


Abstract
In 2007 and 2009, the regulatory approval of the first-in-class complement inhibitor eculizumab revolutionized the clinical management of 2 rare, life-threatening clinical conditions: paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS). Although being completely distinct diseases affecting blood cells and the glomerulus, PNH and aHUS remarkably share several features in their etiology and clinical presentation. An imbalance between complement activation and regulation at host surfaces underlies both diseases precipitating in severe thrombotic events that are largely resistant to anticoagulant and/or antiplatelet therapies. Inhibition of the common terminal complement pathway by eculizumab prevents the frequently occurring thrombotic events responsible for the high mortality and morbidity observed in patients not treated with anticomplement therapy. Although many in vitro and ex vivo studies elaborate numerous different molecular interactions between complement activation products and hemostasis, this review focuses on the clinical evidence that links these 2 fields in humans. Several noninfectious conditions with known complement involvement are scrutinized for common patterns concerning a prothrombotic statues and the occurrence of certain complement activation levels. Next to PNH and aHUS, germline-encoded CD59 or CD55 deficiency (the latter causing the disease complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy), autoimmune hemolytic anemia, (catastrophic) antiphospholipid syndrome, and C3 glomerulopathy are considered. Parallels and distinct features among these conditions are discussed against the background of thrombosis, complement activation, and potential complement diagnostic and therapeutic avenues.
Introduction
The biological process of hemostasis seals injuries in blood vessels by thrombus formation and initializes repair achieving conservation of vital blood components and blockage of invading pathogens. Host defense mechanisms by clot formation go beyond simply plugging a hole. Bacteria become cross-linked and thus entrapped in fibrin fiber networks.1 This mechanism is reminiscent of the ones in arthropods in which clot formation of the hemolymph is 1 key mechanism to serve conservation and defense at the same time.2,3 In mammals, blood conservation and pathogen defense are organized in the response units of hemostasis and immunity. However, both units are often required simultaneously when addressing injured vasculature. Because of coevolution, it seems obvious that multiple physiologic links between these 2 units reinforce each other.3-5 At the heart of this intercommunication are the 2 serine protease networks of the coagulation cascade and the complement system that act as first responders to diverse danger signals6-8 (for a topical representation of the complement cascade, see Figure 1A). Crucial cellular surfaces also link these 2 fields. Many studies have identified different complement-based avenues that activate platelets, endothelium, and/or leukocytes to promote prothrombotic cell activation, but diverse findings prohibit to pinpoint which complement effectors are most critically involved in inducing a prothrombotic state (for selected studies, see Table 1). Yet, the contrary direction of events is also feasible. Cell activation (eg, platelets and endothelium), by other means than complement can promote complement activation at the cell surface via P-selectin expression, thus enhancing immune surveillance at the site of vascular injury.9,10 As instrumental as these molecular interactions studies are for our mechanistic understanding, they often cannot precisely predict the impact of selected interactions on inducing a prothrombotic phenotype in vivo and sometimes do not align with the situation in vivo.11,12 In addition to such molecular interaction studies, the overlap between thrombosis and complement is also strongly supported by several clinical phenomena. Rather than focusing on in vitro findings in a bottom-up approach, here we use a top-down approach and dissect the interactions between complement and the induction of a prothrombotic state by mainly evaluating clinical data and their implications by focusing on pathophysiologic conditions substantially driven by aberrant complement activation.
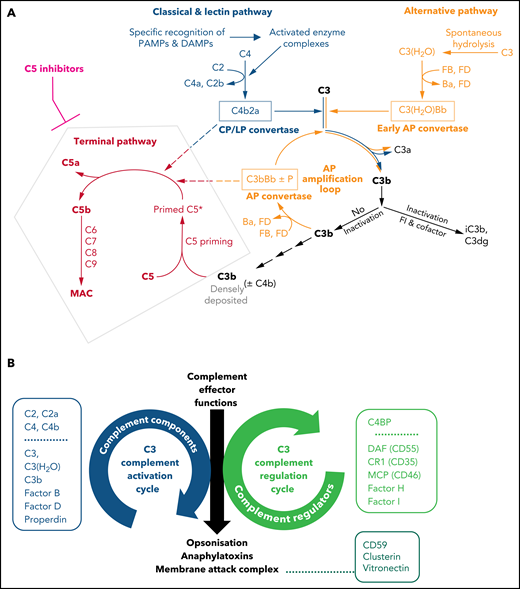
Illustrations of key features of the complement system. (A) Schematic drawing of the complement cascade. The complement initiation pathways of the CP and LP are triggered through recognition of danger signals by soluble pattern recognition molecules in blood cumulating in the assembly of the bimolecular C3 convertase C4b2a. Spontaneous hydrolysis of C3 (tick-over activation) assembles the early AP convertase autonomously of any danger signal. The C3 convertases activate C3 into the anaphylatoxin C3a and the opsonin C3b, which can auto-amplify via the AP amplification loop. Strong activation of CP/LP and/or impaired inactivation of C3b and convertases lead to a high surface density of C3b (±C4b). The latter recruits C5 and induces conformational changes (called priming) that allow the convertases to proteolytically activate primed C5 into the anaphylatoxin C5a and C5b initiating the terminal pathway. C5b launches the assembly of the MAC. Complement inhibitors targeting C5 hinder C5 priming and/or block convertase mediated activation of C5. Although TP is blocked in the presence of C5 inhibitors, the complement routes upstream of C5 continue to function. The figure is reproduced from Mannes et al,122 with modifications. (B) C3 activation and regulation cycles under physiologic conditions. The opposite directionality of the complement activation and regulation cycle maintain a fine-tuned balance controlling the unleashing of the 3 main effector functions of the complement cascade. The C3 activation cycle summarizes all routes and complement components that assemble the complement convertases, whereas the regulatory cycle is built from the convertase-directed regulators that decay the convertase (decay accelerating function) and/or proteolytically inactivate C3b (cofactor activity). Key components of each cycle are indicated in the respective boxes next to the cycles. Components and regulators associated purely with the CP and LP are displayed separately at the top of the boxes. The opsonization with C3 fragments and release of anaphalytoxins are directly effectuated and controlled by the C3 activation and regulation cycles, respectively. The formation of the membrane attack complex (initiated by C5 activation) is a consequence of strong C3 deposition by convertases and hence is indirectly regulated by the C3 regulation cycle. However, direct regulators of C5b-9 assemble exist in forms of the GPI-anchored CD59 on host surfaces and the 2 soluble plasma proteins Clusterin and Vitronectin (also called S protein) as indicated.
Illustrations of key features of the complement system. (A) Schematic drawing of the complement cascade. The complement initiation pathways of the CP and LP are triggered through recognition of danger signals by soluble pattern recognition molecules in blood cumulating in the assembly of the bimolecular C3 convertase C4b2a. Spontaneous hydrolysis of C3 (tick-over activation) assembles the early AP convertase autonomously of any danger signal. The C3 convertases activate C3 into the anaphylatoxin C3a and the opsonin C3b, which can auto-amplify via the AP amplification loop. Strong activation of CP/LP and/or impaired inactivation of C3b and convertases lead to a high surface density of C3b (±C4b). The latter recruits C5 and induces conformational changes (called priming) that allow the convertases to proteolytically activate primed C5 into the anaphylatoxin C5a and C5b initiating the terminal pathway. C5b launches the assembly of the MAC. Complement inhibitors targeting C5 hinder C5 priming and/or block convertase mediated activation of C5. Although TP is blocked in the presence of C5 inhibitors, the complement routes upstream of C5 continue to function. The figure is reproduced from Mannes et al,122 with modifications. (B) C3 activation and regulation cycles under physiologic conditions. The opposite directionality of the complement activation and regulation cycle maintain a fine-tuned balance controlling the unleashing of the 3 main effector functions of the complement cascade. The C3 activation cycle summarizes all routes and complement components that assemble the complement convertases, whereas the regulatory cycle is built from the convertase-directed regulators that decay the convertase (decay accelerating function) and/or proteolytically inactivate C3b (cofactor activity). Key components of each cycle are indicated in the respective boxes next to the cycles. Components and regulators associated purely with the CP and LP are displayed separately at the top of the boxes. The opsonization with C3 fragments and release of anaphalytoxins are directly effectuated and controlled by the C3 activation and regulation cycles, respectively. The formation of the membrane attack complex (initiated by C5 activation) is a consequence of strong C3 deposition by convertases and hence is indirectly regulated by the C3 regulation cycle. However, direct regulators of C5b-9 assemble exist in forms of the GPI-anchored CD59 on host surfaces and the 2 soluble plasma proteins Clusterin and Vitronectin (also called S protein) as indicated.
Selected studies demonstrating a mechanistic link between complement activation and induction of coagulation and/or prothrombotic cell activation in vitro or in animal models
| First author/reference | Main finding | Prothrombotic state induced via activation of | Comment |
|---|---|---|---|
| Zimmerman130 |
| C5-C9 | However, such effects were not reproduced in blood form a C6 deficient and a C7 deficient patient.131,132 |
| Polley133 |
| C3 and C5-C9 Also Purified C5-C9 components alone (to a smaller extent) | The activated platelet surface can acquire complement proteins even in absence of convertases leading to increased platelet activation. C3 was probably acquired as C3(H2O). More recently contact activation of C3134-136 has been described that could explains this phenomenon. |
| Polley137 |
| C3 | Suggests the presence of C3a receptors on human platelets that upon ligation by C3a/C3a-desArg lead to platelet activation |
| Wiedmer138,139 |
| C5-C9 | C5b-9 assembly on platelets accelerate platelet-catalyzed thrombin generation |
| Sims140 |
| C5-C9 | Properties previously allocated to the activated platelet surface may rather or in addition be caused by platelet microparticles |
| Hattori141 |
| C5-C9 | C5b-9 stimulate endothelial cells to secrete prothrombotic, platelet adhesive vWF protein |
| Foreman142 |
| C5 | By promoting adhesive interactions between neutrophils and endothelial cells C5a induces acute inflammatory responses |
| Ritis143 |
| C5 | Neutrophils may link complement activation to coagulation pathways; TF is not upregulated on monocytes in response to C5a alone144 |
| Gushiken145 |
| C3 (and possibly* C5-C9) | C3 deficiency in mice leads to abnormal platelet function |
| Subramaniam146 Asfarhar-Kharghan147 |
| Mainly via C3 regarding platelets Via C5 regarding leukocytes | Suggests a prominent role for C3 and its activation products in platelet activation independent of TP activation. C5 activation acts procoagulantly by inducing TF release |
| Sauter148 |
| C3 | A functional role for C3aR on platelets is proposed |
| First author/reference | Main finding | Prothrombotic state induced via activation of | Comment |
|---|---|---|---|
| Zimmerman130 |
| C5-C9 | However, such effects were not reproduced in blood form a C6 deficient and a C7 deficient patient.131,132 |
| Polley133 |
| C3 and C5-C9 Also Purified C5-C9 components alone (to a smaller extent) | The activated platelet surface can acquire complement proteins even in absence of convertases leading to increased platelet activation. C3 was probably acquired as C3(H2O). More recently contact activation of C3134-136 has been described that could explains this phenomenon. |
| Polley137 |
| C3 | Suggests the presence of C3a receptors on human platelets that upon ligation by C3a/C3a-desArg lead to platelet activation |
| Wiedmer138,139 |
| C5-C9 | C5b-9 assembly on platelets accelerate platelet-catalyzed thrombin generation |
| Sims140 |
| C5-C9 | Properties previously allocated to the activated platelet surface may rather or in addition be caused by platelet microparticles |
| Hattori141 |
| C5-C9 | C5b-9 stimulate endothelial cells to secrete prothrombotic, platelet adhesive vWF protein |
| Foreman142 |
| C5 | By promoting adhesive interactions between neutrophils and endothelial cells C5a induces acute inflammatory responses |
| Ritis143 |
| C5 | Neutrophils may link complement activation to coagulation pathways; TF is not upregulated on monocytes in response to C5a alone144 |
| Gushiken145 |
| C3 (and possibly* C5-C9) | C3 deficiency in mice leads to abnormal platelet function |
| Subramaniam146 Asfarhar-Kharghan147 |
| Mainly via C3 regarding platelets Via C5 regarding leukocytes | Suggests a prominent role for C3 and its activation products in platelet activation independent of TP activation. C5 activation acts procoagulantly by inducing TF release |
| Sauter148 |
| C3 | A functional role for C3aR on platelets is proposed |
“Possibly” is written when an involvement because of the known complement pathways is logical and likely could have occurred but was not formally tested or controlled for.
Noninfectious diseases driven by complement dysregulation
Next to strong complement activation by microbial invaders, complement also activates mildly on host particles like apoptotic bodies to maintain body homeostasis, thereby keeping effector functions restricted to C3 opsonization in absence of terminal pathway (TP) activation.13 To accommodate for such different activation levels, the complement cascade is fine-tuned, keeping the delicate balance between 2 opposing C3 regulatory circuits14: the complement C3 activation and regulation cycles (Figure 1B). At the heart of the activation cycle are the complement convertases that are assembled via any of the 3 initiating pathways. Convertases proteolytically activate the central complement components C3 and consecutively C5 inducing their effector functions. At the core of the inactivation cycle is a set of preformed, convertase-directed regulators that inhibit the convertases in a catalytic fashion (by decay acceleration and cofactor activities; other inhibitors are also available to inhibit other complement effector molecules15,16). Thus, too much activation or too little regulation can independently result in inappropriate and damaging complement effector functions. Overdrive of the complement activation pathways also happens in infectious diseases such as COVID-19. It is conceivable that during SARS-CoV-2 infections, next to signaling in response to pathogenic patterns, exuberant complement activation has a role in prothrombotic cell activation.17-21 However, this review focuses on noninfectious diseases with aberrant complement activation/regulation.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria (PNH) has been described as the “most vicious acquired thrombophilic state known in medicine.”22 Mechanistically, a hyperreactivity of the complement alternative pathway (AP) against blood cells drives the pathophysiology of classical PNH. Because of somatic mutations in the PIG-A gene, affected HSCs and their progeny either lack or have diminished numbers of Glycosylphosphatidylinositol (GPI)-anchored proteins at their surfaces.23,24 This includes the 2 important GPI-anchored complement regulators CD55 (or DAF) and CD59 leading to aberrant AP and consecutively TP activation on affected cells resulting in cytolysis (Figure 2A). Leading clinical features of classical PNH are, among others, anemia caused by hemolysis, smooth muscle dystonia, and thrombosis. Next to the different complement activation products and their impact on inducing a prothrombotic phenotype (Table 1), hemolysis-associated hemoglobin release diminishes free nitric oxide. Depletion of nitric oxide induces smooth muscles contraction and activation and aggregation of platelets.25,26 All plausible mechanisms inducing a prothrombotic phenotype in PNH are summarized here.27 Lately, it was demonstrated that complement mediated hemoglobin release may propagate a vicious cycle as free hemoglobin activates complement.28 Overall, the question remains whether complement activation products, cytolysis, or both events together are the main drivers of thrombotic complications in PNH. Cytolysis by itself may be sufficient to induce a prothrombotic state (eg, via injured cells exposing phosphatidylserine aiding the formation of the tenase complex),29 release of hemoglobin from erythrocytes, and/or leaking of the intracellular danger signals like ADP.30
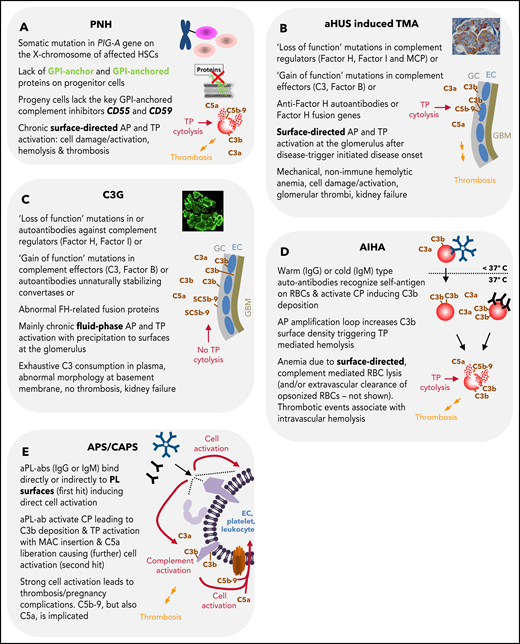
Summary of features for diseases with substantial complement involvement. (A) PNH. Acquired somatic mutations in the PIG-A gene (encoding a glycosyl transferase essential for the synthesis of GPI anchors) of hematopoietic stem cells result in a loss of or in reduced numbers of GPI anchored proteins on the surface of progenitor cells (erythrocytes, platelets, leukocytes, lymphocytes). Consequences thereof are highlighted in the figure. (B) aHUS-induced TMA. Different heterozygous mutations are linked to causing the disease. Gain-of-function mutations in complement components, loss of function mutilations in complement regulators, loss of function because of hybrid genes of the regulator factor H, or autoantibodies directed against factor H are the main contributors to aHUS inducing an imbalance in complement activation/regulation after a complement activation trigger (eg, infection, surgery; DGKE type aHUS is not covered). Key events in aHUS-induced TMA are summarized in the figure. GC, glycocalyx; EC, endothelial cells; GBM, glomerular basement membrane. (C) C3G. Like in aHUS, gain-of-function mutations of complement effector proteins or loss of function mutations in regulators are described. In addition, FH-related fusion proteins and autoantibodies either stabilizing the intrinsic short-lived convertases complexes or impairing complement regulators are reported. This leads to AP dysregulation in the fluid phase, which precipitates onto surfaces apparently without inflicting widespread cytolysis. Although TP activation products are identified in the glomeruli of patients, the C5b-9 complexes appear to be inhibited by the fluid phase inhibitors vitronectin (S protein) and/or clusterin yielding SC5b-9, which cannot insert into membrane bilayers. (D) AIHA. Cold-type autoantibodies bind to erythrocytes at temperatures lower than 37°C and induce covalent C3b deposition that is amplified via the AP amplification loop. At 37°C, the low-affinity IgM antibodies regularly dissociate. If high enough C3b densities are reached, TP-mediated cytolysis occurs via MAC. The sequence of events is similar for warm-type antibodies (usually IgG); however, these (high-affinity) antibodies bind and remain bound to red blood cells (RBCs) at 37°C. Not all IgG antibodies can activate the CP (strong enough) to lead to MAC formation and intravascular hemolysis. If not lysed, IgG and/or C3-fragment positive RBCs are recognized by phagocytes of the reticuloendothelial system (RES), resulting in extravascular removal of such RBCs without intravascular hemolysis (data not shown). Erythrocyte lysis within the vasculature is associated with thrombotic events. (E) APS/CAPS. aPL-abs are a heterogenous group of autoantibodies (mostly IgG and IgM, but IgA also occur) targeting complexes of PLs and PL-binding proteins. Occurrence of aPL-ab– and antibody binding–induced cell activation is considered first hit. Complement activation and signaling via TP activation products C5b-9 and C5a are considered second hit. The role of complement activation in augmenting thrombotic is well documented but may not explain all thrombotic events in APS.
Summary of features for diseases with substantial complement involvement. (A) PNH. Acquired somatic mutations in the PIG-A gene (encoding a glycosyl transferase essential for the synthesis of GPI anchors) of hematopoietic stem cells result in a loss of or in reduced numbers of GPI anchored proteins on the surface of progenitor cells (erythrocytes, platelets, leukocytes, lymphocytes). Consequences thereof are highlighted in the figure. (B) aHUS-induced TMA. Different heterozygous mutations are linked to causing the disease. Gain-of-function mutations in complement components, loss of function mutilations in complement regulators, loss of function because of hybrid genes of the regulator factor H, or autoantibodies directed against factor H are the main contributors to aHUS inducing an imbalance in complement activation/regulation after a complement activation trigger (eg, infection, surgery; DGKE type aHUS is not covered). Key events in aHUS-induced TMA are summarized in the figure. GC, glycocalyx; EC, endothelial cells; GBM, glomerular basement membrane. (C) C3G. Like in aHUS, gain-of-function mutations of complement effector proteins or loss of function mutations in regulators are described. In addition, FH-related fusion proteins and autoantibodies either stabilizing the intrinsic short-lived convertases complexes or impairing complement regulators are reported. This leads to AP dysregulation in the fluid phase, which precipitates onto surfaces apparently without inflicting widespread cytolysis. Although TP activation products are identified in the glomeruli of patients, the C5b-9 complexes appear to be inhibited by the fluid phase inhibitors vitronectin (S protein) and/or clusterin yielding SC5b-9, which cannot insert into membrane bilayers. (D) AIHA. Cold-type autoantibodies bind to erythrocytes at temperatures lower than 37°C and induce covalent C3b deposition that is amplified via the AP amplification loop. At 37°C, the low-affinity IgM antibodies regularly dissociate. If high enough C3b densities are reached, TP-mediated cytolysis occurs via MAC. The sequence of events is similar for warm-type antibodies (usually IgG); however, these (high-affinity) antibodies bind and remain bound to red blood cells (RBCs) at 37°C. Not all IgG antibodies can activate the CP (strong enough) to lead to MAC formation and intravascular hemolysis. If not lysed, IgG and/or C3-fragment positive RBCs are recognized by phagocytes of the reticuloendothelial system (RES), resulting in extravascular removal of such RBCs without intravascular hemolysis (data not shown). Erythrocyte lysis within the vasculature is associated with thrombotic events. (E) APS/CAPS. aPL-abs are a heterogenous group of autoantibodies (mostly IgG and IgM, but IgA also occur) targeting complexes of PLs and PL-binding proteins. Occurrence of aPL-ab– and antibody binding–induced cell activation is considered first hit. Complement activation and signaling via TP activation products C5b-9 and C5a are considered second hit. The role of complement activation in augmenting thrombotic is well documented but may not explain all thrombotic events in APS.
Evidence from clinical practice indicates the cytolytic TP as major driver of thrombosis. Thromboembolism was mainly responsible for the mortality in patients with PNH (up to 67%) before anti-C5 therapy with eculizumab (Table 2). Diagnosis of thrombosis at first presentation of patients with PNH was associated with survival rates at 4 years as low as 40%.27 Thrombotic complications can manifest at various sites alone or simultaneously and include cerebral, hepatic, mesenteric, lower extremities, pulmonary, and dermal veins,31 but also cerebral and coronary arteries.32,33 Although platelet activation (or even a hyperreactivity) is supported by in vitro analysis and seems to coincide with the rate of hemolysis in patients,34 antiplatelet therapy was not usually used. As a result, no conclusive clinical data exist on the effect of antiplatelet drugs in PNH. In contrast, anticoagulation by heparin preparations and/or vitamin K antagonists was regularly administered to patients with PNH characterized by a high risk of or a known history of thromboses.35 However, thrombotic complications often occurred despite therapeutic anticoagulation.27 Only therapeutic intervention at the level of C5 with eculizumab could efficiently prevent thromboembolic events, establishing eculizumab and later ravulizumab (an eculizumab derivative with superior pharmacokinetic features) as the standard of care.27,36-43 Diagnostic ex vivo studies support these findings demonstrating that commencement of eculizumab treatment led to a substantial decrease in thrombin and endothelial cell activation markers in patients with PNH, which coincides with a decrease in the hemolysis marker lactate dehydrogenase (LDH).44,45
Studies demonstrating a reduction in thrombotic events in PNH and aHUS
| First author/ reference | Disease | Rate of TE under best supportive therapy (number of patients) | Rate of TE under complement inhibition by eculizumab (number of patients) | Comment |
|---|---|---|---|---|
| Hillman38 | PNH prevalence: ∼13 per million149 | 7.37 events/ 100 patient-years (195) 10.61 events/100 patient-years on antithrombotic therapy (103) | 1.07 events/100 patient-years (195) 0.62 events/100 patient-years on antithrombotic therapy (103) |
|
| Kelly40 | 5.6 events per 100 patient-years (79) | 0.8 events per 100 patient-years (eculizumab) | ||
| Loschi151 | 27% of patients (191) | 4% of patients (123) | ||
| Legendre152 | aHUS prevalence: ∼5 per million153 | 100% since diagnosis of TMA is per se a thrombotic event (37 of 37 patients) | 19% (7 of 37) did not reach complete TMA-free statues during 62-64 wk of treatment |
|
| First author/ reference | Disease | Rate of TE under best supportive therapy (number of patients) | Rate of TE under complement inhibition by eculizumab (number of patients) | Comment |
|---|---|---|---|---|
| Hillman38 | PNH prevalence: ∼13 per million149 | 7.37 events/ 100 patient-years (195) 10.61 events/100 patient-years on antithrombotic therapy (103) | 1.07 events/100 patient-years (195) 0.62 events/100 patient-years on antithrombotic therapy (103) |
|
| Kelly40 | 5.6 events per 100 patient-years (79) | 0.8 events per 100 patient-years (eculizumab) | ||
| Loschi151 | 27% of patients (191) | 4% of patients (123) | ||
| Legendre152 | aHUS prevalence: ∼5 per million153 | 100% since diagnosis of TMA is per se a thrombotic event (37 of 37 patients) | 19% (7 of 37) did not reach complete TMA-free statues during 62-64 wk of treatment |
|
TE, thromboembolism.
Overall, these observations concerning PNH stress that thrombophilia coincides with cytolytic complement activation and is resistant to treatment with anticoagulants but can be prevented by complement TP inhibition blocking C5 activation.
Isolated germline deficiencies of CD59 or CD55
Isolated CD59 (MIM: 612300) or CD55 (MIM: 226300) deficiency is each caused by homozygous germline mutations in the corresponding genes, giving rise to 2 distinct autosomal recessive disorders characterized by infantile onset and association with consanguineous parents.
Typical symptoms of CD59 deficiency include, among others, recurrent intravascular hemolysis, polyneuropathy, and frequently severe/fatal thromboembolic events,46-49 showing substantial overlap with PNH (Table 3). This is further underlined by a remarkable clinical benefit in these patients through C5 inhibition by eculizumab.49,50 CD59 is a well-characterized and crucial membrane fixed regulator inhibiting membrane attack complex (MAC) formation on host cells (Figure 1B). Thus, absence of CD59 predisposes to TP-mediated cell damage. However, with all proximal convertase-directed complement regulators still in place, the question arises how complement is dysregulated in the proximal pathways to arrive at C5 activation. Possible explanations include complement-amplifying conditions like infections or trauma or a hypothetical indirect role for CD59 in modulating CR1 function, because both regulators have been found to colocalize on ectosomes.51 Thrombotic events next to a pronounced neurologic phenotype were also described for patients with inherited PIG-M deficiency, resulting in global lack of GPI anchors.52,53
Studies analyzing the coincidence of cytolysis and thrombotic events in CD59-deficient patients
| First author/reference | Number of patients | Clinic | Thromboembolic events | Comment |
|---|---|---|---|---|
| Yamashina46,154 | 1 | Intravascular hemolytic anemia | TE was communicated to have occurred | Untypical presentation in comparison with other patients with CD59 because of lack of polyneuropathy as symptom |
| Nevo,47 Ben-Zeev,48 Mevorach49 | 5 (plus 2 deceased historic cases) | Intravascular hemolytic anemia and polyneuropathy | Two of 7 had fatal cerebrovascular events | Four patients treated with eculizumab |
| Höchsmann50 | 1 | Intravascular hemolytic anemia and polyneuropathy | TE documented | EEculizumab herapy initiated with overall improvement of the patient; within 78 mo after onset of anticomplement therapy no further TE* |
| Haliloglu155 | 3 | Intravascular hemolytic anemia and polyneuropathy | Three of 3 had cerebrovascular events (1 fatal) | EEculizumab herapy initiated with overall improvement of the patient |
| Klemann156 | 1 | Intravascular hemolytic anemia and polyneuropathy | Fatal recurrent cerebrovascular events |
| First author/reference | Number of patients | Clinic | Thromboembolic events | Comment |
|---|---|---|---|---|
| Yamashina46,154 | 1 | Intravascular hemolytic anemia | TE was communicated to have occurred | Untypical presentation in comparison with other patients with CD59 because of lack of polyneuropathy as symptom |
| Nevo,47 Ben-Zeev,48 Mevorach49 | 5 (plus 2 deceased historic cases) | Intravascular hemolytic anemia and polyneuropathy | Two of 7 had fatal cerebrovascular events | Four patients treated with eculizumab |
| Höchsmann50 | 1 | Intravascular hemolytic anemia and polyneuropathy | TE documented | EEculizumab herapy initiated with overall improvement of the patient; within 78 mo after onset of anticomplement therapy no further TE* |
| Haliloglu155 | 3 | Intravascular hemolytic anemia and polyneuropathy | Three of 3 had cerebrovascular events (1 fatal) | EEculizumab herapy initiated with overall improvement of the patient |
| Klemann156 | 1 | Intravascular hemolytic anemia and polyneuropathy | Fatal recurrent cerebrovascular events |
Disease: isolated germline encoded homozygous CD59 deficiency (prevalence: too low to calculate). TE, thrombotic event.
From then onward, no further information available (B. Höchsmann, Institute of Transfusion Medicine, University of Ulm, personal communication via email, 12 February 2021).
Complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy (CHAPLE) is caused by CD55 deficiency and predominantly is a gut condition characterized by abdominal pain and diarrhea, primary intestinal lymphangiectasia, hypoproteinemic edema, and malabsorption.54,55 This leads to bowel inflammation and hypogammaglobulinemia, followed by recurrent respiratory infections. Frequently, severe, often fatal, angiopathic thromboembolic events occur (Table 4). Not all people with CD55 deficiency develop CHAPLE.30,56 Clinically significant in vivo hemolysis has not been reported in individuals with the Inab phenotype (ie, CD55 deficiency), although erythrocytes lack the convertase directed regulator CD55.57-59 However, under laboratory conditions, erythrocytes of the Inab phenotype were demonstrated to be slightly more susceptible to complement mediated lysis.59,60 However, 1 case with an acquired transient Inab phenotype was reported, characterized by intravascular hemolysis and thromboembolic disease (splenic infarctions).30 Substantial complement-mediated hemolysis appears unlikely to be the driver of thromboembolism in CHAPLE, although it cannot be completely excluded that during strong complement amplifying conditions (eg, infection or surgery), some hemolysis may occur. A prothrombotic status in patients with CHAPLE may also be induced by enteric loss of anticoagulation factors and hypoalbuminemia.54 However, several lines of evidence show that complement TP activation and cytolyses occur (independently of hemolysis) in patients with CHAPLE: (1) duodenal biopsy specimens revealed MAC complexes in submucosal arterioles61; (2) elevated C5a and sC5b-9 levels were detected in plasma55; and (3) C3 fragments and MAC were detected on peripheral blood leukocytes.54,62 Supported by these biomarkers, treatment with eeculizumab esulted in fast and sustained improvement of symptoms. Thrombotic events did not reoccur during eeculizumab reatment in 1 study.54 In another study, the proportion of patients with thromboembolism was reduced alongside an overall reduction of D-dimers averaged across all patients, but thromboembolic disease was not corrected for all.55
Studies on thrombotic events in CD55-deficient patients
| First author/reference | Number of patients | Clinical presentation | Thromboembolic events | Rate of TE under complement inhibition by eculizumab | Comment |
|---|---|---|---|---|---|
| Matthes30 | 1 | Intravascular hemolytic anemia Abdominal pain | Multiple Splenic infarctions | NA |
|
| Kurolap54,62 | 6 | Chaple No hemolysis, but elevated MAC deposition on white blood cells | Three of 6 with repeated TEs | Only 3 patients treated, but within 18 mo of follow-up, hyper-coagulopathy events did not reoccur after treatment initiation |
|
| Ozen61 | 11 (plus 2 deceased historic cases) | Chaple No hemolysis, but elevated MAC deposition on submucosal arterioles | Five of 13 with multiple thrombi at different sites; in total 3 fatalities caused by thrombosis | NA |
|
| Hagin158 | 1 | Chaple Evidence of hemolysis | Repeated mesenteric vein thrombosis | NA | Eculizumab herapy improved abdominal symptoms, increased hemoglobin levels; LDH levels did not normalize completely; months after start of Ecu a malignancy was diagnosed and Ecu stopped; patient died of sepsis coinciding with antineoplastic therapy |
| Ozen55 | 16 | Chaple No hemolysis, but elevated MAC deposition on submucosal arterioles | Six of 16 (38%) with thrombotic complications | Four of 16 (25%) with thrombotic complications | Eculizumab herapy
|
| First author/reference | Number of patients | Clinical presentation | Thromboembolic events | Rate of TE under complement inhibition by eculizumab | Comment |
|---|---|---|---|---|---|
| Matthes30 | 1 | Intravascular hemolytic anemia Abdominal pain | Multiple Splenic infarctions | NA |
|
| Kurolap54,62 | 6 | Chaple No hemolysis, but elevated MAC deposition on white blood cells | Three of 6 with repeated TEs | Only 3 patients treated, but within 18 mo of follow-up, hyper-coagulopathy events did not reoccur after treatment initiation |
|
| Ozen61 | 11 (plus 2 deceased historic cases) | Chaple No hemolysis, but elevated MAC deposition on submucosal arterioles | Five of 13 with multiple thrombi at different sites; in total 3 fatalities caused by thrombosis | NA |
|
| Hagin158 | 1 | Chaple Evidence of hemolysis | Repeated mesenteric vein thrombosis | NA | Eculizumab herapy improved abdominal symptoms, increased hemoglobin levels; LDH levels did not normalize completely; months after start of Ecu a malignancy was diagnosed and Ecu stopped; patient died of sepsis coinciding with antineoplastic therapy |
| Ozen55 | 16 | Chaple No hemolysis, but elevated MAC deposition on submucosal arterioles | Six of 16 (38%) with thrombotic complications | Four of 16 (25%) with thrombotic complications | Eculizumab herapy
|
Disease: isolated germline encoded homozygous CD55 deficiency (prevalence: too low to calculate). NA, not applicable; TE, thrombotic events.
Atypical hemolytic uremic syndrome
In contrast to the more common typical hemolytic uremic syndrome (HUS) triggered by bacterial infections, the rare atypical form (approximately 10% of cases) is associated with heterozygous germline mutations mostly affecting genes of the complement cascade (Figure 2B).63,64 Hallmarks of atypical HUS (aHUS) include nonimmune microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury often characterized by glomerular thrombotic microangiopathy. Although predominantly a renal disease, patients with aHUS can also present with multiorgan involvement including seizures, strokes, and gastrointestinal manifestations, among others. Before anti-C5 therapy, end-stage renal disease developed in about half of the patients within 5 years after onset. Thrombotic microangiopathy (TMA) develops in aHUS mostly through AP dysregulation on endothelial cell surfaces. Fluid phase AP regulation is not greatly perturbed. These circumstances lead to MAC deposition and damage on the endothelium. Plasma complement levels (eg, C3, C5a, SC5b-9) are only substantially affected in half of the patients, making their diagnostic analysis impracticable for diagnosis.65,66 An elegant murine aHUS study identified the C5a pathway to be instrumental for macrovascular thrombosis and chronic inflammation, whereas the C5b-9 pathway drives the especially fatal renal thrombotic microangiopathy.67 With both C5 activation products being implicated in fueling the thrombotic phenotype, it is not surprising that C5 inhibition is a very effective treatment of aHUS for stopping thrombotic complications (Table 2).
C3 glomerulopathy
C3 glomerulopathy (C3G) is a rare renal disease driven by dysregulation of the complement AP, ultimately inducing glomerulonephritis featuring proteinuria, hematuria, and hypertension.68 C3G is an umbrella term for the 2 slightly different subgroups of C3G (ie, dense deposit disease and C3 glomerulonephritis).69 In both subtypes, chronic AP activation fosters progressive glomerular inflammation leading to irreversible tissue damage and end-stage renal disease. In contrast to aHUS where AP dysregulation progresses mainly at the surfaces of the glomerular microenvironment (ie, glomerular endothelial cells, endothelial glycocalyx, and extracellular matrix components), in C3G, AP dysregulation sets off in the fluid phase and secondarily precipitates into the glomerular microenvironment. Several acquired or genetic factors either overdrive the C3 activation cycle or choke off its regulatory counterpart (Figure 1B). Causes for the former frequently include autoantibodies against complement convertases or gain-of-function mutations in convertase components unnaturally stabilizing these short-lived enzymatic complexes. Impairment of AP regulation results from loss-of-function mutations in the crucial regulators factor I and factor H (FH), autoantibodies against these regulators or from genetic rearrangements that produce unnatural FH-related fusion proteins affecting AP control by FH (Figure 2C).68 The molecular pathophysiology of FH exemplifies that AP dysregulation in aHUS is skewed toward impairment of regulation at the surface, whereas in C3G, fluid phase control is affected. In aHUS, associated autoantibodies and mutations affecting FH map mostly to the C-terminal FH domains, which are crucial for positioning the soluble plasma protein FH on host surfaces.70-75 In contrast, in patients with C3G, mutations and autoantibodies often affect the N-terminal FH domains harboring the AP regulatory functions and thus substantially impair fluid phase AP regulation next to control at host surfaces.76-78
In agreement with major AP dysregulation in the fluid phase in C3G, C3 consumption in plasma and prominent C3 deposition in glomeruli (in absence of immunoglobulin deposits) are observed commonly cumulating into TP activation.79,80 Sethi et al79 found that all analyzed glomeruli from patients with dense deposit disease contained TP proteins in addition to C3. The TP component C9 was prominently detected and, importantly, coincided with the detection of the soluble TP inhibitors clusterin and vitronectin (ie, S protein). These 2 regulators are plasma proteins and bind to assembling membrane attack complexes (C5b-7, C5b-8, C5b-9) inhibiting the formation of a functional, lipid-bilayer penetrating pore by blocking C9 polymerization. Bound by clusterin, vitronectin, or both inhibitors simultaneously, the hydrophobic patches in C5b-9 are shielded, and thus membrane insertion, but also protein aggregation in plasma, is prevented.81,82 This results in soluble terminal complement complexes or SC5b-9 (which stand for S protein solubilized C5b-9), which cannot inflict cytolysis. These findings are congruent to a diagnostic test in which sera from patients with aHUS but not from patients with C3G lead to MAC insertion on cultured endothelial cells.66
Taken together, fluid-phase dysregulation of the AP in C3G leads to substantial fluid phase C3 consumption and C3 deposition in the glomerular microenvironment, likely involving the glycocalyx and the glomerular basement membrane, which lack lipid bilayer–fixed complement regulators. Speculatively, this may provide the microenvironment where C5 activation occurs and where forming C5b-9 complexes are quenched by the soluble regulators clusterin and vitronectin before substantial amounts of C5b-9 precipitate to the endothelium.
C5 activation seems to contribute to glomerular inflammation in C3G, but the most prominent pathophysiologic driver is fluid phase C3 activation causing C3 deposition in the glomerular microenvironment and eventually inducing morphologic changes to basement membrane proteins. To what exact extent the anaphylatoxin and chemoattractant C5a and the cytolytic-inactive SC5b-9 complexes contribute to the glomerular inflammation in human C3G remains unclear thus far. Two murine studies clearly indicated a more prominent role for C5a rather than for terminal complement complexes or MAC: (1) absence of C5, but not C6, alleviated glomerular pathology in a C3G model indicating C5a as driver of disease83; and (2) C5a in absence of C5b-9 drove chronic inflammatory injury resulting in glomerular disease reminiscent of C3 glomerulonephritis while presence of the C5b-9 pathway caused renal TMA reminiscent of aHUS.67 This argues that MAC-inflicted cytolysis and not C5a liberation is the major driver of thrombosis in these animal models. In a therapeutic approach in the former murine C3G model, C5 activity was blocked,83 ameliorating the disease severity in mice but failing to prevent C3 glomerulopathy. Overall, these murine studies align with clinical findings. Although no disease-specific treatment options are available yet, several case studies involving a few patients with C3G investigated the therapeutic potential of C5 inhibition (off-label) with mixed results. Although some patients benefited from anti-C5 therapy to various degrees, a huge proportion had no clinical response.68
Taken together, preclinical and initial clinical data suggest that C3 deposition in the glomerular microenvironment is the driver of C3G pathophysiology with contributions from C5a liberation, whereas soluble and noncytolytic SC5b-9 complexes probably do not contribute substantially.
The comparison of the disease phenotypes across the 3 AP-mediated diseases PNH, aHUS, and C3G provides for a compelling difference. Although AP dysregulation leading to C5 activation is a common pathophysiologic feature early in the course of all 3 diseases, only in PNH and aHUS does complement activation cause thrombotic events associated with severe morbidity and mortality, whereas thrombotic complications in C3G are not a typical feature.
Autoimmune hemolytic anemia
Autoimmune hemolytic anemia (AIHA) is a rare disease characterized by autoantibodies against erythrocytes with diverse clinical/serologic presentations.84,85 AIHA is idiopathic in about half of cases or secondary to conditions such as lymphoproliferative diseases, autoimmune disorders (often systemic lupus erythematosus [SLE]), (viral) infections, or nonlymphoid neoplasms.86,87 Depending on the autoantibody, which often are of polyclonal origin,84 AIHA is classified by warm (wAIHA) or cold (called: cold agglutinins disease [CAD]) antibodies according to their thermal amplitude or into a mixed type if cold (mostly immunoglobulin M [IgM]) and warm (mostly IgG) are present simultaneously. Although complement clearly drives the pathophysiology of CAD, its involvement in wAIHA is less well understood. The direct antiglobulin test (DAT; or Coombs test) for wAIHA is regularly positive detecting either autoantibodies alone or together with the complement activation fragment C3dg/C3d (Figure 1A). In CAD, often only C3d deposits are detected. Such antibodies bind only in the cold and lead to classical pathway (CP) activation inducing C3 deposition. Then cold-type antibodies detach when affected erythrocytes transit from the extremities back to systemic circulation, but C3d remains covalently fixed on the erythrocyte. Atypical AIHA characteristics are as follows: DAT negativity, DAT positivity for IgA only, warm IgM, or mitogen-stimulated DAT.86
Both intravascular and/or extravascular hemolysis occur in AIHA depending on if and to what extent autoantibodies can activate the CP, which may be tested in a human system in vitro as described by Anliker et al.88 Cells of the reticuloendothelial system recognize IgG and C3 activation products fixed on erythrocytes and clear those in a comparatively slow process.89 Potent CP activation, however, pushes the C3 activation cycle into a strong overdrive and overwhelms the preformed erythroid complement regulators leading to dense C3b deposition and TP-mediated intravascular hemolysis (Figures 1A-B and 2D).
Thrombotic events are evident in patients with AIHA86,90 and correlate with splenectomy91 and antiphospholipid antibodies (aPL-abs; such as lupus anticoagulants and anticardiolipin autoantibodies).29,85,92 Patients with CAD have an increased risk of thromboembolisms compared with a matched non-CAD population.93 Thrombotic complications in patients with AIHA also occur in absence of aPL-abs or splenectomy, underlining that AIHA per se is a risk factor for thromboembolism (Table 5).85-87,94-96 Strikingly, thromboembolism is associated with severe active intravascular hemolysis in these patients, also leading to fatal outcomes.86 Multiple organs are affected by thrombi including venous and arterial sites.86,96 In a first prospective trial, eeculizumab as administered to 13 patients with CAD and reduced hemolysis, transfusion dependency, and the median D-dimer concertation in most patients.97 No thromboembolic events occurred during the therapy period of 26 weeks, whereas thromboembolism had occurred in several patients within months before enrollment. This indicates that blocking of the TP may be sufficient to stop thromboembolic events in CAD, although the proximal complement cascade continues to run unaltered. Firm conclusions, however, necessitate trials with longer duration irrespective of C5, C1s, or other complement inhibitors being investigated.97-99
Selected studies analyzing the coincidence of cytolysis and thrombotic events in patients with AIHA
| First author/reference | AIHA type | Rate of TE under best supportive therapy (number of patients) | Signs for cytolysis during thromboembolic events | Comment |
|---|---|---|---|---|
| Pullarkat29 | Not defined | 27% (8 of 30) | TE associated with hemolysis (in 7 of 8) | Of 8 patients with TE
|
| Hendrick95 | w 80%, c 10%, m 10% | 21% (6 of 28) | 100% with signs of intravascular hemolysis (i.e, bilirubin, haptoglobin) | Of 6 patients with TE
|
| Baek94 | w 97%, c 3% | 16% (5 of 32) | >90% of patients with TE had intravascular hemolysis (LDH, bilirubin) | Most patients with AIHA are found to have SLE during follow-up |
| Barcellini86 | w 60%, c 27%, m 8%, a 5% | 11% (33 of 308) | TE associated with intravascular hemolysis (LDH) | Of 33 patients with TE
|
| Roumier96 | w 54%, m 46% | 20% (12 of 60) | TE occurred with active hemolysis in all (haptoglobin/LDH/bilirubin) | Of 12 patients with TE
|
| Lecouffe-Desprets85 | w 100% (inclusion criterium) | 20% (8 of 40) | TE associated with severe hemolytic flare’ | Of 8 patients with TE
|
| Prabhu87 | w 48%, c 46%, a 6% | 6% with life-threating TE (2 of 33) | NS | Of 2 patients with TE
|
| First author/reference | AIHA type | Rate of TE under best supportive therapy (number of patients) | Signs for cytolysis during thromboembolic events | Comment |
|---|---|---|---|---|
| Pullarkat29 | Not defined | 27% (8 of 30) | TE associated with hemolysis (in 7 of 8) | Of 8 patients with TE
|
| Hendrick95 | w 80%, c 10%, m 10% | 21% (6 of 28) | 100% with signs of intravascular hemolysis (i.e, bilirubin, haptoglobin) | Of 6 patients with TE
|
| Baek94 | w 97%, c 3% | 16% (5 of 32) | >90% of patients with TE had intravascular hemolysis (LDH, bilirubin) | Most patients with AIHA are found to have SLE during follow-up |
| Barcellini86 | w 60%, c 27%, m 8%, a 5% | 11% (33 of 308) | TE associated with intravascular hemolysis (LDH) | Of 33 patients with TE
|
| Roumier96 | w 54%, m 46% | 20% (12 of 60) | TE occurred with active hemolysis in all (haptoglobin/LDH/bilirubin) | Of 12 patients with TE
|
| Lecouffe-Desprets85 | w 100% (inclusion criterium) | 20% (8 of 40) | TE associated with severe hemolytic flare’ | Of 8 patients with TE
|
| Prabhu87 | w 48%, c 46%, a 6% | 6% with life-threating TE (2 of 33) | NS | Of 2 patients with TE
|
Prevalence: 170 per 1 million.159 a, atypical; c, cold; m, mixed; NS, not specified; TE, thrombotic events; w, warm.
(Catastrophic) antiphospholipid syndrome
Antiphospholipid syndrome (APS) is a systemic autoimmune disease characterized by arterial and/or venous thrombosis or pregnancy morbidity in the presence of persistent aPL-abs (ie, anticardiolipin, anti–β-2 glycoprotein I, or lupus anticoagulant).100-103 There can be an overlap between APS and the HELLP (ie, hemolysis, elevated liver enzymes and low platelets) syndrome,104,105 which recently was also linked to complement dysregulation (but is not covered here).106 Thrombotic complications in APS are frequently resistant to anticoagulation.107 In most cases, APS is either idiopathic or develops on the background of SLE. In about one third of patients with catastrophic APS (CAPS), there is evidence for hemolysis.101 About 1% of patients with APS develop the often fatal (>30% mortality despites therapy101) CAPS, affecting multiple organs simultaneously within a short time frame.108-110 aPL-abs bind (directly or indirectly) to phospholipid surfaces inducing multifactorial mechanisms (eg, aPL-abs were described to interfere with natural anti- and procoagulant regulators and/or lead to direct prothrombotic activation of endothelial cells, platelets, monocytes, and neutrophils).111 Also, complement activation has a well-documented role in the pathogenesis of aPL-abs inducing thrombosis and/or pregnancy morbidity (key findings are summarized in Table 6). The precise mechanism how aPL-abs induce complement activation remains incompletely understood, but blocking this interaction, which potentially is upstream of prothrombotic cell activation, is an attractive avenue. Clinical observations and results from animal models indicate that in addition to aPL-abs, a second hit in the form of a complement-amplifying condition (like pregnancy, surgery, or infection) is necessary to trigger thrombotic complications.112,113 Supported by many animal studies, patient-derived diagnostic data indicate TP in APS/CAPS pathophysiology (Table 6). EEculizumab as successfully used as salvage therapy in several life-threating refractory cases.114,115 This provides circumstantial clinical evidence for the TP- and MAC-mediated cell damage propagating morbidity in APS/CAPS. However, the experimental nature of anti-C5 therapy needs to be acknowledged. Yelnik et al116 reported a small cohort of patients with CAPS responding inconsistently to eculizumab, with responders typically suffering from lower platelet counts and more frequent microangiopathic hemolytic anemia than nonresponders. To conclude, complement and especially TP activation are playing a critical role in obstetric or/and thrombotic APS, at least in a substantial subset of patients. Further clinical investigations on anticomplement therapy are warranted.
Selected studies describing complement activation in the pathogenesis of aPL-induced pregnancy morbidity or thrombosis
| First author/reference | Experimental/clinical setting | Important findings | Comment |
|---|---|---|---|
| Davis160 | Analysis of complement activation levels in stroke patients with and without aPL-abs | C5b-9 levels were higher in sera of patients with aPL and stroke when compared with stroke in absence of aPL-abs | TP activation seems to be implicated in the pathophysiology of aPL-ab–associated stroke |
| Holers161 |
| C3 activation is required for aPL-ab induced intrauterine growth restriction and/or fetal loss in mice | C3 KO mice or mice treated with complement inhibitor Crry are protected from fetal loss |
| Girardi162 |
| Protected from fetal loss by
|
|
| Girardi163 | Murine model of aPL-ab induced fetal loss treated with anticoagulants | Heparin but not other anticoagulants (like fonda-parinux or hirudin) prevents aPL-ab–induced fetal loss | unfractionated or low-molecular-weight heparin prevented complement activation in vitro and in vivo while other anticoagulants failed to do so |
| Fischetti164 |
| Thrombus formation induced by antibodies to β2-glycoprotein I requires complement TP activation and a priming factor |
|
| Carrera-Marín165 |
| C6 knock-out mice are protected from thrombophilia mediated by aPL-abs | Indicates MAC (and not C5a) as a main driver of pathophysiology in addition to direct cell activating effects of aPL-abs |
| Agostinis166 |
| An engineered antibody that efficiently binds PL but cannot activate complement fails to induce vascular thrombosis and fetal loss (even after LPS priming) |
|
| Meroni167 | Patient with recurrent thrombotic complications (despite anticoagulation) was treated with Ecu to prevent rethrombosis after vascular surgery | First report demonstrating colocalization of aPL-ab and complement (C1q; C4, C3 and C5b-9 or MAC) in the arterial wall of an APS patient | Suggests activation of complement classical pathway and involvement of MAC in pathophysiology |
| Arachchillage168 | Complement activation markers were compared in APS patients treated with anticoagulants | Complement activation markers were elevated in warfarin anticoagulated thrombotic APS patients, but decreased when switching to rivaroxaban | sC5b-9 plasma levels decreased when warfarin was substituted with rivaroxaban indicating that FXa may activate complement proteins fueling the cross-activation of inflammatory and prothrombotic pathways |
| Rand169 | Novel 2-stage assay that detects the complement activation potential in patients with aPL-abs | Highly sensitive assay system using patient plasma to distinguish APS from other inflammatory and thrombotic disorders that are not associated with aPL-abs |
|
| Kim170 | Complement activation markers were determined in pregnant patients with SLE and/or antiphospholipid antibodies | Complement activation levels predict adverse pregnancy outcome | sC5b-9 plasma levels detectable early in pregnancy are strongly predictive of adverse pregnancy outcomes |
| Tinit114 Skoczynska115 Nauseel171 | Each: a case report of CAPS and optional systematic review of the literature for other case reports | TP inhibition by Ecu can be a salvage therapy for CAPS refractory to conventional therapy |
|
| Chaturvedi172 | Sera and genetic information were investigated for the propensity to activate complement in patients suffering from APS, CAPS, and SLE |
| Anti–β2-GP-I antibodies trigger
|
| First author/reference | Experimental/clinical setting | Important findings | Comment |
|---|---|---|---|
| Davis160 | Analysis of complement activation levels in stroke patients with and without aPL-abs | C5b-9 levels were higher in sera of patients with aPL and stroke when compared with stroke in absence of aPL-abs | TP activation seems to be implicated in the pathophysiology of aPL-ab–associated stroke |
| Holers161 |
| C3 activation is required for aPL-ab induced intrauterine growth restriction and/or fetal loss in mice | C3 KO mice or mice treated with complement inhibitor Crry are protected from fetal loss |
| Girardi162 |
| Protected from fetal loss by
|
|
| Girardi163 | Murine model of aPL-ab induced fetal loss treated with anticoagulants | Heparin but not other anticoagulants (like fonda-parinux or hirudin) prevents aPL-ab–induced fetal loss | unfractionated or low-molecular-weight heparin prevented complement activation in vitro and in vivo while other anticoagulants failed to do so |
| Fischetti164 |
| Thrombus formation induced by antibodies to β2-glycoprotein I requires complement TP activation and a priming factor |
|
| Carrera-Marín165 |
| C6 knock-out mice are protected from thrombophilia mediated by aPL-abs | Indicates MAC (and not C5a) as a main driver of pathophysiology in addition to direct cell activating effects of aPL-abs |
| Agostinis166 |
| An engineered antibody that efficiently binds PL but cannot activate complement fails to induce vascular thrombosis and fetal loss (even after LPS priming) |
|
| Meroni167 | Patient with recurrent thrombotic complications (despite anticoagulation) was treated with Ecu to prevent rethrombosis after vascular surgery | First report demonstrating colocalization of aPL-ab and complement (C1q; C4, C3 and C5b-9 or MAC) in the arterial wall of an APS patient | Suggests activation of complement classical pathway and involvement of MAC in pathophysiology |
| Arachchillage168 | Complement activation markers were compared in APS patients treated with anticoagulants | Complement activation markers were elevated in warfarin anticoagulated thrombotic APS patients, but decreased when switching to rivaroxaban | sC5b-9 plasma levels decreased when warfarin was substituted with rivaroxaban indicating that FXa may activate complement proteins fueling the cross-activation of inflammatory and prothrombotic pathways |
| Rand169 | Novel 2-stage assay that detects the complement activation potential in patients with aPL-abs | Highly sensitive assay system using patient plasma to distinguish APS from other inflammatory and thrombotic disorders that are not associated with aPL-abs |
|
| Kim170 | Complement activation markers were determined in pregnant patients with SLE and/or antiphospholipid antibodies | Complement activation levels predict adverse pregnancy outcome | sC5b-9 plasma levels detectable early in pregnancy are strongly predictive of adverse pregnancy outcomes |
| Tinit114 Skoczynska115 Nauseel171 | Each: a case report of CAPS and optional systematic review of the literature for other case reports | TP inhibition by Ecu can be a salvage therapy for CAPS refractory to conventional therapy |
|
| Chaturvedi172 | Sera and genetic information were investigated for the propensity to activate complement in patients suffering from APS, CAPS, and SLE |
| Anti–β2-GP-I antibodies trigger
|
Disease: APS/CAPS (prevalence APS: 500 per 1 million173; CAPS about 1% of APS).
Ecu, eculizumab.
Discussion
Basic and clinical evidence demonstrate that exuberant complement activation can lead to severe thromboembolic complications. Almost all mechanistic studies implicate TP activation in promoting a prothrombotic phenotype (Table 1). Clinical findings also demonstrate an instrumental role for TP in complement-mediated thromboembolism. In PNH, isolated CD59 deficiency and AIHA thrombotic complications associate with complement-mediated hemolysis. C5 inhibition by eeculizumab educes erythrocyte lysis and thrombotic events simultaneously (of note, definitive clinical data are not available for AIHA and CD59 deficency; Tables 2, 3, and 5). Thrombotic events in patients with PNH are drastically reduced but not completely absent during anti-C5 therapy (Table 2). This could imply a role for C3 activation products in inducing prothrombotic cell activation, because during therapeutic C5 block, C3 activation proceeds unhindered in patients with PNH.117-119 An alternative explanation is that C5 inhibition is not complete in some patients with PNH, which has already been shown for various C5 inhibitors.120-123 In PNH, this is clinically documented by residual but chronic C5 activation and sporadic but severe hemolytic events during complement-amplifying conditions like infections.124-126 Severe hemolytic events in presence of excess amounts of C5 inhibitors are explained by a pharmacodynamic breakthrough mechanism.122,124,127
Other diseases discussed here either do not exhibit hemolysis as a cardinal feature (ie, CHAPLE and APS/CAPS101), or hemolysis is caused by mechanical stress (aHUS) rather than by MAC assembly. However, thrombotic events still associated with TP activation are corrected or mitigated by therapeutic inhibition of C5 activation. In CHAPLE, elevated MAC deposition on white blood cells and submucosal arterioles was demonstrated, and circumstantial clinical evidence suggests benefits by C5 inhibition including the rate of thromboembolic events (Table 4). In APS/CAPS, there is strong preclinical evidence linking TP activation to vascular thrombosis, particularly indicating MAC in the pathophysiology. This is supported by successful eeculizumab reatment (as salvage therapy) for patients with refractory CAPS (Table 6). However, it has been shown that not all refractory patients with CAPS benefit from C5 inhibition, calling for further and more detailed clinical investigations.116 In aHUS, complement dysregulation mainly affects the glomerular capillaries leading to MAC deposition and TMA. Next to PNH, there is also definitive clinical evidence that C5 activation and cytolysis are critical drivers of the pathophysiology in aHUS. Inhibition of C5 prevents the fatal thrombotic events in aHUS, despite proximal complement not being inhibited (Table 2).
Comparison of the 3 AP-mediated diseases PNH, aHUS, and C3G illustrates that TP activation per se is not sufficient to induce prothrombotic cell activation. In patients with C3G, TP activation is evident but appears not to result in the formation of cytolytic MAC complexes. Instead C5b-9 complexes detected in the glomerular microenvironment were reported to be complexed by vitronectin, rendering the resultant SC5b-9 complexes inactive for membrane penetration. This situation is reminiscent of the molecular events induced by cobra venom factor (CVF) treatment. In animal models, CVF is used to deplete animals of complement activity. Depending on the type of CVF involved, consumptive activation of C3 and/or C5 proceeds in the fluid phase, without ill effect (including thrombotic complications) on the organism. Safe complement depletion by CVF has been achieved for sustained duration up to 30 days and includes tests in several mammals such as mice, rats, and nonhuman primates.128,129 The unifying features of complement-induced thromboembolism, among diseases discussed here, are TP activation and MAC-inflicted damage of plasma membranes involving the endothelium and/or cells within the vasculature. Such cell surface–directed activation, in contrast to fluid phase complement dysregulation, does not regularly lead to exhaustive consumption/activation of complement components. Thus, the global plasma concentrations of nonactivated components (eg, C3, C5) or activation products (eg, C3a, C5a, SC5b.9) are often normal or only slightly altered, rendering those plasma markers useless for diagnostic purposes because they fail to securely report whether devastating but locally confined complement activation occurs on cell surfaces in a patient. This point is emphasized by the fact that some diseases discussed here require a second trigger (ie, a complement-amplifying condition such as during infections, vaccination, pregnancy, and surgery) in addition to the underlying complement dysregulation before severe complement damage is inflicted. Recognized by different groups, sensitive assays were developed to assess whether patient plasma/serum can inflict severe surface-directed complement damage (Table 7): a strategy that may provide an additional rational for starting anticomplement therapy.
Studies describing assay systems that can detect cellular/surface directed complement activation
| First author/reference | Setup of test | Main finding | Other interesting findings | Comment |
|---|---|---|---|---|
| Noris66 | Cultured and stimulated HMEC-1 are exposed to patient sera; C5b-9 (or MAC) deposition is detected |
|
| These assays are not expected to detect aHUS associated with alterations in DGKE, MCP or THBD genes or function |
| Gavriilaki174 | Engineered PIG-A-null reagent cell line is exposed to patient sera and viability (or insertion of C5b-9) is detected |
|
| These assays are not expected to detect aHUS associated with alterations in DGKE, MCP or THBD genes or function |
| Rand170 | Phospholipid vesicles pre-incubated with APS plasmas (1st step) are analyzed for C5b-9 insertion when exposed to NHS (2nd step). | Distinguishes APS from other inflammatory and thrombotic disorders not associated with aPL-abs | Plasma levels of C5a and sC5b-9 were higher in APS patients but not significantly increased when compared with healthy controls | Artificially produced PL vesicles appear especially suitable and are easily available |
| Anliker88 | RBCs of patients with a PNH III RBC clone >20% exposed to plasma with allo- or autoantibodies (first step) and analyzed for MAC mediated hemolysis in NHS (second step) |
| The C5 inhibition abrogated mild but not strong complement TP activation in vitro. However, residual TP activity could be further depressed by combining C5 inhibitors with proximal C inhibitors | Limiting applicability: ABO matched reagents (or RBCs with ABO ‘O’) are needed; PNH RBCs can be frozen, but supply of PNH RBCs is sparse |
| First author/reference | Setup of test | Main finding | Other interesting findings | Comment |
|---|---|---|---|---|
| Noris66 | Cultured and stimulated HMEC-1 are exposed to patient sera; C5b-9 (or MAC) deposition is detected |
|
| These assays are not expected to detect aHUS associated with alterations in DGKE, MCP or THBD genes or function |
| Gavriilaki174 | Engineered PIG-A-null reagent cell line is exposed to patient sera and viability (or insertion of C5b-9) is detected |
|
| These assays are not expected to detect aHUS associated with alterations in DGKE, MCP or THBD genes or function |
| Rand170 | Phospholipid vesicles pre-incubated with APS plasmas (1st step) are analyzed for C5b-9 insertion when exposed to NHS (2nd step). | Distinguishes APS from other inflammatory and thrombotic disorders not associated with aPL-abs | Plasma levels of C5a and sC5b-9 were higher in APS patients but not significantly increased when compared with healthy controls | Artificially produced PL vesicles appear especially suitable and are easily available |
| Anliker88 | RBCs of patients with a PNH III RBC clone >20% exposed to plasma with allo- or autoantibodies (first step) and analyzed for MAC mediated hemolysis in NHS (second step) |
| The C5 inhibition abrogated mild but not strong complement TP activation in vitro. However, residual TP activity could be further depressed by combining C5 inhibitors with proximal C inhibitors | Limiting applicability: ABO matched reagents (or RBCs with ABO ‘O’) are needed; PNH RBCs can be frozen, but supply of PNH RBCs is sparse |
C, complement; DGKE, diacylglycerolkinase-ɛ; HMEC-1, human microvascular endothelial cells; MCP, membrane cofactor protein or CD46; THBD, thrombomodulin; TTP, thrombotic thrombocytopenic purpura.
In aggregate, TP inhibition is a proven or a promising strategy to prevent thromboembolism driven by exuberant complement activation associated with C5 activation and cytolysis. It should be noted that one does not necessarily need to inhibit the TP to prevent it from being activated. Next to C5 inhibitors, other more proximal complement inhibitors are in clinical development99 and are expected to efficiently block C5 activation. The key role of TP and MAC in PNH, CHAPLE, CD59 deficiency, aHUS, AIHA, and APS/CAPS does not necessarily mean that proximal complement activation products do not have a role in hemostasis at all. For example, it may be possible that in thrombotic conditions driven by other means than complement activation (eg, vascular stenosis), proximal complement activation products could have an augmenting prothrombotic effect. Finally, despite the established cross talk between complement and hemostasis, the former is not strictly necessary for stopping blood loss via hemostasis. This is consistent with complement inhibition not leading to bleeding complications during clinical trials or practice.
Acknowledgment
This work was supported by Deutsche Forschungsgemeinschaft grant SCHM 3018/2-2 to C.Q.S.
Authorship
Contribution: C.Q.S., H.S., and D.K. discussed the concepts and wrote the manuscript.
Conflict-of-interest disclosure: C.Q.S. and H.S. are inventors of patent application(s) that describes the use of complement inhibitors for therapeutic applications. C.Q.S. has received research funding from Takeda Pharmaceutical. C.Q.S. and H.S. received honoraria for speaking at symposia organized by Alexion Pharmaceuticals. H.S. served on an advisory committee for and received research funding from Alexion Pharmaceuticals. H.S. served on an advisory committee for Ra Pharmaceuticals, Sanofi, Novartis, Apellis and Alnylam. D.K. is a director of and scientific advisor to Gyroscope Therapeutics. D.K. received advisory board payments from Idorsia, Novartis, ChemoCentryx, Alexion, Apellis, Biomarin, and Sarepta. D.K.’s spouse works for GSK.
Correspondence: Christoph Q. Schmidt, The Institute of Pharmacology of Natural Products and Clinical Pharmacology, Ulm University, Helmholtzstraße 20, D-89081 Ulm, Germany; e-mail: christoph.schmidt@uni-ulm.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal