

Key Points
ERp46-αIIbβ3 interactions play a critical role in platelet aggregation and platelet thrombus formation.
ERp46 reduces disulfide bonds in β3 in the absence of ligand binding.

Abstract
Although several members of protein disulfide isomerase (PDI) family support thrombosis, other PDI family members with the CXYC motif remain uninvestigated. ERp46 has 3 CGHC redox-active sites and a radically different molecular architecture than other PDIs. Expression of ERp46 on the platelet surface increased with thrombin stimulation. An anti-ERp46 antibody inhibited platelet aggregation, adenosine triphosphate (ATP) release, and αIIbβ3 activation. ERp46 protein potentiated αIIbβ3 activation, platelet aggregation, and ATP release, whereas inactive ERp46 inhibited these processes. ERp46 knockout mice had prolonged tail-bleeding times and decreased platelet accumulation in thrombosis models that was rescued by infusion of ERp46. ERp46-deficient platelets had decreased αIIbβ3 activation, platelet aggregation, ATP release, and P-selectin expression. The defects were reversed by wild-type ERp46 and partially reversed by ERp46 containing any of the 3 active sites. Platelet aggregation stimulated by an αIIbβ3-activating peptide was inhibited by the anti-ERp46 antibody and was decreased in ERp46-deficient platelets. ERp46 bound tightly to αIIbβ3 by surface plasmon resonance but poorly to platelets lacking αIIbβ3 and physically associated with αIIbβ3 upon platelet activation. ERp46 mediated clot retraction and platelet spreading. ERp46 more strongly reduced disulfide bonds in the β3 subunit than other PDIs and in contrast to PDI, generated thiols in β3 independently of fibrinogen. ERp46 cleaved the Cys473-Cys503 disulfide bond in β3, implicating a target for ERp46. Finally, ERp46-deficient platelets have decreased thiols in β3, implying that ERp46 cleaves disulfide bonds in platelets. In conclusion, ERp46 is critical for platelet function and thrombosis and facilitates αIIbβ3 activation by targeting disulfide bonds.
Introduction
Several members of the protein disulfide isomerase (PDI) family, PDI, ERp5, ERp57, ERp72, and most recently TMX1, have been shown to have distinct functions in regulating thrombosis. These enzymes are secreted or expressed by platelets or endothelial cells at the site of vascular injury and thus are available to catalyze critical reactions in hemostasis and thrombosis. In mammals, there are over 20 members of the PDI family, 12 of which contain thioredoxin-like domain CXYC motifs that catalyze the formation, reduction, or rearrangement of disulfide bonds.1,2 Platelets express at least 8 members.3,4 Using specific inhibitory antibodies, targeted knockout mice, and mutant isomerases, we and others have shown that 4 secreted members of the PDI family, PDI, ERp57, ERp5, and ERp72, play critical roles in supporting platelet accumulation and fibrin generation in vivo.5-15 In contrast, the transmembrane PDI family member TMX1 negatively regulates platelet function.4
Thiols in αIIbβ3 are required for activation of this integrin and platelet aggregation.16,17 One mechanism by which the prothrombotic PDIs can support platelet function and thrombosis is by reduction of disulfide bonds in αIIbβ3 and regulation of thiol-disulfide exchange reactions.18 The transmembrane TMX1 converts thiols to disulfide bonds in αIIbβ3, inhibiting its activation.4 Thus, these PDIs form a redox network that regulates thrombosis. Whether additional CXYC-containing members of the PDI family are involved in thrombosis remains a critical unanswered question.
ERp46 is a member of the PDI family with 3 surface exposed CGHC motifs in the a°, a, and a′ domains separated by flexible linker loops.19 This is in contrast to the more compact U-shaped structures of PDI, ERp57, and ERp72, in which the active sites face each other across the substrate binding cleft. The active sites of ERp46 act independently, whereas the active sites of PDI act cooperatively.19 ERp46 introduces disulfide bonds into unfolded substrates rapidly and promiscuously, followed by a slower, more precise PDI-mediated disulfide bond formation.20 ERp46 protects against apoptosis21 and functions in immunoglobulin22 and insulin production23 and growth of prostate cancer cells.24 Although Holbrook et al found that human megakaryocytes and human umbilical vein endothelial cells express ERp46,3 the role of ERp46 in thrombosis is unknown.
We generated a new model of ERp46-targeted knockout mice and found that ERp46 plays an important role in platelet aggregation, secretion, and platelet thrombus formation in vivo. ERp46 is expressed by platelets and is the fifth member of the PDI family found to act extracellularly on the platelet fibrinogen receptor, the αIIbβ3 integrin. Each CGHC motif supports platelet aggregation and secretion. ERp46 reduces disulfide bonds in αIIbβ3 in the absence of fibrinogen, suggesting that ERp46 acts prior to ligand binding to αIIbβ3. Differential labeling of thiols in the β3 subunit identified disulfide bonds targeted by ERp46, suggesting a mechanism by which ERp46 regulates activation of this integrin.
Methods
Materials and the methods for polymerase chain reaction (PCR), western blotting, bleeding time analysis, flow cytometry and aggregation and secretion studies of human and mouse platelets, FeCl3-induced thrombosis of the mesenteric artery, and intravital microscopy of laser-induced thrombosis of the cremaster muscle arterioles are all previously described.12,14 The Di-E-GSSG PDI assay,8 3-N-maleimidylpropionyl-biocytin (MPB) labeling of thiols in αIIbβ325,26 or PDI,27 and protein labeling with sulfo-NHS-LC-Biotin (SSB)25 were performed as previously described. The variant ERp46 enzymes were generated by GenScript, with cysteine residues of 1 active site mutated to alanine residues representing the Cys (C) change to Ala (A) as described.14 Surface plasmon resonance (SPR) studies on ERp46 and αIIbβ3,28 coimmunoprecipitation experiments,5 and statistical analysis11,12 were performed as described. Detailed methods for these techniques are included with minor revisions in supplemental Methods.
Labeling of αIIbβ3 with iodoTMT and determination of relative amounts of iodoTMT label per αIIb and β3 protein by blotting
Purified human αIIbβ3 (0.5 μM) (Abcam) was incubated with ERp46 or another PDI (1 μM) for 20 minutes at 37°C. Subsequently, the samples were labeled with 400 μM iodo tandem mass tag (iodoTMT) in 1% sodium dodecyl sulfate (SDS) and 5 mM ethylenediaminetetraacetic acid for 1 hour at 37°C in the dark according to the manufacturer’s protocol (ThermoFisher). In experiments in which reduced glutathione (GSH) was used, the concentration of iodoTMT was increased by an amount equivalent to the GSH added. Relative amounts of iodoTMT label per protein were determined by blotting using the IRDye 800CW (green) goat anti-mouse secondary antibody to detect the primary mouse anti-TMT antibody, and IRDye 680RD (red) goat anti-rabbit secondary antibody to detect the primary rabbit antibody against β3. Blots were visualized with the ODYSSEY infrared imaging system. The intensity of each band was calculated using the Image J program, with the ratio of iodoTMT to protein reported as β3 TMT/β3.
IodoTMT labeling of thiols in αIIbβ3 treated with ERp46 for LC-MS/MS analysis
Purified human αIIbβ3 was obtained from Abcam in glycerol free buffer (0.02M Tris-HCl; 0.1M NaCl; 0.1% Triton X-100; 0.001M CaCl2; pH 7.4) (supplemental Figure 27). αIIbβ3 (1.5 μM) with GSH (300 μM) was incubated with or without ERp46 (3 μM). The samples were then concentrated from ∼600 μL to 100 μL using a lyophilizer prior to the iodoTMT labeling. Thiols in the proteins in non-ERp46– or ERp46-treated samples were labeled with either iodoTMT-126 or iodoTMT-130 reagent, respectively. The excess iodoTMT reagents were quenched with 20 mM of dithiothreitol (DTT) by incubation at 55°C for 30 minutes, which also reduced disulfide bonds. The remaining DTT was removed via methanol/chloroform precipitation. The protein pellets were resuspended in 8 M guanidine hydrochloride in 50 mM triethylammonium bicarbonate (pH8.5). To ensure thorough reduction of the disulfide bonds, Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was added to a final concentration of 5 mM in each sample and incubated at 55°C for 30 minutes. The newly generated thiols in the non-ERp46– or ERp46-treated samples were labeled with iodoTMT-127 or iodoTMT-131, respectively. Both iodoTMT-labeled samples were combined and digested with trypsin at 37°C overnight. The resulting peptides were desalted via C18 cartridges for liquid chromatography with tandem mass spectrometry (LC-MS/MS) and data analysis (supplemental Methods).
Results
Expression of ERp46 on platelets increases with activation
We generated a rabbit anti-ERp46 antibody that reacts with a single band the size of ERp46 in platelets and does not crossreact with other CGHC-containing PDIs on western blots (supplemental Figure 1). Using this antibody, ERp46 was found on the surface of nonactivated platelets and, following platelet activation, its presence on the platelet surface and in the supernatant increased (Figure 1A-C; supplemental Figure 2). ERp46 is likely externalized from the platelet–dense tubular system as other PDIs.29 The reduced form of ERp46 was found on the surface of nonactivated platelets, and thiols in ERp46 increased with platelet activation to a greater extent than the ERp46 protein (Figure 1D-E).
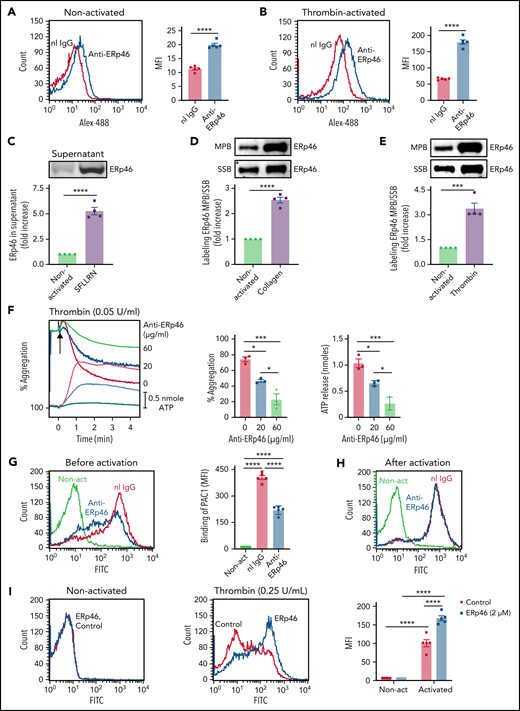
Functional role of ERp46 in human platelets. Expression of ERp46 on the surface of nonactivated platelets (A) and thrombin (1 U/mL)-activated platelets (B). Mean fluorescent intensity (MFI) ± standard error of the mean (SEM), n = 5, ****P < .0001, Student t test. Normal rabbit IgG (nI IgG, 30 μg/mL, red) and anti-ERp46 antibody (30 μg/mL, blue) were both labeled with Alexa-488. (C) SFLLRN (100 μM)-induced platelet activation/aggregation releases ERp46 into the supernatant. Shown is the fold increase of ERp46 protein in the supernatant with platelet activation. ERp46 was immunoprecipitated from the supernatant and analyzed by immunoblotting. (D-E) Platelet activation increases thiol labeling in surface ERp46. After activation by collagen (10 μg/mL) (D) or thrombin (1 U/mL) (E), platelets were labeled with MPB or SSB as described in “Methods.” After immunoblotting with the anti-ERp46 antibody, the band intensities were used to calculate the ratio of MPB/SSB in ERp46 on the surface of activated platelets relative to ERp46 on the surface of nonactivated platelets. (F) The polyclonal anti-ERp46 antibody inhibits platelet aggregation and ATP release. Representative aggregation and ATP release tracings (left) and combined results (right) showing inhibition by the anti-ERp46 antibody of human platelets activated with thrombin (0.05 U/mL); mean ± SEM, n = 3, *P < .05, ***P < .001, analysis of variance (ANOVA). Washed human platelets were preincubated with normal rabbit IgG and polyclonal anti-ERp46 antibody at the indicated concentration. Aggregation and ATP secretion were monitored in the lumi-aggregometer. (G-I) Anti-ERp46 antibody inhibits αIIbβ3 activation measured by PAC1 binding in platelets when added before (G) but not after (H) thrombin (0.025 U/mL) activation. MFI ± SEM, n = 5, ****P < .0001, ANOVA. In these experiments, the anti-ERp46 antibody (30 μg/mL) was incubated with the human platelets for 10 minutes before activation (G) or after activation (H). The red and blue histograms represent thrombin-activated platelets. (I) Addition of ERp46 (2 μM) (blue) potentiates thrombin (0.25 U/mL) activation of αIIbβ3 measured by PAC1 binding on platelets but does not itself induce αIIbβ3 activation. PBS control (red). Left panels, representative histograms; right panels, bar graph of combined results; mean ± SEM, n = 5, ****P < .0001, ANOVA. Non-act, nonactivated platelets.
Functional role of ERp46 in human platelets. Expression of ERp46 on the surface of nonactivated platelets (A) and thrombin (1 U/mL)-activated platelets (B). Mean fluorescent intensity (MFI) ± standard error of the mean (SEM), n = 5, ****P < .0001, Student t test. Normal rabbit IgG (nI IgG, 30 μg/mL, red) and anti-ERp46 antibody (30 μg/mL, blue) were both labeled with Alexa-488. (C) SFLLRN (100 μM)-induced platelet activation/aggregation releases ERp46 into the supernatant. Shown is the fold increase of ERp46 protein in the supernatant with platelet activation. ERp46 was immunoprecipitated from the supernatant and analyzed by immunoblotting. (D-E) Platelet activation increases thiol labeling in surface ERp46. After activation by collagen (10 μg/mL) (D) or thrombin (1 U/mL) (E), platelets were labeled with MPB or SSB as described in “Methods.” After immunoblotting with the anti-ERp46 antibody, the band intensities were used to calculate the ratio of MPB/SSB in ERp46 on the surface of activated platelets relative to ERp46 on the surface of nonactivated platelets. (F) The polyclonal anti-ERp46 antibody inhibits platelet aggregation and ATP release. Representative aggregation and ATP release tracings (left) and combined results (right) showing inhibition by the anti-ERp46 antibody of human platelets activated with thrombin (0.05 U/mL); mean ± SEM, n = 3, *P < .05, ***P < .001, analysis of variance (ANOVA). Washed human platelets were preincubated with normal rabbit IgG and polyclonal anti-ERp46 antibody at the indicated concentration. Aggregation and ATP secretion were monitored in the lumi-aggregometer. (G-I) Anti-ERp46 antibody inhibits αIIbβ3 activation measured by PAC1 binding in platelets when added before (G) but not after (H) thrombin (0.025 U/mL) activation. MFI ± SEM, n = 5, ****P < .0001, ANOVA. In these experiments, the anti-ERp46 antibody (30 μg/mL) was incubated with the human platelets for 10 minutes before activation (G) or after activation (H). The red and blue histograms represent thrombin-activated platelets. (I) Addition of ERp46 (2 μM) (blue) potentiates thrombin (0.25 U/mL) activation of αIIbβ3 measured by PAC1 binding on platelets but does not itself induce αIIbβ3 activation. PBS control (red). Left panels, representative histograms; right panels, bar graph of combined results; mean ± SEM, n = 5, ****P < .0001, ANOVA. Non-act, nonactivated platelets.
ERp46 on the platelet surface is required for platelet aggregation and activation of αIIbβ3
Preincubation of platelets with the anti-ERp46 antibody inhibited thrombin-induced platelet aggregation and adenosine triphosphate (ATP) release (Figure 1F). The antibody inhibited the reductase activity of recombinant ERp46, but not of other PDIs, and did not inhibit aggregation of ERp46-null platelets (supplemental Figures 3 and 4), confirming its specificity for ERp46. Anti-ERp46 inhibited activation of the αIIbβ3 fibrinogen receptor (measured by binding of an activation-specific antibody, PAC-1) when added before (Figure 1G) but not after activation (Figure 1H). This implies that inhibition of activation of αIIbβ3 was by inhibition of ERp46 and not from steric effects on PAC-1 binding. Adding ERp46 to resting platelets did not activate αIIbβ3 (Figure 1I); however, ERp46 potentiated thrombin-induced activation of αIIbβ3. When fibrinogen binding to αIIbβ3 on platelets was blocked with eptifibatide, adding ERp46 potentiated ATP release and P-selectin expression in the absence of aggregation, suggesting that ERp46 also targets a non-αIIbβ3 substrate (supplemental Figure 5).
Generation and characterization of conditional ERp46 knockout mice
We generated conditional ERp46 knockout mice by introducing loxP sites flanking the ERp46 gene encoding exon 3 using cloned stem cells (supplemental Figure 6). Genotyping of Pf4-Cre/ERp46fl/fl mice showed the homozygous floxed allele and the presence of the Cre gene (Figure 2A). Pf4-Cre/ERp46fl/fl mice did not express ERp46 protein in platelets (Figure 2B). The levels of PDI, ERp57, ERp72, and ERp5 were comparable with WT mice (Figure 2B), indicating successful targeting of ERp46. Complete blood counts in Pf4-Cre/ERp46fl/fl mice revealed no abnormalities. Pf4-Cre/ERp46fl/fl mice had normal platelet counts, platelet size (MPV: 5.3 ± 0.07 fL vs 5.2 ± 0.06 fL, P = NS, n = 11), and expression of platelet αIIbβ3, GpIbα, and GpVI (supplemental Figure 7A-B). Leukocytes from Pf4-Cre/ERp46fl/fl mice had normal ERp46 levels (supplemental Figure 7C-D).
![Platelet ERp46 is required for hemostasis, thrombosis, and platelet accumulation into a growing thrombus. (A-B) Characterization of Pf4-Cre/ERp46fl/fl mice. (A) PCR products of tail DNA from Cre− ERp46fl/fl mice and Pf4-Cre/ERp46fl/fl mice. The bands represent PCR product from the ERp46-floxed allele (upper panel, 431-bp) and the Cre gene (lower panel, 420-bp), respectively. (B) Western blots of platelet lysates using a polyclonal rabbit anti-ERp46 antibody and antibodies against PDI, ERp57, ERp72, and ERp5. Shown are the actin loading controls. Left panel representative blot; right panel, quantitative analysis of protein level by densitometry of band density of PDIs relative to the ERp46fl/fl wild-type (WT) control, which was set at 100%; mean ± SEM, ****P < .0001, n = 4, Student t test. (C) Tail bleeding times; mean ± SEM, n = 16 for each group, ****P < .001, Student t test. (D-E) Incorporation of platelets into a growing thrombus in ERp46fl/fl mice and Pf4-Cre/ERp46fl/fl mice was detected by Alexa 488 anti-CD41 using FeCl3-induced mesenteric arterial injury. Mean artery diameters were 140.2 ± 3.35 μm (SEM) in ERp46fl/fl mice, 136.0 ± 3.73 μm in Pf4-Cre/ERp46fl/fl mice (P = not significant [ns]), 136.3 ± 3.24 μm in Pf4-Cre/ERp46fl/fl mice plus rERp46 (P = ns), 132.6 ± 5.21 μm in Pf4-Cre/ERp46fl/fl mice plus rERp46(aaaaaa), P = ns, ANOVA. (D) Images at 7, 11, and 15 minutes. Dotted lines mark the vessel wall. Scale bar, 200 μm. Composite of fluorescence intensity (FI) per area analyzed (FI/μm2) in ERp46fl/fl (21 thrombi from 8 mice), Pf4-Cre/46fl/fl (20 thrombi from 8 mice), Pf4-Cre/46fl/fl plus rERp46 (24 thrombi from 8 mice), and Pf4-Cre/ERp46fl/fl plus rERp46(aaaaaa) (25 thrombi from 9 mice); mean ± SEM, *P < .05, ****P < .0001, ANOVA. (F-H) Cremaster laser injury in arterioles of Pf4-Cre/ERp46fl/fl mice and their Cre− ERp46fl/fl littermate control mice. Platelets at the site of injury were detected using anti-CD41 F(ab)2 fragments conjugated to Alexa Fluor 647. (F) Representative fluorescence images from widefield intravital microscopy for platelet accumulation (red) at the indicated time points after injury. (G) The mean ± SEM integrated FIs of anti-CD41 F(ab)2 fragments over 240 seconds from ERp46fl/fl (37 thrombi from 4 mice) and Pf4-Cre/ERp46fl/fl (26 thrombi from 4 mice). (H) The areas under the FI curves over 240 seconds were analyzed with a Mann-Whitney U test; **P < .01. (I) Time to occlusion of FeCl3-induced carotid artery thrombosis in Pf4-Cre/ERp46fl/fl mice compared with ERp46fl/fl littermate controls; ***P < .001, Student t test. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/13/10.1182_blood.2021012055/5/m_bloodbld2021012055f2.png?Expires=1771333923&Signature=lZJUwS-s4vu85LDap3nLQOZrKRex2jYyqzlwtS~Vgm86INAECVfcnRsCK3F9F137hjevcK7XtTPAENrSW16x-uXfcWCiGWo-Zji9-lDjFmp4zenml1EC86yeZIo3JkWz-V5WJ~q5YE8hsPMHbMCxrXzM4HMvr3xAsDub6pk8FgHLELzwc7aZrZGIqnfP2eaPRVAfLl7AsFWNb1hMNPwiXCfnn37Poh-4294WdGyHXjMBgigUlaB-zJPW01DgYnlL~MownntOhE7VYNYJhCufVUprv~8tLnMCAJy15TTZCR6zlVh7Sgucda2h4leRHqWeRBGu1o0YyaNN6obRunFtIw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Platelet ERp46 is required for hemostasis, thrombosis, and platelet accumulation into a growing thrombus. (A-B) Characterization of Pf4-Cre/ERp46fl/fl mice. (A) PCR products of tail DNA from Cre− ERp46fl/fl mice and Pf4-Cre/ERp46fl/fl mice. The bands represent PCR product from the ERp46-floxed allele (upper panel, 431-bp) and the Cre gene (lower panel, 420-bp), respectively. (B) Western blots of platelet lysates using a polyclonal rabbit anti-ERp46 antibody and antibodies against PDI, ERp57, ERp72, and ERp5. Shown are the actin loading controls. Left panel representative blot; right panel, quantitative analysis of protein level by densitometry of band density of PDIs relative to the ERp46fl/fl wild-type (WT) control, which was set at 100%; mean ± SEM, ****P < .0001, n = 4, Student t test. (C) Tail bleeding times; mean ± SEM, n = 16 for each group, ****P < .001, Student t test. (D-E) Incorporation of platelets into a growing thrombus in ERp46fl/fl mice and Pf4-Cre/ERp46fl/fl mice was detected by Alexa 488 anti-CD41 using FeCl3-induced mesenteric arterial injury. Mean artery diameters were 140.2 ± 3.35 μm (SEM) in ERp46fl/fl mice, 136.0 ± 3.73 μm in Pf4-Cre/ERp46fl/fl mice (P = not significant [ns]), 136.3 ± 3.24 μm in Pf4-Cre/ERp46fl/fl mice plus rERp46 (P = ns), 132.6 ± 5.21 μm in Pf4-Cre/ERp46fl/fl mice plus rERp46(aaaaaa), P = ns, ANOVA. (D) Images at 7, 11, and 15 minutes. Dotted lines mark the vessel wall. Scale bar, 200 μm. Composite of fluorescence intensity (FI) per area analyzed (FI/μm2) in ERp46fl/fl (21 thrombi from 8 mice), Pf4-Cre/46fl/fl (20 thrombi from 8 mice), Pf4-Cre/46fl/fl plus rERp46 (24 thrombi from 8 mice), and Pf4-Cre/ERp46fl/fl plus rERp46(aaaaaa) (25 thrombi from 9 mice); mean ± SEM, *P < .05, ****P < .0001, ANOVA. (F-H) Cremaster laser injury in arterioles of Pf4-Cre/ERp46fl/fl mice and their Cre− ERp46fl/fl littermate control mice. Platelets at the site of injury were detected using anti-CD41 F(ab)2 fragments conjugated to Alexa Fluor 647. (F) Representative fluorescence images from widefield intravital microscopy for platelet accumulation (red) at the indicated time points after injury. (G) The mean ± SEM integrated FIs of anti-CD41 F(ab)2 fragments over 240 seconds from ERp46fl/fl (37 thrombi from 4 mice) and Pf4-Cre/ERp46fl/fl (26 thrombi from 4 mice). (H) The areas under the FI curves over 240 seconds were analyzed with a Mann-Whitney U test; **P < .01. (I) Time to occlusion of FeCl3-induced carotid artery thrombosis in Pf4-Cre/ERp46fl/fl mice compared with ERp46fl/fl littermate controls; ***P < .001, Student t test. ns, not significant.
Platelet ERp46 is required for hemostasis, thrombosis, and platelet accumulation into a growing thrombus. (A-B) Characterization of Pf4-Cre/ERp46fl/fl mice. (A) PCR products of tail DNA from Cre− ERp46fl/fl mice and Pf4-Cre/ERp46fl/fl mice. The bands represent PCR product from the ERp46-floxed allele (upper panel, 431-bp) and the Cre gene (lower panel, 420-bp), respectively. (B) Western blots of platelet lysates using a polyclonal rabbit anti-ERp46 antibody and antibodies against PDI, ERp57, ERp72, and ERp5. Shown are the actin loading controls. Left panel representative blot; right panel, quantitative analysis of protein level by densitometry of band density of PDIs relative to the ERp46fl/fl wild-type (WT) control, which was set at 100%; mean ± SEM, ****P < .0001, n = 4, Student t test. (C) Tail bleeding times; mean ± SEM, n = 16 for each group, ****P < .001, Student t test. (D-E) Incorporation of platelets into a growing thrombus in ERp46fl/fl mice and Pf4-Cre/ERp46fl/fl mice was detected by Alexa 488 anti-CD41 using FeCl3-induced mesenteric arterial injury. Mean artery diameters were 140.2 ± 3.35 μm (SEM) in ERp46fl/fl mice, 136.0 ± 3.73 μm in Pf4-Cre/ERp46fl/fl mice (P = not significant [ns]), 136.3 ± 3.24 μm in Pf4-Cre/ERp46fl/fl mice plus rERp46 (P = ns), 132.6 ± 5.21 μm in Pf4-Cre/ERp46fl/fl mice plus rERp46(aaaaaa), P = ns, ANOVA. (D) Images at 7, 11, and 15 minutes. Dotted lines mark the vessel wall. Scale bar, 200 μm. Composite of fluorescence intensity (FI) per area analyzed (FI/μm2) in ERp46fl/fl (21 thrombi from 8 mice), Pf4-Cre/46fl/fl (20 thrombi from 8 mice), Pf4-Cre/46fl/fl plus rERp46 (24 thrombi from 8 mice), and Pf4-Cre/ERp46fl/fl plus rERp46(aaaaaa) (25 thrombi from 9 mice); mean ± SEM, *P < .05, ****P < .0001, ANOVA. (F-H) Cremaster laser injury in arterioles of Pf4-Cre/ERp46fl/fl mice and their Cre− ERp46fl/fl littermate control mice. Platelets at the site of injury were detected using anti-CD41 F(ab)2 fragments conjugated to Alexa Fluor 647. (F) Representative fluorescence images from widefield intravital microscopy for platelet accumulation (red) at the indicated time points after injury. (G) The mean ± SEM integrated FIs of anti-CD41 F(ab)2 fragments over 240 seconds from ERp46fl/fl (37 thrombi from 4 mice) and Pf4-Cre/ERp46fl/fl (26 thrombi from 4 mice). (H) The areas under the FI curves over 240 seconds were analyzed with a Mann-Whitney U test; **P < .01. (I) Time to occlusion of FeCl3-induced carotid artery thrombosis in Pf4-Cre/ERp46fl/fl mice compared with ERp46fl/fl littermate controls; ***P < .001, Student t test. ns, not significant.
Pf4-Cre/ERp46fl/fl mice have prolonged tail bleeding times and decreased platelet incorporation into a growing thrombus
Tail bleeding times of Pf4-Cre/ERp46fl/fl mice that lack platelet ERp46 were doubled compared with their littermate controls (Figure 2C), with no sex differences noted (supplemental Figure 8). Incorporation of ERp46-negative platelets into thrombi in FeCl3-injured mesenteric arteries was substantially decreased (Figure 2D-E). The platelet accumulation defect was rescued by infusing WT ERp46 but not inactivated ERp46. The platelet accumulation defect in the FeCl3-induced arterial injury was greater in Pf4-Cre/ERp46fl/fl mice than in previously studied PDI-targeted knockout mice12 as measured by the area under the platelet incorporation curves (supplemental Figure 9). Pf4-Cre/ERp46fl/fl mice also had decreased platelet accumulation in a laser-induced injury model, confirming the role of ERp46 in platelet accumulation (Figure 2F-H). ERp46 accumulated at the site of injury near the developing platelet thrombus (supplemental Figure 10). The time to complete occlusion was prolonged in Pf4-Cre/ERp46fl/fl mice after FeCl3 injury of the carotid artery (Figure 2I), without detectable sex differences (supplemental Figure 11).
Infusion of the anti-ERp46 antibody almost eliminated the residual platelet accumulation in Pf4-Cre/ERp46 mice in Figure 2G of the laser injury model (supplemental Figure 12). Because we did not detect prolongation of the time to occlusion in the carotid injury model with infusion of the anti-ERp46 antibody into Tie2-Cre/ERp46fl/fl mice (lacking both endothelial cell and platelet ERp46) (supplemental Figure 13), the antibody is specific for ERp46 in vivo without major off-target effects. Therefore, the elimination of platelet accumulation in Pf4-Cre/ERp46fl/fl mice by the anti-ERp46 antibody likely reflects inhibition of ERp46 supplied from other vascular sources at the site of injury, such as endothelial cells or plasma that partially support platelet accumulation.
ERp46 deficiency impairs platelet function
Because prolonged tail bleeding times and impaired platelet thrombus formation in mice whose platelets lack ERp46 suggest a role for ERp46 in platelet responses, we analyzed the function of ERp46-null platelets. Thrombin-induced activation of αIIbβ3 and P-selectin expression and thrombin and collagen-related peptide (CRP)-induced platelet aggregation and ATP release were decreased in ERp46-null platelets (Figure 3A-D). Aggregation and ATP release induced by the thromboxane A2 analog, U46619 and calcium ionophore (supplemental Figure 14A-B), and adenosine diphosphate (ADP)-induced aggregation were also defective (Figure 3E). Higher doses of agonists overcame these defects (supplemental Figure 14C-G).
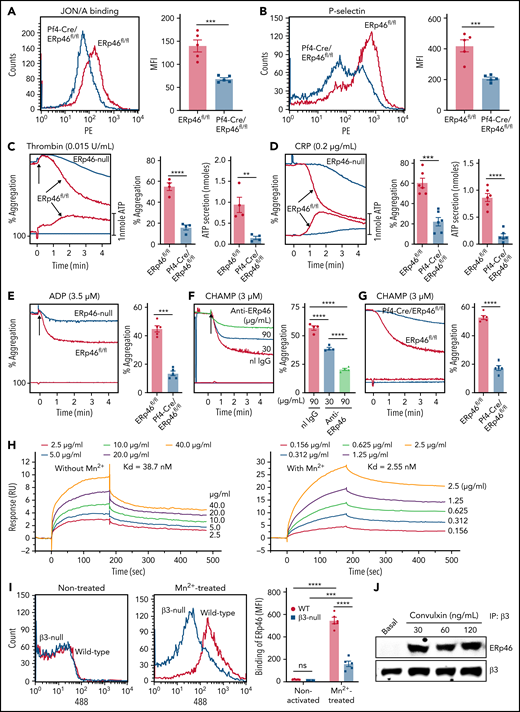
ERp46 is critical for aggregation of mouse platelets and interacts with αIIbβ3 (A) ERp46-deficient platelets have defective thrombin (0.02 U/mL-induced activation of αIIbβ3 detected by the JON/A activation-dependent antibody). (B) P-selectin expression is decreased in thrombin-stimulated ERp46-null platelets. (A-B) left panels, representative histogram; right panels, combined results; mean ± SEM, n = 5 for each group, ***P < .001, Student t test. (C-E) Representative aggregation and ATP release tracings (left panels) and combined results (right) showing the defects in ERp46-deficient platelets using (C) thrombin, (D) CRP, or aggregation for (E) ADP; mean ± SEM, n = 4 (thrombin), n = 6 (CRP), n = 5 (ADP), **P < .01, ***P < .001, ****P < .0001, Student t test. Aggregation and ATP secretion were monitored in the lumi-aggregometer. (F) Anti-ERp46 inhibits CHAMP-induced aggregation of human platelets; mean ± SEM, n = 4, ****P < .0001, ANOVA. Normal rabbit IgG (90 μg/mL) was used as a control. (G) CHAMP-induced aggregation of ERp46-null mouse platelets is decreased; mean ± SEM, n = 5, ****P < .0001, Student t test. CHAMP was added to final concentration of 3 μM and aggregation was performed in the presence of indomethacin (100 µM) and Apyrase (4 U/mL). (H) ERp46 interaction with αIIbβ3 by surface plasmon resonance. Recombinant full-length human αIIbβ3 (25 μg/mL) was immobilized on the surface of a CM5 chip. Different concentrations of WT ERp46 without or with MnCl2 (1 mM) were infused over the chip in the running buffer. The equilibrium dissociation constant was calculated based on the Kon and Koff values with Biacore T200 evaluation software. (I) ERp46 interacts with β3 integrins on mouse platelets. Binding of Alexa Fluor 488-conjugated ERp46 to Mn2+-treated WT and β3-null mouse platelets. Representative histogram (left panels); cumulative data (right panels); mean ± SEM, n = 5 for each group, ***P < .001, ****P < .0001, ANOVA. Washed mouse platelets (3 ×108/mL) were preincubated with Alexa Fluor 488 ERp46 (30 μg/mL) for 10 minutes at room temperature and then treated with Mn2+ (12 mM) for 5 minutes at room temperature. Surface binding of Alexa Fluor 488 ERp46 was detected by flow cytometry. (J) Stimulation-dependent association of ERp46 with integrin β3. Platelets (1 × 109/mL) were stimulated in the presence of EGTA, apyrase, and indomethacin at varying concentrations of convulxin (30, 60, or 120 ng/mL) for 90 seconds. Following sample lysis, proteins were precipitated with mouse anti-β3 antibody SZ21 and protein G–resin. Immunoblotting with goat anti-ERp46 antibody and polyclonal rabbit anti-β3 antibody (Abcam) showed interacting proteins.
ERp46 is critical for aggregation of mouse platelets and interacts with αIIbβ3 (A) ERp46-deficient platelets have defective thrombin (0.02 U/mL-induced activation of αIIbβ3 detected by the JON/A activation-dependent antibody). (B) P-selectin expression is decreased in thrombin-stimulated ERp46-null platelets. (A-B) left panels, representative histogram; right panels, combined results; mean ± SEM, n = 5 for each group, ***P < .001, Student t test. (C-E) Representative aggregation and ATP release tracings (left panels) and combined results (right) showing the defects in ERp46-deficient platelets using (C) thrombin, (D) CRP, or aggregation for (E) ADP; mean ± SEM, n = 4 (thrombin), n = 6 (CRP), n = 5 (ADP), **P < .01, ***P < .001, ****P < .0001, Student t test. Aggregation and ATP secretion were monitored in the lumi-aggregometer. (F) Anti-ERp46 inhibits CHAMP-induced aggregation of human platelets; mean ± SEM, n = 4, ****P < .0001, ANOVA. Normal rabbit IgG (90 μg/mL) was used as a control. (G) CHAMP-induced aggregation of ERp46-null mouse platelets is decreased; mean ± SEM, n = 5, ****P < .0001, Student t test. CHAMP was added to final concentration of 3 μM and aggregation was performed in the presence of indomethacin (100 µM) and Apyrase (4 U/mL). (H) ERp46 interaction with αIIbβ3 by surface plasmon resonance. Recombinant full-length human αIIbβ3 (25 μg/mL) was immobilized on the surface of a CM5 chip. Different concentrations of WT ERp46 without or with MnCl2 (1 mM) were infused over the chip in the running buffer. The equilibrium dissociation constant was calculated based on the Kon and Koff values with Biacore T200 evaluation software. (I) ERp46 interacts with β3 integrins on mouse platelets. Binding of Alexa Fluor 488-conjugated ERp46 to Mn2+-treated WT and β3-null mouse platelets. Representative histogram (left panels); cumulative data (right panels); mean ± SEM, n = 5 for each group, ***P < .001, ****P < .0001, ANOVA. Washed mouse platelets (3 ×108/mL) were preincubated with Alexa Fluor 488 ERp46 (30 μg/mL) for 10 minutes at room temperature and then treated with Mn2+ (12 mM) for 5 minutes at room temperature. Surface binding of Alexa Fluor 488 ERp46 was detected by flow cytometry. (J) Stimulation-dependent association of ERp46 with integrin β3. Platelets (1 × 109/mL) were stimulated in the presence of EGTA, apyrase, and indomethacin at varying concentrations of convulxin (30, 60, or 120 ng/mL) for 90 seconds. Following sample lysis, proteins were precipitated with mouse anti-β3 antibody SZ21 and protein G–resin. Immunoblotting with goat anti-ERp46 antibody and polyclonal rabbit anti-β3 antibody (Abcam) showed interacting proteins.
ERp46 interacts with αIIbβ3
Because platelet aggregation depends on αIIbβ3 activation, we examined the interaction of ERp46 with αIIbβ3. The computed helical anti-membrane protein (CHAMP) anti-αIIb TM is a peptide that inserts in platelet membranes to specifically interact with the transmembrane domain of αIIb and triggers activation of αIIbβ3.30,31 The anti-ERp46 antibody inhibited human platelet aggregation induced by anti-αIIb TM (Figure 3F). Anti-αIIb TM-induced aggregation was also decreased in ERp46-null platelets (Figure 3G). Addition of indomethacin and apyrase did not affect aggregation, indicating secreted thromboxane A2 or ADP did not contribute to aggregation (supplemental Figure 15) as previously reported.30,31 Because ERp46 is on resting platelets (Figure 1A,D-E), it is available to support anti-αIIb TM-induced conformational changes in αIIbβ3 and aggregation in the absence of platelet activation induced by standard agonists. As shown by SPR, ERp46 binds directly to αIIbβ3 with a dissociation constant (Kd) of 38.7 nM in the absence of Mn2+ and 2.55 nM in the presence of Mn2+ (Figure 3H). To study ERp46 binding to activated αIIbβ3 in the absence of platelet activation, we treated platelets with Mn2+.32 Binding of ERp46 to Mn2+-treated β3-null platelets was substantially decreased compared with binding to WT platelets (Figure 3I). Moreover, ERp46 associated with αIIbβ3 upon platelet activation using coimmunoprecipitation of either αIIbβ3 or ERp46 followed by detection of ERp46 or αIIbβ3, respectively (Figure 3J; supplemental Figure 16). Together, these data suggest that ERp46 interacts with αIIbβ3 and regulates its activation. ERp46(aa-aa-aa) and anti-ERp46 inhibit platelet adhesion and spreading on the GFOGER peptide, a well-characterized specific ligand for platelet integrin α2β1,33-36 (supplemental Figure 17), suggesting that ERp46 regulates other integrins in addition to αIIbβ3.
The a°, a, and a′ active sites of ERp46 support platelet aggregation
Using recombinant mutant proteins in genetic mouse models, we previously found that the CGHC-containing a′ active sites of PDI and ERp57 and the a and a′ active sites, but not the a° active site, of ERp72 selectively support thrombosis.10,12,14 To determine the relative importance of the 3 ERp46 active sites in affecting platelet function, we generated the ERp46 active site mutants (Figure 4A). One mutant, ERp46(cc-cc-aa), had the a′ CGHC motif inactivated. Two others, ERp46(cc-aa-aa) and ERp46(aa-aa-aa), had the a and a′ CGHC motifs or all 3 active sites inactivated, respectively. The addition of active sites to ERp46 progressively restored ERp46 activity in the Di-E-GSSG assay (Figure 4B; supplemental Figure 18A). Completely inactivated ERp46 [ERp46(aa-aa-aa)] did not restore aggregation and ATP release of ERp46-null platelets, whereas the addition of active sites restored aggregation in a stepwise fashion (Figure 4C). Mutants with only a single functional active site in any domain partially recovered aggregation and ATP release of ERp46-null platelets, whereas mutants having 2 functional active sites gave more full recovery (Figure 4D-E).
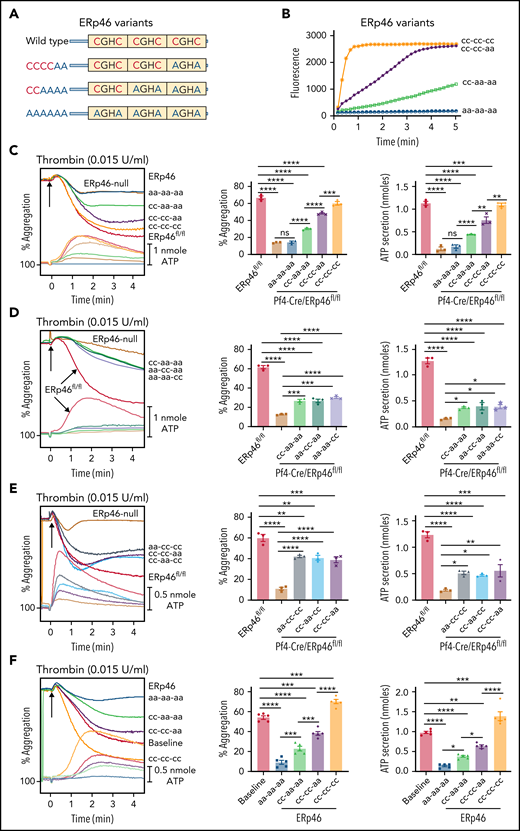
The a°, a, and a′ active sites of ERp46 each contribute to aggregation and ATP release of mouse and human platelets. (A) Schematic diagram of the ERp46 variants. (B) Characterization of variant ERp46 proteins in the Di-E-GSSG assay. ERp46(cc-cc-cc), WT ERp46; ERp46(cc-cc-aa) had the a′ CGHC motif inactivated; ERp46(cc-aa aa) had the a and a′ active sites inactivated; ERp46(aa-aa-aa) had all 3 sites inactivated. (C-E) Correction of the aggregation and secretion defects of ERp46-null platelets (2 × 108 platelets/mL) by ERp46 variants (100 nM). (F) Effect of preincubating human platelets (2 × 108 platelets/mL) with ERp46 variants (1 μM). Submaximal aggregation (baseline) was stimulated with thrombin (0.015 U/mL). The ERp46 variants were added 5 minutes prior to the addition of thrombin. Representative aggregation and ATP release tracings (left panels) and combined results (right); mean ± SEM, n = 3, mouse platelets; n = 5, human platelets, *P < .05, **P < .01, ***P < .001, ****P < .0001, ANOVA. ns, not significant.
The a°, a, and a′ active sites of ERp46 each contribute to aggregation and ATP release of mouse and human platelets. (A) Schematic diagram of the ERp46 variants. (B) Characterization of variant ERp46 proteins in the Di-E-GSSG assay. ERp46(cc-cc-cc), WT ERp46; ERp46(cc-cc-aa) had the a′ CGHC motif inactivated; ERp46(cc-aa aa) had the a and a′ active sites inactivated; ERp46(aa-aa-aa) had all 3 sites inactivated. (C-E) Correction of the aggregation and secretion defects of ERp46-null platelets (2 × 108 platelets/mL) by ERp46 variants (100 nM). (F) Effect of preincubating human platelets (2 × 108 platelets/mL) with ERp46 variants (1 μM). Submaximal aggregation (baseline) was stimulated with thrombin (0.015 U/mL). The ERp46 variants were added 5 minutes prior to the addition of thrombin. Representative aggregation and ATP release tracings (left panels) and combined results (right); mean ± SEM, n = 3, mouse platelets; n = 5, human platelets, *P < .05, **P < .01, ***P < .001, ****P < .0001, ANOVA. ns, not significant.
Completely inactivated ERp46 [ERp46(aa-aa-aa)] inhibited aggregation of human platelets (Figure 4F), presumably by competing with endogenous ERp46. Stepwise recovery occurred with the addition of each functional active site, and WT ERp46 potentiated aggregation, presumably by targeting additional surface substrates. Mutants with 2 nonfunctional active sites more strongly inhibited aggregation of human platelets than mutants with 1 nonfunctional site (supplemental Figure 18B-C). Binding of completely inactive ERp46(aa-aa-aa) to Mn2+-treated mouse platelets and to αIIbβ3 by SPR was comparable to WT ERp46 (supplemental Figure 18D-E), indicating that the functional consequences of the different mutants were not due to decreased binding. These results imply that each of the 3 active sites contribute to platelet aggregation and ATP release. Although ERp46(aa-aa-aa) inhibited aggregation, it did not affect the association of αIIbβ3 with talin-1 (supplemental Figure 19).
Distinct roles of ERp46, ERp72, PDI, and ERp57 in platelet function
Platelets specifically lacking ERp46 have a major aggregation defect (Figures 3C-E and 4C-E), suggesting that ERp46 has a distinct role from the other PDIs in αIIbβ3 activation. Because it is possible that the combined redox function of the PDIs is limiting and the modes of action redundant, we tested whether PDI, ERp57, or ERp72 could recover aggregation of ERp46-null platelets. Although addition of ERp46 restores the aggregation response of ERp46-null platelets, addition of PDI, ERp57, or ERp72 to ERp46-null platelets does not restore aggregation (Figure 5A). Moreover, ERp46 was unable to restore aggregation of PDI or ERp57-null platelets (Figure 5B-C), implying that each enzyme plays a different role in αIIbβ3 activation and platelet aggregation.
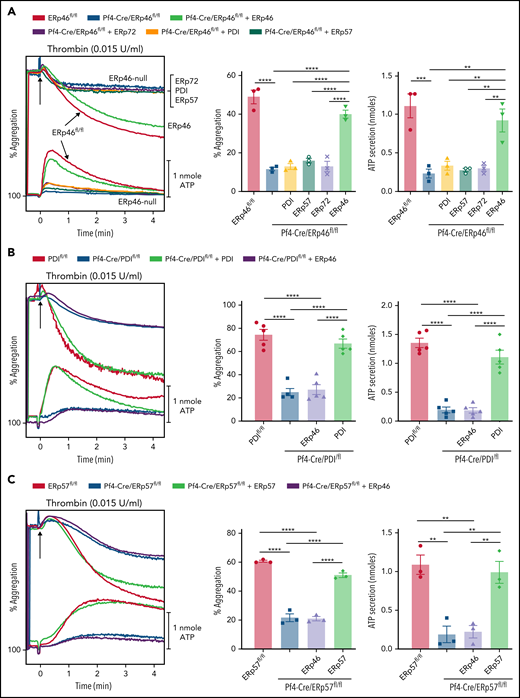
ERp46 but not other PDIs recover aggregation and ATP release of ERp46-null platelets. (A) ERp46-null platelets were incubated with 100 nM PDI, ERp57, ERp72, or ERp46. (B) PDI-null platelets were incubated with 100 nM ERp46 or PDI. (C) ERp57-null platelets were incubated with 100 nM ERp46 or ERp57. Platelet aggregation was stimulated with thrombin (0.015 U/mL); representative aggregation and ATP release tracings (left panel) and combined results (right); mean ± SEM, n = 3 (A); n = 5 (B); n = 3 (C); **P < .01, ***P < .001, ****P < .0001, ANOVA.
ERp46 but not other PDIs recover aggregation and ATP release of ERp46-null platelets. (A) ERp46-null platelets were incubated with 100 nM PDI, ERp57, ERp72, or ERp46. (B) PDI-null platelets were incubated with 100 nM ERp46 or PDI. (C) ERp57-null platelets were incubated with 100 nM ERp46 or ERp57. Platelet aggregation was stimulated with thrombin (0.015 U/mL); representative aggregation and ATP release tracings (left panel) and combined results (right); mean ± SEM, n = 3 (A); n = 5 (B); n = 3 (C); **P < .01, ***P < .001, ****P < .0001, ANOVA.
Addition of anti-PDI to ERp46-null platelets inhibits activation of αIIbβ3, P-selectin expression, and platelet aggregation (supplemental Figure 20), consistent with these enzymes having distinct functions. These data suggest that dual targeting of ERp46 and PDI would modulate the level of inhibition of thrombosis.
ERp46 mediates clot retraction and platelet spreading
Because ERp46 regulates inside-out signaling through αIIbβ3 and fibrinogen binding, it may impact outside-in signaling responsible for clot retraction and platelet spreading. The anti-ERp46 antibody or inactivated ERp46(aa-aa-aa) decreased clot size and inhibited platelet spreading (supplemental Figures 21 and 22), implicating ERp46 in these processes.
ERp46 generates thiols in αIIbβ3
ERp46 contains more thiols than other PDIs, with most of its thiols found in the 6 active site cysteines (supplemental Figures 23 and 24), and ERp46 cleaves disulfide bonds in the Di-E-GSSG assay much better than PDI, ERp57, ERp72, or ERp5 (Figure 6A). Because ERp46 interacts with αIIbβ3, supporting activation of this receptor, we studied the effect of ERp46 on disulfide bonds.
![ERp46 generates thiols in the β3 subunit of αIIbβ3. (A) Reductase activity of ERp46 compared with ERp57, PDI, ERp5, ERp72, and inactivated ERp46 [ERp46(aaaaaa)] (30 nM each) in the Di-E-GSSG assay. (B-C) ERp46 generates thiols in β3 more effectively than PDI, ERp57, ERp72, or ERp5. ERp46 or other PDIs (1 μM) were incubated with αIIbβ3 (0.5 μM) (Abcam) for 20 minutes at 37°C. IodoTMT (400 μM) was then added for 1 hour at 37°C and labeling of thiols performed in 1% SDS with 5 mM ethylenediaminetetraacetic acid . These studies were performed in the absence of added GSH. (D-E) The active site cysteines of ERp46 generate thiols in αIIbβ3. Shown is the labeling of β3 with WT ERp46 or inactivated ERp46 [ERp46(aaaaaa)]. ERp46 or ERp46(aaaaaa) (1 μM) was incubated with αIIbβ3 (0.5 μM) and GSH (100 μM), and labeling was performed with iodoTMT (500 μM). (F-G) Effect of fibrinogen (Fbgn) on thiol generation by ERp46 and PDI in αIIbβ3. ERp46 or PDI (1 μM) were incubated with αIIbβ3 (0.5 μM) with GSH (100 μM). In some samples, fibrinogen (0.5 μM) was added. Thiols were labeled with iodoTMT (500 μM). Blotting for iodoTMT was performed using the anti-TMT antibody. (H-I) Effect of fibrinogen on thiol generation by ERp46 and PDI in αIIbβ3 in platelets. ERp46 or PDI (1 μM) were incubated with human platelets (4 × 108 platelets/mL) without GSH. In some samples, fibrinogen (1 μM) was added. Thiols were labeled with MPB and blotting was performed as described in supplemental Methods. The intensity of each band was calculated using the Image J program, and the ratio of iodoTMT (TMT) or MPB to β3 protein was compared with the untreated sample. Shown are the blots for β3 (H-96 or B-7 for D), PDI and ERp46 (rabbit antibodies), and the β chain of fibrinogen; mean ± SEM, n = 4 (B-C), n = 3 (D-E), n = 3 (F-G), n = 7 (H-I); *P < .05, **P < .01, ***P < .001, ****P < .0001, ANOVA. (J-K) Addition of ERp46 followed by PDI (ERp46/PDI) maximally generates thiols in β3. αIIbβ3 was treated with 1 μM PDI or ERp46 for 40 minutes, except when added sequentially they were added for 20 minutes each. Addition of PDI followed by ERp46 (PDI/ERp46) gave similar results to ERp46 alone or PDI plus ERp46 added simultaneously. Labeling was with iodoTMT (400 μM) in the absence of GSH; mean ± SEM, n = 4; ****P < .0001, ANOVA. (L-M) Addition of ERp46 followed by PDI (ERp46/PDI) provides maximal enhancement of platelet aggregation. 1.5 μM PDI or ERp46 were added for 10 minutes to platelets, except when added sequentially they were added for 5 minutes each. Aggregation was induced with thrombin (0.012 U/mL). The aggregation tracings for ERp46 alone, PDI plus ERp46 added simultaneously, or PDI followed by ERp46 (PDI/ERp46) are overlapping; mean ± SEM, n = 5; *P < .05, ****P < .0001, ANOVA. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/13/10.1182_blood.2021012055/5/m_bloodbld2021012055f6.png?Expires=1771333923&Signature=2BOzEdIUWseqNHbG2aW10tGcgNAUVa3PmP33O2vba1ZwQcnJll9tUVypXUDpTSwZKXHz59X7uGy6X1orvF1zDl8JFoEF80gSfY2dhdiks-nvoWx0kXOZENObgR4YxdKqmYWa~AAuCMwx83gsf-uxDGx4ci6Xri5Pe0eibEXknqBqu1hkjvYHpxiicBFk7ltiNxuRya5F1ton75B45zfXjknNyTmzhwx9BdGGZlprYMYD1jjVwWBoYP8yJtHgyZo24a1xLDavxScO695-EibMEp3fpLF59rJZaqdUQ4MTNgroLqLUBCsPFWAmHDZibUTI02Z0H7HS6zWfIjtLQq6FQQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ERp46 generates thiols in the β3 subunit of αIIbβ3. (A) Reductase activity of ERp46 compared with ERp57, PDI, ERp5, ERp72, and inactivated ERp46 [ERp46(aaaaaa)] (30 nM each) in the Di-E-GSSG assay. (B-C) ERp46 generates thiols in β3 more effectively than PDI, ERp57, ERp72, or ERp5. ERp46 or other PDIs (1 μM) were incubated with αIIbβ3 (0.5 μM) (Abcam) for 20 minutes at 37°C. IodoTMT (400 μM) was then added for 1 hour at 37°C and labeling of thiols performed in 1% SDS with 5 mM ethylenediaminetetraacetic acid . These studies were performed in the absence of added GSH. (D-E) The active site cysteines of ERp46 generate thiols in αIIbβ3. Shown is the labeling of β3 with WT ERp46 or inactivated ERp46 [ERp46(aaaaaa)]. ERp46 or ERp46(aaaaaa) (1 μM) was incubated with αIIbβ3 (0.5 μM) and GSH (100 μM), and labeling was performed with iodoTMT (500 μM). (F-G) Effect of fibrinogen (Fbgn) on thiol generation by ERp46 and PDI in αIIbβ3. ERp46 or PDI (1 μM) were incubated with αIIbβ3 (0.5 μM) with GSH (100 μM). In some samples, fibrinogen (0.5 μM) was added. Thiols were labeled with iodoTMT (500 μM). Blotting for iodoTMT was performed using the anti-TMT antibody. (H-I) Effect of fibrinogen on thiol generation by ERp46 and PDI in αIIbβ3 in platelets. ERp46 or PDI (1 μM) were incubated with human platelets (4 × 108 platelets/mL) without GSH. In some samples, fibrinogen (1 μM) was added. Thiols were labeled with MPB and blotting was performed as described in supplemental Methods. The intensity of each band was calculated using the Image J program, and the ratio of iodoTMT (TMT) or MPB to β3 protein was compared with the untreated sample. Shown are the blots for β3 (H-96 or B-7 for D), PDI and ERp46 (rabbit antibodies), and the β chain of fibrinogen; mean ± SEM, n = 4 (B-C), n = 3 (D-E), n = 3 (F-G), n = 7 (H-I); *P < .05, **P < .01, ***P < .001, ****P < .0001, ANOVA. (J-K) Addition of ERp46 followed by PDI (ERp46/PDI) maximally generates thiols in β3. αIIbβ3 was treated with 1 μM PDI or ERp46 for 40 minutes, except when added sequentially they were added for 20 minutes each. Addition of PDI followed by ERp46 (PDI/ERp46) gave similar results to ERp46 alone or PDI plus ERp46 added simultaneously. Labeling was with iodoTMT (400 μM) in the absence of GSH; mean ± SEM, n = 4; ****P < .0001, ANOVA. (L-M) Addition of ERp46 followed by PDI (ERp46/PDI) provides maximal enhancement of platelet aggregation. 1.5 μM PDI or ERp46 were added for 10 minutes to platelets, except when added sequentially they were added for 5 minutes each. Aggregation was induced with thrombin (0.012 U/mL). The aggregation tracings for ERp46 alone, PDI plus ERp46 added simultaneously, or PDI followed by ERp46 (PDI/ERp46) are overlapping; mean ± SEM, n = 5; *P < .05, ****P < .0001, ANOVA. ns, not significant.
ERp46 generates thiols in the β3 subunit of αIIbβ3. (A) Reductase activity of ERp46 compared with ERp57, PDI, ERp5, ERp72, and inactivated ERp46 [ERp46(aaaaaa)] (30 nM each) in the Di-E-GSSG assay. (B-C) ERp46 generates thiols in β3 more effectively than PDI, ERp57, ERp72, or ERp5. ERp46 or other PDIs (1 μM) were incubated with αIIbβ3 (0.5 μM) (Abcam) for 20 minutes at 37°C. IodoTMT (400 μM) was then added for 1 hour at 37°C and labeling of thiols performed in 1% SDS with 5 mM ethylenediaminetetraacetic acid . These studies were performed in the absence of added GSH. (D-E) The active site cysteines of ERp46 generate thiols in αIIbβ3. Shown is the labeling of β3 with WT ERp46 or inactivated ERp46 [ERp46(aaaaaa)]. ERp46 or ERp46(aaaaaa) (1 μM) was incubated with αIIbβ3 (0.5 μM) and GSH (100 μM), and labeling was performed with iodoTMT (500 μM). (F-G) Effect of fibrinogen (Fbgn) on thiol generation by ERp46 and PDI in αIIbβ3. ERp46 or PDI (1 μM) were incubated with αIIbβ3 (0.5 μM) with GSH (100 μM). In some samples, fibrinogen (0.5 μM) was added. Thiols were labeled with iodoTMT (500 μM). Blotting for iodoTMT was performed using the anti-TMT antibody. (H-I) Effect of fibrinogen on thiol generation by ERp46 and PDI in αIIbβ3 in platelets. ERp46 or PDI (1 μM) were incubated with human platelets (4 × 108 platelets/mL) without GSH. In some samples, fibrinogen (1 μM) was added. Thiols were labeled with MPB and blotting was performed as described in supplemental Methods. The intensity of each band was calculated using the Image J program, and the ratio of iodoTMT (TMT) or MPB to β3 protein was compared with the untreated sample. Shown are the blots for β3 (H-96 or B-7 for D), PDI and ERp46 (rabbit antibodies), and the β chain of fibrinogen; mean ± SEM, n = 4 (B-C), n = 3 (D-E), n = 3 (F-G), n = 7 (H-I); *P < .05, **P < .01, ***P < .001, ****P < .0001, ANOVA. (J-K) Addition of ERp46 followed by PDI (ERp46/PDI) maximally generates thiols in β3. αIIbβ3 was treated with 1 μM PDI or ERp46 for 40 minutes, except when added sequentially they were added for 20 minutes each. Addition of PDI followed by ERp46 (PDI/ERp46) gave similar results to ERp46 alone or PDI plus ERp46 added simultaneously. Labeling was with iodoTMT (400 μM) in the absence of GSH; mean ± SEM, n = 4; ****P < .0001, ANOVA. (L-M) Addition of ERp46 followed by PDI (ERp46/PDI) provides maximal enhancement of platelet aggregation. 1.5 μM PDI or ERp46 were added for 10 minutes to platelets, except when added sequentially they were added for 5 minutes each. Aggregation was induced with thrombin (0.012 U/mL). The aggregation tracings for ERp46 alone, PDI plus ERp46 added simultaneously, or PDI followed by ERp46 (PDI/ERp46) are overlapping; mean ± SEM, n = 5; *P < .05, ****P < .0001, ANOVA. ns, not significant.
The function of cysteines in the β3 subunit has been a subject of previous investigation. Although most of the β3 cysteines are in the disulfide form,37 thiols in β3 increase with platelet activation.25,37 Additionally, mutations of cysteines in disulfide bonds in the epidermal growth factor (EGF) domains of the β3 subunit enhance αIIbβ3 activation.38,39 Moreover, using a differential labeling mass spectrometry method, Passam et al identified a disulfide bond in β3 targeted by ERp5.37 Using iodoTMT to label thiols, we found that ERp46 cleaved β3 disulfide bonds much better than other PDIs (Figure 6B-C), a function of its active site thiols (Figure 6D-E). Using either purified proteins or intact platelets, we found that ERp46 generated thiols in β3 in the absence of fibrinogen, whereas PDI generation of thiols was fibrinogen-dependent (Figure 6F-I).
Sequential addition of ERp46 followed by PDI to purified αIIbβ3 (ERp46/PDI) resulted in maximal thiol generation in β3 and maximal platelet aggregation (Figure 6J-M). The sequential effect on thiol generation and platelet aggregation was substantially greater than addition of either PDI or ERp46 alone, addition of PDI and ERp46 simultaneously, or addition of PDI followed by ERp46 (PDI/ERp46). (The weaker thiol generation when both PDI and ERp46 were incubated together with αIIbβ3 suggests PDI may interfere with the ERp46 catalyzed reactions. Under the experimental conditions used [absence of Mn2+], PDI binds to αIIbβ3 by SPR stronger than ERp46 [Kd of 15.7 nM] and inhibits Alexa-ERp46 binding to platelets better than unlabeled ERp46 [supplemental Figure 25A-C].) Overall, these experiments suggest that the sequence in which ERp46 and PDI act on αIIbβ3 is a determinant of αIIbβ3 activation and that these enzymes are nonredundant.
Mapping of disulfide bonds cleaved by ERp46 in the β3 subunit of αIIbβ3
To analyze the disulfide bond(s) regulated by ERp46, we incubated αIIbβ3 with or without ERp46 and performed differential alkylation and mass spectrometry. Briefly, thiols in human αIIbβ3 samples were alkylated with iodoTMT-126 in the sample without ERp46 and with iodoTMT-130 in the sample with ERp46 (supplemental Figure 26). Following reduction with dithiothreitol and TCEP, the newly formed thiols were alkylated with iodoTMT-127 and iodoTMT-131, respectively. Representative MS spectra of a Cys-containing peptide in the EGF-2 domain are shown (Figure 7A-C). The percentage of thiols in β3 of the initial samples to total thiols after reduction was calculated.
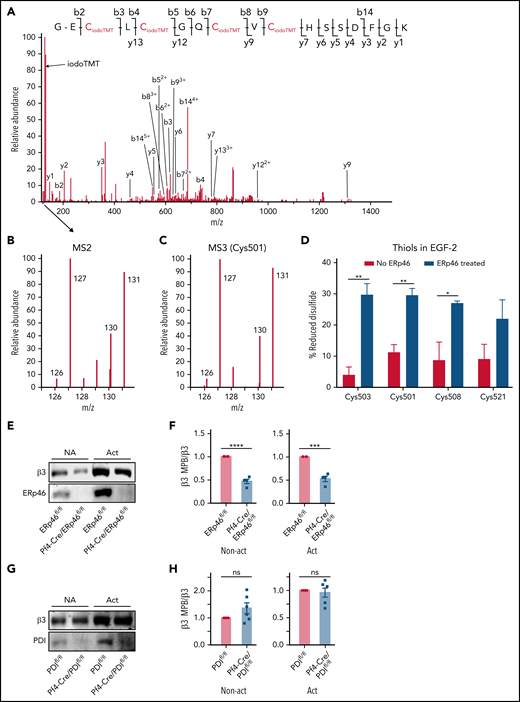
ERp46 generates thiols in the EGF-2 domain of the β3 subunit of the αIIbβ3 integrin. (A) The MS/MS spectrum of a 5+ charged ion (m/z 618.72) corresponding to the peptide GECLCGQCVCHSSDFGK from integrin β3 with all 4 cysteines alkylated by iodoTMT reagent. The y- and b-ion series in (A) enabled the confident identification of the peptide sequence. During the MS/MS stage of acquisition to derive fragment ions and sequence information, a unique reporter ion mass is also generated. These reporter ions are in the low mass region of the MS/MS spectrum. The intensity of iodoTMT tag (126, 127, 130, and 131) in (B) indicate the relative quantitation of redox changes. IodoTMT-126 or iodoTMT-130 represent the initial abundance of thiols in the peptide, whereas iodoTMT-127 and iodoTMT-131 represent the thiols in the peptide labeled after reduction of disulfide bonds in each sample with DTT/TCEP. (C) Representative MS3 spectrum of the first Cys (Cys501) in the peptide showing the increase in iodoTMT-130 induced by ERp46 relative to iodoTMT-126 in the sample without ERp46. IodoTMT-127 and iodoTMT-131 represent the thiols labeled after reduction of disulfide bonds in the samples. (D) ERp46 generates thiols in Cys503, Cys501, Cys508, and Cys521 of the EGF-2 domain as measured by MS3; mean ± SEM, n = 3; *P < .05, **P < .01, t test. These cysteines form the Cys473-Cys503, Cys486-Cys501, and Cys508-Cys521 disulfide bonds. (E-G) Thiols are decreased in the β3 subunit of nonactivated (NA) and activated (Act) ERp46-null platelets but are unchanged in PDI-null platelets. Platelets from Pf4-Cre/ERp46fl/fl mice, Pf4-Cre/PDIfl/fl mice, or Cre− littermates were activated with collagen (10 μg/mL) for 10 minutes without stirring followed by MPB labeling. After platelet lysis, the MPB was pulled down with streptavidin-agarose beads. The samples were analyzed by blotting with rabbit anti-β3 and anti-ERp46 or anti-PDI antibodies and the band densities calculated by densitometry using Odyssey V3.0. (E and G) Representative blots (F and H) cumulative data for nonactivated (NA) and activated platelets; mean streptavidin SEM, n = 4 (E-F), n = 6 (G-H), ***P < .001, ****P < .0001, Student t test.
ERp46 generates thiols in the EGF-2 domain of the β3 subunit of the αIIbβ3 integrin. (A) The MS/MS spectrum of a 5+ charged ion (m/z 618.72) corresponding to the peptide GECLCGQCVCHSSDFGK from integrin β3 with all 4 cysteines alkylated by iodoTMT reagent. The y- and b-ion series in (A) enabled the confident identification of the peptide sequence. During the MS/MS stage of acquisition to derive fragment ions and sequence information, a unique reporter ion mass is also generated. These reporter ions are in the low mass region of the MS/MS spectrum. The intensity of iodoTMT tag (126, 127, 130, and 131) in (B) indicate the relative quantitation of redox changes. IodoTMT-126 or iodoTMT-130 represent the initial abundance of thiols in the peptide, whereas iodoTMT-127 and iodoTMT-131 represent the thiols in the peptide labeled after reduction of disulfide bonds in each sample with DTT/TCEP. (C) Representative MS3 spectrum of the first Cys (Cys501) in the peptide showing the increase in iodoTMT-130 induced by ERp46 relative to iodoTMT-126 in the sample without ERp46. IodoTMT-127 and iodoTMT-131 represent the thiols labeled after reduction of disulfide bonds in the samples. (D) ERp46 generates thiols in Cys503, Cys501, Cys508, and Cys521 of the EGF-2 domain as measured by MS3; mean ± SEM, n = 3; *P < .05, **P < .01, t test. These cysteines form the Cys473-Cys503, Cys486-Cys501, and Cys508-Cys521 disulfide bonds. (E-G) Thiols are decreased in the β3 subunit of nonactivated (NA) and activated (Act) ERp46-null platelets but are unchanged in PDI-null platelets. Platelets from Pf4-Cre/ERp46fl/fl mice, Pf4-Cre/PDIfl/fl mice, or Cre− littermates were activated with collagen (10 μg/mL) for 10 minutes without stirring followed by MPB labeling. After platelet lysis, the MPB was pulled down with streptavidin-agarose beads. The samples were analyzed by blotting with rabbit anti-β3 and anti-ERp46 or anti-PDI antibodies and the band densities calculated by densitometry using Odyssey V3.0. (E and G) Representative blots (F and H) cumulative data for nonactivated (NA) and activated platelets; mean streptavidin SEM, n = 4 (E-F), n = 6 (G-H), ***P < .001, ****P < .0001, Student t test.
We identified ∼40 of the 56 Cys in the β3 subunit by iodoTMT labeling of thiols (supplemental Table 1). ERp46 increased the thiol label of multiple cysteines by more than twofold. Because ERp46 appears to target multiple cysteines and because preliminary results suggested the Cys473-Cys503 allosteric disulfide bond was targeted by ERp46, we focused on the EGF-2 domain containing this disulfide. ERp46 generated thiols in Cys503, Cys501, Cys508 (Figure 7D), and Cys521, which forms a disulfide pair with Cys508.40 These findings suggest that ERp46 also cleaves the Cys486-Cys501 and Cys508-Cys521 disulfide bonds. ERp46 may affect additional Cys in the EGF-3 or other domains (supplemental Table 1) but did not increase iodoTMT label in several cysteines in the β3 subunit (Cys386, Cys601, and Cys604), implying that ERp46 does not target all the cysteines in β3. Thiol labeling was decreased in β3 in nonactivated and activated ERp46-null platelets, suggesting that platelet ERp46 has reductase activity (Figure 7E-F). Thiol labeling was not decreased in β3 in PDI-null platelets (Figure 7G-H), further implying these enzymes act by distinct mechanisms in platelets.
Discussion
In this study, we show that ERp46 is necessary for hemostasis, thrombosis, and platelet function. ERp46 is expressed on the platelet surface and potentiates αIIbβ3 activation. Both the anti-ERp46 antibody and inactivated ERp46 protein inhibited platelet aggregation and dense granule release, whereas WT ERp46 potentiated αIIbβ3 activation and platelet aggregation (Figures 1I and 4F). ERp46-null platelets showed decreased αIIbβ3 activation and reduced platelet aggregation and granule release that was recovered by the addition of ERp46. These findings demonstrate a central role for extracellular ERp46 in platelet function and that the substrate of ERp46 is on the platelet surface. ERp46 deficiency prolonged the tail bleeding time and decreased platelet accumulation in FeCl3 and laser-induced arterial injury models. The platelet accumulation defect was rescued by infusion of WT ERp46 but not inactivated ERp46, indicating that the catalytic activity of extracellular ERp46 is required for platelet accumulation. Thus, ERp46 is the fifth member of the PDI family on the platelet surface that supports platelet aggregation, hemostasis, and thrombosis.
Several lines of evidence implicate αIIbβ3 as a substrate for ERp46. The CHAMP peptide anti-αIIb TM binds directly to the transmembrane domain of the αIIb subunit of αIIbβ3, causing separation of the αIIbβ3 heterodimer resulting in fibrinogen binding and platelet aggregation.30,31 CHAMP-induced aggregation of human platelets was inhibited by the anti-ERp46 antibody and was decreased in ERp46-null platelets (Figure 3F-G), suggesting that a physical interaction of ERp46 with αIIbβ3 is required for aggregation.
Moreover, coimmunoprecipitation studies showed that ERp46 associated with αIIbβ3 upon platelet activation. Additionally, the affinity of ERp46 binding to αIIbβ3 in the presence of Mn2+ by SPR (Kd = 2.55 nM) is the strongest reported for prothrombotic PDIs that bind to αIIbβ3.13,28,41 ERp46 binds to αIIbβ3 by SPR with a low Kd even in the absence of Mn2+, suggesting ERp46 can bind to minimally activated αIIbβ3. Moreover, ERp46 binding to Mn2+-treated platelets lacking αIIbβ3 was decreased consistent with a direct association of ERp46 with αIIbβ3 on platelets.32 ERp46 also generates thiols in αIIbβ3 as detected by thiol labeling and mass spectrometry, providing additional evidence for a functional interaction between the 2 proteins. Other potential extracellular substrates for ERp46 on the platelet surface include proteins that regulate ATP release and P-selectin expression (Figure 3B-D; supplemental Figure 5). ERp46 may also be part of a redox chain of thiol isomerases on the platelet surface.42,43
Each active site of ERp46 has a similar effect on recovery of aggregation of ERp46-null platelets, in contrast to PDI, ERp57, and ERp72.10,12,14 Each active site of ERp46 also functions similarly in protein folding19 in contrast to other PDIs.44-46 This is compatible the radically different molecular architecture of ERp46 with 3 solvent-exposed active sites located separately on the protein surface.19,20
The thiol form of ERp46 that reduces disulfide bonds is found on resting platelets, and with activation the thiols increase (Figure 1D-E). ERp46 cleaves disulfide bonds in GSSG and in αIIbβ3 much better than other prothrombotic PDIs, and ERp46 was previously reported to have higher reductase than oxidase/isomerase activity compared with PDI and ERp57.47 Moreover, ERp46-deficient platelets have decreased thiols in β3, indicating that ERp46 cleaves disulfide bonds in platelets (Figure 7E-F). ERp46 has strong activity against purified αIIbβ3 and αIIbβ3 in platelets in the absence of fibrinogen, whereas the ability of PDI to cleave disulfide bonds in αIIbβ3 is dependent upon fibrinogen (Figure 6F-I). Also, addition of ERp46 to platelets followed by PDI provided maximal thiol generation in β3 and maximal enhancement of platelet aggregation (Figure 6J-M). The concept that these enzymes catalyze sequential events is consistent with the role of ERp46 in other studies where ERp46 acts prior to PDI.19,20,48 Together, these data suggest that ERp46 acts earlier in activation of αIIbβ3 before ligand binding, whereas PDI catalyzes a subsequent reaction. ERp46 cleavage of disulfide bonds in αIIbβ3 could generate thiols allowing another prothrombotic PDI to catalyze a thiol-disulfide exchange resulting in full activation of αIIbβ3.
ERp46 cleaves the Cys473-Cys503, Cys486-Cys501, and Cys508-Cys521 disulfide bonds of the EGF-2 domain. In contrast to the broader disulfide cleavage of ERp46, ERp5 targets a single disulfide bond, Cys177-Cys184.37 The notion that the PDIs differentially target disulfide bonds provides mechanistic insight into the distinct functions of the PDIs in platelets.
Several lines of evidence suggest that ERp46 has a distinct role in αIIbβ3 activation. First, the defect in ERp46-deficient platelet aggregation was only recovered by ERp46, but not other PDIs. Second, the effect of PDI on αIIbβ3 requires fibrinogen, whereas the effect of ERp46 does not (Figure 6F-I). Third, there is a different thiol labeling pattern in the β3 subunit of αIIbβ3 in ERp46 versus PDI-null platelets (Figure 7E-H). Fourth, there is a substantial difference in thiol generation in β3 and potentiation of platelet aggregation depending on the sequence in which ERp46 and PDI are added to αIIbβ3 or platelets (Figure 6J-M). Fifth, ERp46 targets the Cys473-Cys503 disulfide bond (Figure 7D), whereas PDI and ERp5 do not.37 The different thiol labeling pattern in β3 of ERp46-null versus PDI-null platelets, the sequence of the reactions these enzymes catalyze, the differences in fibrinogen dependence of these enzymes, and the disulfide bonds they target, provide evidence for nonredundancy of these enzymes.
The defects in ERp46-null platelets appear greater than for other PDIs. Aggregation is decreased by ∼80% in ERp46-null platelets (Figures 4C-E and 5A) compared with the ∼65% decrease in PDI and ERp57-null platelets (Figure 5B-C).10,12,14 The platelet accumulation defect in FeCl3-induced arterial injury of the mesenteric artery was greater in Pf4-Cre/ERp46fl/fl mice than in previously studied PDI targeted knockout mice.12 These findings suggest that ERp46 is of major importance in platelet function and are consistent with ERp46 acting prior to other PDIs whose function requires an ERp46-catalyzed reaction. Therefore, therapeutic targeting of ERp46 may be more efficacious than targeting other PDIs.
Cys177-Cys184 is the only disulfide bond with an allosteric conformation identified from the protein database40,49 shown to date to be targeted by a PDI.37 Here, we found the Cys473-Cys503 disulfide bond that also has an allosteric conformation is targeted by ERp46. Although the Cys486-Cys501 and Cys508-Cys521 disulfide bonds are considered structural,40 the conformation of a disulfide bond may change while the protein is in solution.49,50 Moreover, structural disulfide bonds can also be functional.51 Thus, the functional targets of ERp46 are not necessarily limited to disulfide bonds with allosteric configurations.18,49 The concept that ERp46 targets multiple disulfide bonds in β3 is consistent with ERp46 targeting multiple cysteines in proteins in the endoplasmic reticulum.20
The “yin and yang” regulation of thrombosis by the PDI family provides a novel concept that both pro- and antithrombotic PDIs regulate platelet function and thrombosis.18 This thiol isomerase redox network maintains vascular homeostasis by constituting off-on redox switches controlling activation of hemostatic factors as αIIbβ3. This study uncovers ERp46 as a novel prothrombotic PDI family member that appears to serve as an early regulator for αIIbβ3 activation before ligand binding. The role of ERp46 in platelets offers novel insight into the redox network controlling hemostasis and thrombosis and will stimulate further investigation of its role in disease processes.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01HL118526 (D.W.E.), R35HL150698 (M.P.), and HL155694 (S.P.K.). The mass spectrometry study was funded in part by NIH P30NS046593 and S10OD025047 (H.L.).
We dedicate this article to our colleague Joel S. Bennett, who recently passed away. Joel made seminal discoveries on the αIIbβ3 integrin, and we valued his insight. He will be missed.
Authorship
Contribution: J.Z. designed and performed research, collected and analyzed data, and corrected portions of the manuscript; L.W. performed research and analyzed data; L.R., G.K., and M.P. assisted with the in vivo hemostasis and thrombosis models and helped with study design and interpretation; T.L. and H.L. performed the mass spectrometry experiments; K.P.F. and J.S.B. provided the CHAMP peptide and helped with the study design and data analysis; S.P.K. helped with the FeCl3 injury models; Y.W. performed research; and D.W.E. and Y.W. designed the project, supervised the research, analyzed the data, and wrote the manuscript.
Joel S. Bennett died on 21 June 2021.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David W. Essex, Temple University School of Medicine, Room 204 MRB, 3430 North Broad St, Philadelphia, PA 19140; e-mail: essex@temple.edu.
Requests for data sharing may be submitted to David W. Essex (essex@temple.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal