

In this issue of Blood, 1 provide evidence that factor VIIa (FVIIa)–released extravascular vesicles (EVs) transfer microRNA (miR) 10a to recipient cells, producing an anti-inflammatory phenotype in monocytes and a barrier protective effect in endothelial cells. These findings suggest a novel view of the biological functions of pharmacological concentrations of FVIIa. The data presented have implications for understanding the therapeutic potential of FVIIa in treating bleeding- and inflammation-associated pathologies.
The primary function of FVIIa is to initiate the activation of the coagulation cascade by binding to tissue factor (TF) after injury.2 However, FVIIa was also found to slowly activate FX in the absence of TF, in a reaction requiring procoagulant phospholipids.3,4 This property has allowed recombinant FVIIa (rFVIIa) to become a safe and effective bypass agent to treat bleeding using inhibitors in patients with hemophilia. rFVIIa is also used to treat bleeding associated with surgery, trauma, or intracranial hemorrhage.5,6
In 2007, Ghosh et al7 reported that FVIIa also binds to endothelial cell protein C receptor (EPCR) on endothelial cells. This opened a new area of investigation to understand the relevance of the FVIIa interaction with EPCR.
In a recent study, Das et al8 identified a new role in the FVIIa-EPCR-PAR1 signaling story. They showed that FVIIa-EPCR-PAR1 signaling releases EVs from endothelial cells, and FVIIa-released EVs are enriched with phosphatidylserine on their outer leaflet. This study provides evidence that FVII-released EVs promote hemostatic effects in therapeutic settings, linking for the first time FVIIa-EPCR-PAR1 signaling with hemostasis.
The current study by Das et al provides important new information on FVIIa-released EVs. This study shows that (1) multiple signaling pathways triggered after endothelial stimulation by FVIIa, particularly the Rho/kinase-dependent pathway, contribute to the biogenesis and release of EVs from endothelial cells; (2) FVIIa-released EVs are taken up by the target cells (leukocytes and endothelial cells), probably by endocytosis, and elicit an anti-inflammatory phenotype in recipient cells; (3) FVIIa upregulates the expression of anti-inflammatory miRs, predominantly miR 10a, in endothelial cells, and FVIIa-released EVs are enriched with miR 10a; (4) delivery of miR 10a from the EV cargo recipient target cells is responsible for the anti-inflammatory and barrier protective effects; (5) delivery of miR 10a to monocytes results in anti-inflammatory effects by downregulating the expression of TAK1 and subsequent NF-κB–mediated signaling, while delivery to endothelial cells provides a barrier protective effect by blocking the downregulation of ZO-1 expression; (6) administration of FVIIa-released EVs from endothelial cells to wild mice attenuates the lipopolysaccharide-induced increase in inflammatory cytokines in plasma, leukocyte infiltration in the lungs, and vascular leakage into vital tissues; (7) as observed in cultured endothelial cells, EVs released into circulation in response to FVIIa in mice were enriched with miR 10a, and these EVs were shown to exert anti-inflammatory and barrier protective effects in ex vivo studies; (8) incorporation of anti–miR 10a into FVIIa-released EVs diminishes the ability of FVIIa-released EVs to elicit cytoprotective effects; and (9) administration of ROCK inhibitor Y27632 to mice, which inhibits FVIIa release of EVs into circulation in vivo, attenuates the cytoprotective effects of FVIIa (see figure).
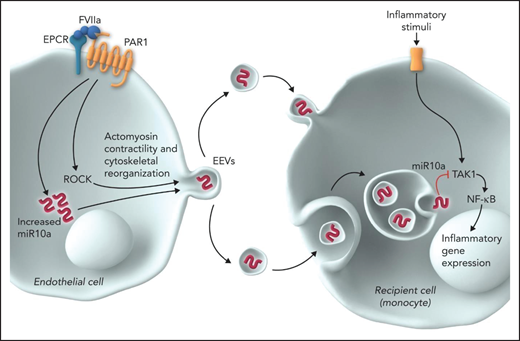
FVIIa-released EVs transmit signals from endothelial cells to monocytes. FVIIa, upon binding to EPCR on endothelial cells, activates PAR1. FVIIa-EPCR-PAR1–mediated cell signaling results in the activation of ROCK and upregulation of miR 10a expression. ROCK mediates actomyosin contractility and cytoskeletal reorganization, which results in the shedding of EVs from the plasma membrane. miR 10a is packaged into the FVIIa-released EVs. Monocytes take up FVIIa-released EVs by either membrane fusion or endocytosis, and miR 10a is delivered into the cytosol. TAK1 is the central signaling molecule activated by a vast array of inflammatory stimuli and the crucial player in the activation of NF-κB and subsequently NF-κB–mediated inflammatory gene expression. miR 10a binds to TAK1 and degrades it and thus inhibits NF-κB activation and suppresses inflammatory gene expression. Professional illustration by Somersault18:24.
FVIIa-released EVs transmit signals from endothelial cells to monocytes. FVIIa, upon binding to EPCR on endothelial cells, activates PAR1. FVIIa-EPCR-PAR1–mediated cell signaling results in the activation of ROCK and upregulation of miR 10a expression. ROCK mediates actomyosin contractility and cytoskeletal reorganization, which results in the shedding of EVs from the plasma membrane. miR 10a is packaged into the FVIIa-released EVs. Monocytes take up FVIIa-released EVs by either membrane fusion or endocytosis, and miR 10a is delivered into the cytosol. TAK1 is the central signaling molecule activated by a vast array of inflammatory stimuli and the crucial player in the activation of NF-κB and subsequently NF-κB–mediated inflammatory gene expression. miR 10a binds to TAK1 and degrades it and thus inhibits NF-κB activation and suppresses inflammatory gene expression. Professional illustration by Somersault18:24.
Earlier studies showed that activated protein C (APC) induces the release of EVs from endothelial cells in EPCR- and PAR1-dependent mechanisms and that the APC-induced EVs exert cytoprotective and anti-inflammatory effects. However, in 1 study, a large amount of APC was associated with the APC-induced EVs, and this APC was responsible for the cytoprotective effects observed.9 This scenario was ruled out with FVIIa-released EVs. An earlier study provided convincing evidence that no measurable or functionally relevant amounts of FVIIa are found in FVIIa-released EVs.8 In this report, the authors showed that FVIIai, which could compete with any traces of FVIIa that may have been associated with EVs, did not affect FVIIa-released EV protective effects. Furthermore, the anti-inflammatory and barrier protective effects of FVIIa-released EVs were shown to come from the transfer of miR 10a from EVs to recipient cells, because anti–miR 10a significantly reduced the protective effect of FVIIa-released EVs. These data suggest that although both FVIIa and APC induce EPCR-dependent PAR1-mediated cytoprotective signaling, they may be using distinct signaling mechanisms. It could be interesting to compare and contrast the cargo of FVIIa- and APC-released EVs, which could provide further insight into their mode of action.
In summary, the current study provides new insight into our understanding of how pharmacological concentrations of FVIIa induce cytoprotective effects. It is the first report to document that FVIIa communicates with various cell types through the release of EVs. Although the current study is limited to monocytes and endothelial cells, it is possible that FVIIa-released endothelial EVs could be taken up by other cell types and could induce phenotypic changes in other cell types in the body. This is important, because not all cell types carry EPCR or PAR1, and therefore, FVIIa cannot directly exert its signaling effect on these cell types. It should also be noted that the present study is only relevant for the therapeutic use of high concentrations of rFVIIa in various bleeding disorders. A detailed characterization of FVIIa-released EVs and elucidation of their role in hemostasis, anti-inflammation, and vascular barrier protection in patients treated with rFVIIa will give us a better understanding of the therapeutic potential of FVIIa. The knowledge gained from the present study may be relevant in understanding the long-term protective effects seen with prophylactic use of FVIIa in patients with hemophilia.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal