = .0026). fXa in this experiment was endogenous, hfXa, which was previously shown to bind to ptfV with appreciable affinity.26
To assess the role of the a1-loop in prothrombin processing in a purified system, thrombin generation assays were run with ptfV or the 2 a1-loop chimeras, ptfXa and human prothrombin (Figure 6C). Comparison of the slopes at the early time points (Figure 6C inset) revealed that swapping the a1-loop with that of hfV increased the rate of thrombin generation by 3.6-fold. In contrast, the a1-loop from hfVIII reduced the rate by 6.6-fold. Prothrombin processing was also monitored by visualizing reaction products on SDS-PAGE (Figure 7A). Under the conditions used in this experiment, wild-type pseutarin C took ∼25 minutes to cleave half of the prothrombin, whereas the hfV a1-loop chimera took only 10 minutes. The hfVIII a1-loop chimera had cleaved only ∼30% of the prothrombin by the end of the experiment (60 minutes; Figure 7B).
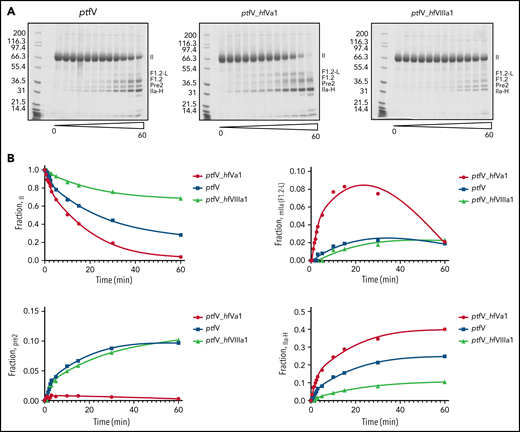
Monitoring of cleavage of prothrombin by ptfV chimeric variants using SDS-PAGE analysis. (A) Prothrombin processing by ptfXa in the presence of ptfV, ptfV_hfVa1, and ptfV_hfVIIIa1 was monitored for up to 60 minutes, and reaction products at selected time points were visualized by SDS-PAGE. (B) Fraction of total for each band was determined by densitometry analysis and plotted against time to assess disappearance of prothrombin (II) and appearance Pre-2 and meizothrombin (mIIa), inferred from F1.2+light chain (F1.2-L) band and thrombin heavy chain (IIa-H), as indicated.
Monitoring of cleavage of prothrombin by ptfV chimeric variants using SDS-PAGE analysis. (A) Prothrombin processing by ptfXa in the presence of ptfV, ptfV_hfVa1, and ptfV_hfVIIIa1 was monitored for up to 60 minutes, and reaction products at selected time points were visualized by SDS-PAGE. (B) Fraction of total for each band was determined by densitometry analysis and plotted against time to assess disappearance of prothrombin (II) and appearance Pre-2 and meizothrombin (mIIa), inferred from F1.2+light chain (F1.2-L) band and thrombin heavy chain (IIa-H), as indicated.
Disappearance of the prothrombin band can occur through initial cleavage at Arg320, resulting in meizothrombin, or at Arg271, resulting in Pre-2. Although the F1.2-L fragment, the hallmark of meizothrombin, was observed for all versions of pseutarin C, only the hfV a1-loop chimera appeared to process exclusively through the meizothrombin intermediate, as indicated by the rapid formation of F1.2-L and the complete absence of the band corresponding to Pre-2 (Figure 7A-B). This suggests increased affinity for prothrombin and/or improved presentation of Arg320 to the active site of ptfXa. It also confirms the importance of the a1-loop of fVa in the docking and correct positioning of prothrombin on pseutarin C.
Discussion
Biochemical and mutagenesis studies conducted over the years have shed some light on prothrombinase assembly and prothrombin binding, although with conflicting results.22,28,30-32 Several models of prothrombinase have also been generated in an attempt to understand the interactions between the components, mostly using partial crystal structures guided by biochemical data.33-36 A major breakthrough in the field came with the availability of the crystal structure of the first assembled prothrombinase complex from P textilis, pseutarin C.24 Close sequence homology with the human components and consistency with much of the available biochemical data suggested that it could serve as a reliable blueprint for human prothrombinase.25,37
However, an important question could not be answered by the structure or resulting models: how does prothrombin bind to assembled prothrombinase? Based on electrostatic complementarity, glycosylation-free patches, and the requirement for presentation of the constrained Arg320 cleavage site into the active site of fXa, we proposed a potential binding location of prothrombin on fVa, as well as a plausible mechanism for the sequential processing of prothrombin, referred to as ratcheting.24 It was proposed that the Gla and first kringle domains of prothrombin bind to the membrane and then interact with the side of the C2 domain of fVa. The long unstructured linker would then thread through an unglycosylated channel to place Pre-1 (second kringle domain and the catalytic domain) on a ledge composed of elements of the A1 and A2 domains and the a1-loop between them.24,38 The highly basic nature of the A1-a1-A2 ledge necessitated interaction with an acidic face of Pre-1, which coincidently placed the Arg320 site in close proximity of the active site of fXa.24 This initial docking interface involved Arg271, rendering it inaccessible to cleavage. The conformational change conferred by cleavage at Arg320 (conversion to the active intermediate meizothrombin) alters the shape and electrostatics of the proposed interface with fVa, reorienting the Arg271 site to present it for cleavage.24 The ultimate test of this hypothesis will be solving crystal or cryo-EM structures of prothrombinase with prothrombin and meizothrombin. However, short of such detailed structural information, we interrogated the proposed binding interface between fVa and Pre-1 by site-directed PEGylation.
The use of pseutarin C in this study is justified by its close sequence homology to human prothrombinase and its conserved prothrombin processing through the meizothrombin intermediate. Indeed, pseutarin C has advantages over human prothrombinase because it does not require membranes and is unable to cleave prothrombin unless ptfXa is bound to ptfV and prothrombin can interact with ptfV. Therefore, blocking the interaction of prothrombin with pseutarin C will result in no thrombin generation or prothrombin cleavage. The positive control for the effect of a PEG moiety on protein-protein interactions was mutation of Ser578, buried by its interaction with ptfXa in the pseutarin C crystal structure. The conservative mutation from Ser to Cys had no effect on prothrombin processing, but labeling Cys578 with PEG completely abolished activity. Similarly, as a negative control, PEGylation of the naturally occurring free cysteine of ptfV, Cys540, located in a heavily glycosylated region on the back of the molecule, had no effect on prothrombin processing. As predicted, residues on the front of the A1 domain (134 and 168) had no effect on thrombin generation when PEGylated, whereas residues at the top of the A1 domain near the interface with the A2 domain (276 and 302) dramatically slowed thrombin generation. Similar reductions in thrombin generation were seen with PEGylation at 441 and 455 at the top of the A2 domain. These results confirm the importance of the ledge formed at the top of the A1-A2 interface for prothrombin docking.
Individual a1-loop mutations are perhaps more difficult to interpret. PEGylation of 310, 311, 314, and 315 resulted in dramatic reductions in rates of thrombin generation, whereas PEGylation at adjacent positions, 312 and 316, had only modest effects. None of these residues could be modeled in the crystal structure of pseutarin C, suggesting a high degree of flexibility for the a1-loop. It is possible that the local conformation of residues 312 and 316 is better able to accommodate the PEG moiety in the context of preserving prothrombin binding. It is, however, clear from the results that the a1-loop forms a critical part of the prothrombin binding site.
The importance of the a1-loop was confirmed by swapping the ptfV a1-loop with those of hfV and hfVIII. The extra length of the hfVIII a1-loop chimera and the electrostatic difference resulted in a significant reduction in the rate of thrombin generation (6.6-fold) and only 30% cleavage of prothrombin after 1 hour when monitoring cleavage by SDS-PAGE. In contrast, swapping the P. textilis a1-loop with that of hfV resulted in a 3.6-fold increase in the rate of thrombin generation, complete cleavage of prothrombin by 1 hour, and, importantly, an apparent increase in the rate of cleavage at the Arg320 site, so the alternative first cleavage at Arg271 leading to formation of Pre-2 was not observed at all by SDS-PAGE. Our modeling places the a1-loop of fVa in close proximity to the linker between the K2 and protease domains of prothrombin. Intriguingly, the major sequence difference between human and rodent (the main diet of the eastern brown snake) prothrombin lies within this very linker. Human prothrombin has a 5-residue insert relative to rodent prothrombin contributing 4 additional acidic amino acids, making the net change for the linker −10 for human and −6 and −7 for rat and mouse, respectively. This is consistent with the net charge difference between the a1-loops of human and ptfV of +7 and may help explain the improved processing of human prothrombin by the human a1-loop chimera of pseutarin C. In addition, the exclusive processing via the meizothrombin intermediate (initial cleavage at Arg320) for this chimera can be similarly explained, because the linker between the K2 and protease domains contains the Arg271 cleavage site. We conclude that the a1-loop of fV forms a critical part of the prothrombin binding site and that specific interactions between it and prothrombin help to present the Arg320 site for cleavage.
Acknowledgment
This work was funded by British Heart Foundation Programme Grant RG/16/9/32391 (J.A.H.).
Authorship
Contribution: J.A.H. and F.I.Ü. conceived of and designed the study; F.I.Ü. conducted all experiments; and J.A.H. and F.I.Ü. interpreted the data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: James A. Huntington, Cambridge Institute for Medical Research, Department of Haematology, University of Cambridge, The Keith Peters Building, Hills Rd, Cambridge CB2 0XY, United Kingdom; e-mail: jah52@cam.ac.uk.
Requests for data sharing may be submitted to James A. Huntington (jah52@cam.ac.uk.).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal