

Key Points
FVIIa induces the expression of anti-inflammatory miR10a by endothelial cells, and FVIIa-released EEVs carry miR10a in high abundance.
FVIIa-released EEVs suppress inflammation and protect barrier integrity in vitro and in vivo via the transfer of miR10a to target cells.

Abstract
Coagulation protease, factor VIIa (FVIIa), binds to endothelial cell protein C receptor (EPCR) and induces anti-inflammatory and endothelial barrier protective responses via protease-activated receptor-1 (PAR1)–mediated, biased signaling. Our recent studies had shown that the FVIIa-EPCR-PAR1 axis induces the release of extracellular vesicles (EVs) from endothelial cells. In the present study, we investigated the mechanism of FVIIa release of endothelial EVs (EEVs) and the contribution of FVIIa-released EEVs to anti-inflammatory and vascular barrier protective effects, in both in vitro and in vivo models. Multiple signaling pathways regulated FVIIa release of EVs from endothelial cells, but the ROCK-dependent pathway appeared to be a major mechanism. FVIIa-released EEVs were enriched with anti-inflammatory microRNAs (miRs), mostly miR10a. FVIIa-released EEVs were taken up readily by monocytes/macrophages and endothelial cells. The uptake of FVIIa-released EEVs by monocytes conferred anti-inflammatory phenotype to monocytes, whereas EEV uptake by endothelial cells resulted in barrier protection. In additional experiments, EEV-mediated delivery of miR10a to monocytes downregulated the expression of TAK1 and activation of the NF-κB–mediated inflammatory pathway. In in vivo experiments, administration of FVIIa-released EEVs to wild-type mice attenuated LPS-induced increased inflammatory cytokines in plasma and vascular leakage into vital tissues. The incorporation of anti-miR10a into FVIIa-released EEVs diminished the ability of FVIIa-released EEVs to confer cytoprotective effects. Administration of the ROCK inhibitor Y27632, which significantly inhibits FVIIa release of EEVs into the circulation, to mice attenuated the cytoprotective effects of FVIIa. Overall, our study revealed novel insights into how FVIIa induces cytoprotective effects and communicates with various cell types.
Introduction
Factor VIIa (FVIIa) initiates blood coagulation after vascular injury upon binding to tissue factor, a procoagulant cofactor.1 Our studies2,3 and those of others4,5 have revealed that FVIIa also binds the anticoagulant cofactor endothelial cell protein C receptor (EPCR). EPCR plays a crucial role in the protein C/activated protein C–mediated anticoagulant pathway by promoting protein C activation by the thrombin-thrombomodulin complex.6 Our recent studies established that FVIIa, similar to activated protein C,7-9 induces EPCR- and protease-activated receptor-1 (PAR1)–dependent cytoprotective signaling in endothelial cells.10-12 However, the mode of their action appears to be different.11
Our recent studies showed that FVIIa induces the release of extracellular vesicles (EVs) from endothelial cells via the EPCR-PAR1 axis.13 FVIIa-released EVs were found to exhibit prohemostatic activity.13 EVs transport complex cargo of bioactive materials, including lipids, proteins, messenger RNA (mRNA), and microRNAs (miRs); therefore, they could play important roles in intercellular communication.14 Most of the EVs detected in the blood of healthy subjects are derived from platelets and red blood cells, and only a small fraction of EVs are from endothelial cells.15-18 Various pathological conditions, including coronary syndrome,19,20 antiphospholipid syndrome,21 and sickle cell disease,22 increase the release of endothelial cell–derived EVs (EEVs) into the circulation. In many cases, EEVs have been found to be detrimental, as they induce inflammation and enhanced endothelial dysfunction.23-26 However, EEVs have also been shown to mediate cytoprotective effects and repair functions.27,28 EEVs, based on the state of endothelial cells from which they originate, have been shown either to activate monocytes and induce inflammation or to suppress monocyte activation and confer an anti-inflammatory phenotype.26,29 As FVIIa is used clinically to treat bleeding disorders in patients with hemophilia with inhibitors and other bleeding disorders,30,31 it is important to determine whether FVIIa-released EEVs exhibit cytoprotective or detrimental effects.
The data presented herein show that FVIIa-released EEVs carry anti-inflammatory miRs, mostly miR10a. The transfer of miR10a from FVIIa-released EEVs to target monocytes and endothelial cells suppressed LPS-induced inflammation and endothelial permeability, in both in vitro and in vivo models. Inhibiting miR10a with anti-miR10a attenuated the ability of FVIIa-released EEVs to confer cytoprotective effects. Our experiments also showed that administration of the ROCK inhibitor Y27632, which significantly reduced FVIIa-induced generation of EVs, markedly reduced FVIIa-induced anti-inflammatory and barrier protective effects in vivo. Overall, our results suggests that FVIIa influences the phenotype of various cell types by releasing EVs from endothelial cells and transferring the cargo of the EVs to target cells. The knowledge gained from the present study may be relevant in understanding the long-term protective effects of prophylactic use of FVIIa in patients with hemophilia32 and in exploring new therapeutic potentials for FVIIa or FVIIa-released EEVs in treating inflammatory disorders.
Materials and methods
Additional methods are described in the supplemental Data (available on the Blood Web site).
Cells
Human umbilical vein endothelial cells (HUVECs) were obtained from Lonza. THP-1 cells and the murine brain endothelial cell line bEND.3 were from American Type Culture Collection. Human periph.eral blood mononuclear cells (PBMCs) were isolated from whole blood by density gradient centrifugation, using Ficoll-Paque. Murine peritoneal macrophages were isolated from C57Bl/6J mice, as described previously.33
Mice
Wild-type (WT) C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME) and bred in-house.
Isolation and quantification of EVs
Analysis of miR expression
The mirVana miR isolation kit (Thermo Fisher) was used to isolate miR. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analyses were performed according to standard protocols. Primers used for analyses and conditions for qRT-PCR can be found in the supplemental Materials and methods.
Transfection of cells with siRNA, miR inhibitors, and miR mimics
Cells were transfected with selective siRNA, miR inhibitors, miR mimic, or the corresponding scrambled (scr) sequences by using the Lipofectamine RNAiMAX reagent (Thermo Fisher) in serum-free conditions. Cells were cultured for 48 hours before they were used in experiments.
Uptake of EVs
EVs generated from endothelial cells stained with a cell-impermeable fluorescent dye (PKH67) were incubated with target cells for 4 hours at 37°C. After 4 hours, the cells were washed twice, and fixed with 4% p-paraformaldehyde, and the nuclei were stained with 4′,6-diamidino-2-phenylindole. (DAPI). Cells were imaged for fluorescent EVs by confocal microscopy (LSM 510 Meta; Zeiss). EEV uptake in THP-1 cells was also monitored by the presence of VE-cadherin, the protein specific to endothelial cells, in an immunoblot analysis.
Measurement of cytokines
Proinflammatory cytokines in cell supernatants and murine plasma were determined with enzyme-linked immunosorbent assay (ELISA) kits from eBioscience.
In vitro endothelial barrier permeability assay
A barrier permeability assay was performed with a Transwell chamber, as mentioned in our previous publication.12
In vivo experiments
In the peritonitis model, saline or EEVs were injected into the peritoneum of C57BL/6J mice, and 4 hours later, mice were injected with LPS (5 mg/kg) intraperitoneally. Two hours after administration of LPS, peritoneal macrophages were isolated and analyzed for tumor necrosis factor α (TNFα) and interleukin 1β (IL-1β) gene expression by qRT-PCR. For the LPS-induced systemic inflammation model, C57BL/6J mice were injected with saline or EEVs via the tail vein. Four hours later, LPS (5 mg/kg) was administered intraperitoneally. Mice were euthanized at 6 or 16 hours after LPS administration to measure the levels of inflammatory cytokines in the plasma and vascular leakage into the tissues,12 respectively. Neutrophil infiltration into the lungs was analyzed as described earlier.11
Results
Mechanism of FVIIa-induced EV biogenesis in endothelial cells
Earlier, we found that FVIIa promotes generation of EVs from the endothelium, both in vitro and in vivo.13 Herein, we elucidate the underlying mechanisms involved in FVIIa-induced EV biogenesis. Earlier studies showed that the β-arrestin1–dependent pathway mediates FVIIa-EPCR-PAR1–induced anti-inflammatory effects.10,11 Hence, we first investigated the role of β-arrestins in the FVIIa release of EVs. HUVECs were transfected with β-arrestin 1 or β-arrestin 2 siRNA to knock down specific β-arrestin isoforms (Figure 1A). The transfected cells were exposed to a control vehicle or FVIIa, and EVs released into the overlying supernatant were quantified by NTA. Knockdown of β-arrestin 1, but not of β-arrestin 2, significantly attenuated FVIIa-induced release of EVs from endothelial cells (Figure 1B). The knockdown of β-arrestin 1 had no significant effect on the internalization of FVIIa, thereby excluding the possibility that generation of β-arrestin 1–dependent EVs is linked to FVIIa endocytosis (supplemental Figure 1). One of our earlier studies showed that FVIIa-induced activation of the β-arrestin 1–dependent pathway leads to activation of both AKT and ERK1/2.11 Others have reported that β-arrestin 1 activates rho-dependent ROCK signaling to induce formation of invadopodia.34 Therefore, we next investigated the role of the AKT, ERK1/2, and ROCK signaling pathways in FVIIa-induced generation of EEVs, by using specific inhibitors. As expected, LY294002, an inhibitor of PI3K, blocked FVIIa-induced activation of AKT (Figure 1C), and the MEK1/2 inhibitor U0126 attenuated FVIIa-induced activation of ERK1/2 (Figure 1D). Treatment of HUVECs with either of those inhibitors reduced FVIIa-induced generation of EEVs to a modest but statistically significant extent (Figure 1E). Inactive analogues of LY294002 and U0126 neither inhibited FVIIa activation of AKT and ERK1/2, respectively, nor FVIIa release of EEVs (supplemental Figure 2). Inhibition of ROCK with a selective inhibitor (Y27632) decreased the FVIIa-induced generation of EEVs in a more pronounced manner, compared with inhibition of AKT or ERK1/2 alone, but not completely (Figure 1E). The use of all 3 inhibitors together showed the highest reduction of FVIIa-released EEVs (Figure 1E). Overall, the data suggest that FVIIa signals through β-arrestin 1 and that the ROCK-dependent pathway plays a major role in FVIIa-induced biogenesis of EVs (Figure 1F).

Mechanism of FVIIa-induced biogenesis of EVs in endothelial cells. (A) HUVECs were transfected with scr RNA or β-arrestin 1 (β-Arr.1) or β-arrestin 2 (β-Arr.2) siRNA (200 nM each) for 48 hours. The extent of β-arrestin knockdown was examined by immunoblot analysis (top) and quantified by densitometry (bottom). (B) β-Arrestin 1– or β-arrestin 2–silenced endothelial cells were serum starved for 1 hour and then treated with a control vehicle (Con) or FVIIa (100 nM) for 24 hours. EVs were isolated from the conditioned medium and quantified by NTA. (C-D) Serum-starved HUVECs were treated for 1 hour with specific inhibitors of AKT, ERK1/2, or ROCK: LY294002 (LY, 25 µM), U0126 (U, 20 µM), or Y27632 (Y, 10 µM), respectively. The cells were treated with a control vehicle or FVIIa (100 nM) for 1 hour (C) or 20 minutes (D), to assess the activation of AKT or ERK1/2, respectively, by immunoblot analysis (top), and the band intensities were quantified by densitometry to determine the extent of activation (bottom). (E) HUVECs were treated with LY294002, U0126, or Y27632, alone or in combination, at the concentrations and for the times described for panels C and D and then exposed to the control vehicle or FVIIa (100 nM). After 24 hours, EVs were isolated from the conditioned medium and quantified by NTA. (F) The potential signaling mechanism involved in FVIIa-induced generation of EVs. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant. GAPDH, glyceraldehyde phosphate dehydrogenase.
Mechanism of FVIIa-induced biogenesis of EVs in endothelial cells. (A) HUVECs were transfected with scr RNA or β-arrestin 1 (β-Arr.1) or β-arrestin 2 (β-Arr.2) siRNA (200 nM each) for 48 hours. The extent of β-arrestin knockdown was examined by immunoblot analysis (top) and quantified by densitometry (bottom). (B) β-Arrestin 1– or β-arrestin 2–silenced endothelial cells were serum starved for 1 hour and then treated with a control vehicle (Con) or FVIIa (100 nM) for 24 hours. EVs were isolated from the conditioned medium and quantified by NTA. (C-D) Serum-starved HUVECs were treated for 1 hour with specific inhibitors of AKT, ERK1/2, or ROCK: LY294002 (LY, 25 µM), U0126 (U, 20 µM), or Y27632 (Y, 10 µM), respectively. The cells were treated with a control vehicle or FVIIa (100 nM) for 1 hour (C) or 20 minutes (D), to assess the activation of AKT or ERK1/2, respectively, by immunoblot analysis (top), and the band intensities were quantified by densitometry to determine the extent of activation (bottom). (E) HUVECs were treated with LY294002, U0126, or Y27632, alone or in combination, at the concentrations and for the times described for panels C and D and then exposed to the control vehicle or FVIIa (100 nM). After 24 hours, EVs were isolated from the conditioned medium and quantified by NTA. (F) The potential signaling mechanism involved in FVIIa-induced generation of EVs. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant. GAPDH, glyceraldehyde phosphate dehydrogenase.
Previous studies have indicated that ROCK plays a major role in EV biogenesis by altering actomyosin dynamics via phosphorylation of myosin light chain 2 (MLC2) and cofilin-1.35-37 Consistent with the concept expressed herein, inhibition of ROCK with Y27632 downregulated FVIIa-induced phosphorylation of both MLC2 and cofilin-1 (supplemental Figure 3A-D). Further experiments showed that inhibition of MLC2 or cofilin-1 phosphorylation with specific inhibitors of MLC kinase and LIM kinase (ML-7 and cucurbitacin E, respectively) significantly inhibited FVIIa-induced release of EEVs (supplemental Figure 3E-F). These data suggest that the FVIIa release of EEVs involves ROCK-dependent activation of MLC2 and cofilin-1.
FVIIa-released EEVs attenuate LPS-induced inflammation in monocytes
To determine whether EVs from endothelial cells fuse with monocytes and affect the monocyte phenotype, an equal number of EEVs (2 × 108) isolated from control vehicle– or FVIIa-treated HUVECs were incubated with monocytic THP-1 cells (2 × 106) for various lengths of time. The fusion of EEVs was analyzed by probing THP-1 cells by immunoblot analysis for the endothelial cell–specific marker VE-cadherin. THP1 cells lack VE-cadherin but acquired VE-cadherin after incubation with EEVs (supplemental Figure 4). The data showed that THP1 cells incorporated EEVs at 1 hour, but maximal incorporation required ∼4 hours. Both control- and FVIIa-released EEVs were taken up by THP-1 cells with similar efficiency. Microscopic analysis of THP-1 cells incubated with PKH67-labeled EEVs confirmed the uptake of EEVs by THP-1 cells (Figure 2A). Next, THP-1 cells were incubated with an equal number of control vehicle– or FVIIa-released EEVs for 4 hours and then challenged with LPS for 12 hours. LPS treatment markedly increased the release of proinflammatory cytokines (TNFα, IL-6, and IL-1β) from the THP-1 cells (Figure 2B-D). Uptake of FVIIa-released EEVs by THP-1 cells markedly reduced LPS-induced expression of inflammatory cytokines (Figure 2B-D). No significant or only a slight decrease in LPS-induced inflammatory cytokines was observed in the THP-1 cells that were exposed to control EEVs (Figure 2B-D). Uptake of EEVs generated from TNFα- or LPS-activated endothelial cells by THP-1 cells resulted in a small but statistically significant increase in IL-6 levels but no change in IL-1β levels in the absence of LPS treatment (supplemental Figure 5A-B). In contrast to FVIIa-EEVs, EEVs generated by activated endothelial cells failed to diminish LPS-induced expression of inflammatory cytokines in monocytic cells (supplemental Figure 5A-B). Additional experiments showed a similar anti-inflammatory effect of FVIIa-EEVs in human PBMCs (Figure 2E-G). It is pertinent to note that our earlier experiments showed no measurable amount of FVIIa (by immunoblot analysis or ELISA; detection limit, 1 ng/mL) in FVIIa-released EEVs, which rules out the possibility that FVIIa associated with EEVs is responsible for the observed anti-inflammatory effects of FVIIa-EEVs. Furthermore, the addition of FVIIa directly to monocytes had no effect on the LPS-induced expression of inflammatory cytokines (supplemental Figure 6).
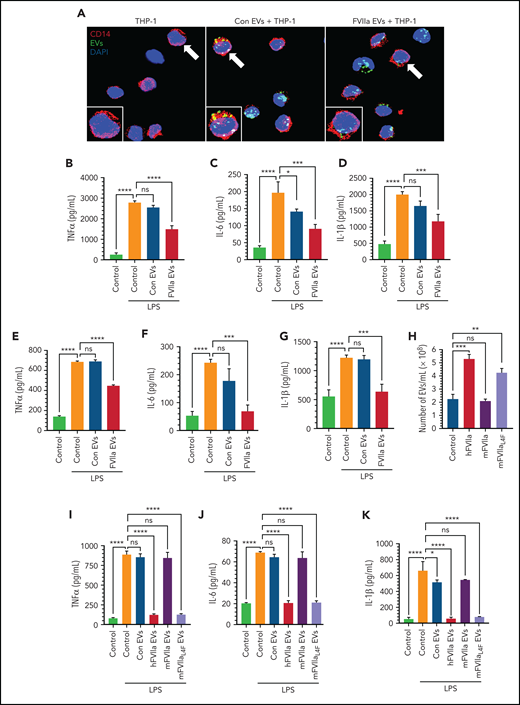
Anti-inflammatory potential of FVIIa-released EEVs. (A) An equal number of EEVs (2 × 108) were isolated from HUVECs labeled with fluorescent PKH67 dye and then treated for 24 hours with a control vehicle (Con EVs) or FVIIa (100 nM). FVIIa-EVs were incubated with THP-1 cells for 4 hours. EV uptake by THP-1 cells was analyzed by monitoring PKH67 fluorescence (green) in the cells. DAPI was used to stain the nuclei, and anti-CD14 antibodies were used to stain the monocytic cell surface marker CD14. Images were taken by a confocal microscope with a 63× objective lens (LSM 510 Meta; Zeiss). (B-D) THP-1 cells were incubated with a control vehicle (Control) or EVs derived from HUVECs treated with a control vehicle (Con EVs) or FVIIa (FVIIa EVs) for 4 hours. After the cells were washed to remove free EVs, they were challenged with LPS (200 ng/mL). After 12 hours, the levels of proinflammatory cytokines, TNF-α (B), IL-6 (C), or IL-1 β (D) in the supernatant medium was determined by ELISA. (E-G) PBMCs isolated from human blood were incubated with a control vehicle or EEVs and challenged with LPS, as described for panels B, C, and D. TNF-α (E), IL-6 (F), and IL-1β (G) in the supernatant medium were measured by ELISA. (H) Murine endothelial cells (bEND.3) were treated with 100 nM of hFVIIa, mFVIIa, or mFVIIaL4F for 24 hours. EEVs released into the supernatant medium were quantified by NTA. (I-K) An equal number of EVs (2 × 108) isolated from bEND.3 cells treated with a control vehicle (Con EVs), hFVIIa (hFVIIa EVs), mFVIIa (mFVIIa EVs), or mFVIIaL4F (mFVIIaL4F EVs) were incubated with murine peritoneal macrophages for 4 hours and then challenged with LPS for 12 hours. The levels of TNF-α (I), IL-6 (J), and IL-1β (K) in the supernatant medium were determined by ELISA. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
Anti-inflammatory potential of FVIIa-released EEVs. (A) An equal number of EEVs (2 × 108) were isolated from HUVECs labeled with fluorescent PKH67 dye and then treated for 24 hours with a control vehicle (Con EVs) or FVIIa (100 nM). FVIIa-EVs were incubated with THP-1 cells for 4 hours. EV uptake by THP-1 cells was analyzed by monitoring PKH67 fluorescence (green) in the cells. DAPI was used to stain the nuclei, and anti-CD14 antibodies were used to stain the monocytic cell surface marker CD14. Images were taken by a confocal microscope with a 63× objective lens (LSM 510 Meta; Zeiss). (B-D) THP-1 cells were incubated with a control vehicle (Control) or EVs derived from HUVECs treated with a control vehicle (Con EVs) or FVIIa (FVIIa EVs) for 4 hours. After the cells were washed to remove free EVs, they were challenged with LPS (200 ng/mL). After 12 hours, the levels of proinflammatory cytokines, TNF-α (B), IL-6 (C), or IL-1 β (D) in the supernatant medium was determined by ELISA. (E-G) PBMCs isolated from human blood were incubated with a control vehicle or EEVs and challenged with LPS, as described for panels B, C, and D. TNF-α (E), IL-6 (F), and IL-1β (G) in the supernatant medium were measured by ELISA. (H) Murine endothelial cells (bEND.3) were treated with 100 nM of hFVIIa, mFVIIa, or mFVIIaL4F for 24 hours. EEVs released into the supernatant medium were quantified by NTA. (I-K) An equal number of EVs (2 × 108) isolated from bEND.3 cells treated with a control vehicle (Con EVs), hFVIIa (hFVIIa EVs), mFVIIa (mFVIIa EVs), or mFVIIaL4F (mFVIIaL4F EVs) were incubated with murine peritoneal macrophages for 4 hours and then challenged with LPS for 12 hours. The levels of TNF-α (I), IL-6 (J), and IL-1β (K) in the supernatant medium were determined by ELISA. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
Unlike human FVIIa (hFVIIa), murine FVIIa (mFVIIa) does not bind to murine EPCR.38 However, a substitution of a single amino acid in murine FVIIa, leucine (L) in the place of phenylalanine (F) at residue 4, enables it to bind EPCR.39 As expected, hFVIIa and mFVIIaL4F, which bind murine EPCR, released EVs from murine endothelial cells, whereas WT mFVIIa did not (Figure 2H). More important, when an equal number of EEVs released from murine endothelial cells was added to murine peritoneal macrophages, hFVIIa- and mFVIIaL4F-released EVs, but not control vehicle– or mFVIIa-released EEVs, markedly attenuated LPS-induced expression of proinflammatory cytokines (Figure 2I-K).
Endothelial barrier protective responses of FVIIa-released EEVs
Next, we investigated the effect of EEVs on LPS-induced endothelial barrier permeability. As shown with THP-1 cells (Figure 2A), both control vehicle– and FVIIa-released EEVs were readily taken up by endothelial cells (Figure 3A). EEVs were readily colocalized with endosomes, indicating that cells took up the EEVs via endocytosis (supplemental Figure 7). Uptake of FVIIa-released EEVs by endothelial cells protected them against LPS-induced endothelial barrier leakage (Figure 3B). These data were further strengthened by additional experiments with murine endothelial cells. As shown in Figure 3C, uptake of EEVs of bEND.3 cells treated with hFVIIa or mFVIIaL4F led to a significant decrease in LPS-induced barrier permeability. In contrast, uptake of control vehicle– or mFVIIa-EEVs had no significant effect on LPS-induced endothelial barrier permeability (Figure 3C). Similarly, EEVs released from activated endothelial cells also had no significant effect on vascular permeability (supplemental Figure 5C).
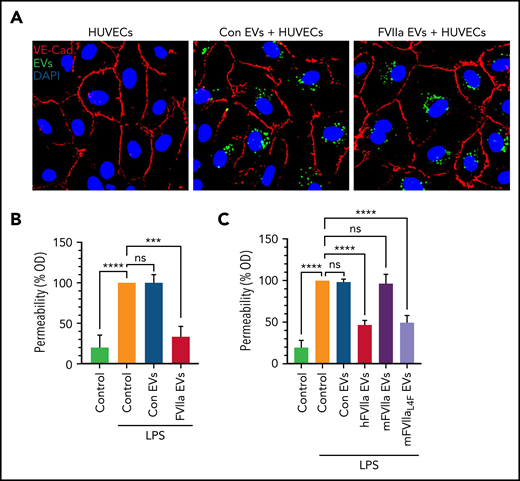
FVIIa-released EEVs induce barrier protection in target endothelial cells. (A) An equal number of EVs (2 × 108), released from HUVECs prelabeled with PKH67 dye and treated with a control vehicle (Con EVs) or FVIIa (FVIIa EVs), were incubated with naive HUVECs for 4 hours. The uptake of EEVs by target naive endothelial cells was determined by analyzing PKH67 fluorescence (green). Endothelial cells were immunostained for VE-cadherin to identify the cell periphery. Nuclei were stained with DAPI. (B) HUVECs grown to confluence in transwells were incubated with a control vehicle (Control) or EVs (2 × 108) released from HUVECs treated with control vehicle (Con EVs) or FVIIa (FVIIa EVs) for 4 hours. After 4 hours, the monolayer was washed twice and challenged with LPS (200 ng/mL). Barrier permeability was measured 12 hours after the addition of LPS, as described in “Materials and methods.” The barrier permeability (OD readings) observed in cells treated with LPS that were not exposed to EVs were taken as 100%. (C) bEND.3 cells grown to confluence in transwells were exposed to control vehicle (Control) or EVs (2 × 108) released from bEND.3 cells treated with a control vehicle (Con EVs), hFVIIa (hFVIIa EVs), mFVIIa (mFVIIa EVs), or mFVIIaL4F (mFVIIaL4F EVs). LPS-induced barrier permeability was evaluated as described in panel B. ***P < .001; ****P < .0001; ns, not significant.
FVIIa-released EEVs induce barrier protection in target endothelial cells. (A) An equal number of EVs (2 × 108), released from HUVECs prelabeled with PKH67 dye and treated with a control vehicle (Con EVs) or FVIIa (FVIIa EVs), were incubated with naive HUVECs for 4 hours. The uptake of EEVs by target naive endothelial cells was determined by analyzing PKH67 fluorescence (green). Endothelial cells were immunostained for VE-cadherin to identify the cell periphery. Nuclei were stained with DAPI. (B) HUVECs grown to confluence in transwells were incubated with a control vehicle (Control) or EVs (2 × 108) released from HUVECs treated with control vehicle (Con EVs) or FVIIa (FVIIa EVs) for 4 hours. After 4 hours, the monolayer was washed twice and challenged with LPS (200 ng/mL). Barrier permeability was measured 12 hours after the addition of LPS, as described in “Materials and methods.” The barrier permeability (OD readings) observed in cells treated with LPS that were not exposed to EVs were taken as 100%. (C) bEND.3 cells grown to confluence in transwells were exposed to control vehicle (Control) or EVs (2 × 108) released from bEND.3 cells treated with a control vehicle (Con EVs), hFVIIa (hFVIIa EVs), mFVIIa (mFVIIa EVs), or mFVIIaL4F (mFVIIaL4F EVs). LPS-induced barrier permeability was evaluated as described in panel B. ***P < .001; ****P < .0001; ns, not significant.
EEV-mediated transfer of miR10a promotes anti-inflammation and endothelial barrier protection in vitro
To obtain clues on the driving factors behind the anti-inflammatory and barrier protective effect of FVIIa-EEVs, an equal number of EEVs (1 × 109), isolated from control vehicle– and FVIIa-treated HUVECs were subjected to microRNA analysis by deep sequencing. The data revealed that 974 different miRNAs were present in both the control- and FVIIa-EEVs, with an abundance varying from 1 400 000 to 0.13 transcripts, and 233 of them were present in 10 or more transcripts. Among them, 26 were upregulated in FVIIa-EEVs by twofold or more, whereas 56 were downregulated by twofold or more. Among the upregulated miRs, 18 were anti-inflammatory, miR10a-5p being the most abundant (control-EEVs, 499 435; FVIIa-EEVs, 1 354 485), followed by miR126-3p (control-EEVs, 26 370; FVIIa-EEVs, 59 331), miR-21-5p (control-EEVs, 22 380; FVIIa-EEVs, 45 041), and miR10b-5p (control-EEVs, 2723; FVIIa-EEVs, 6525). Among the 56 downregulated miRs, 14 were proinflammatory.
The differential expression of anti-inflammatory miRs, miR10a-5p, miR126-3p, and miR-21-5p in control- and FVIIa-EEVs and their relative abundance was validated by RT-PCR (supplemental Figure 8A). Because the abundance of miR10a-5p was strikingly higher compared with other differentially regulated anti-inflammatory miRs, we focused our experiments on miR10a-5p. To determine whether increased levels of miR10a in FVIIa-EEVs over control-EEVs were due to differential sorting of miR10a into FVIIa-EEVs, the result of enhanced expression of miR10a in cells treated with FVIIa, or both, we analyzed the expression of miR10a in HUVECs treated with a control vehicle or FVIIa. The data showed that FVIIa treatment significantly upregulated miR10a expression in the HUVECs in a time-dependent manner, reaching a peak at 8 hours (an approximate sixfold increase over control vehicle; supplemental Figure 8B). Increased miR10a levels in FVIIa-EEVs (supplemental Figure 8C) follow the time course of increased miR10a expression of FVIIa-treated endothelial cells. The levels of miR10a, even after normalizing to the number of EVs, were higher in FVIIa-EEVs than in control EEVs.
Next, we investigated whether the transfer of miR10a from EEVs to target monocytes or endothelial cells confers anti-inflammatory or barrier protective phenotype, respectively. THP-1 cells expressed very low levels of miR10a, but miR10a levels were markedly higher in THP-1 cells after their incubation with FVIIa-released EEVs, compared with control-EEVs (Figure 4A). Actinomycin D treatment did not block the increase in miR10a levels in THP-1 cells after the incorporation of EEVs (Figure 4B), indicating that increased miR10a levels in THP1 cells are not related to de novo synthesis of miR10a but are the result of EEV uptake. Actinomycin D treatment completely blocked expression of the transient gene c-myc, indicating the effective inhibition of transcription by actinomycin D (supplemental Figure 9).
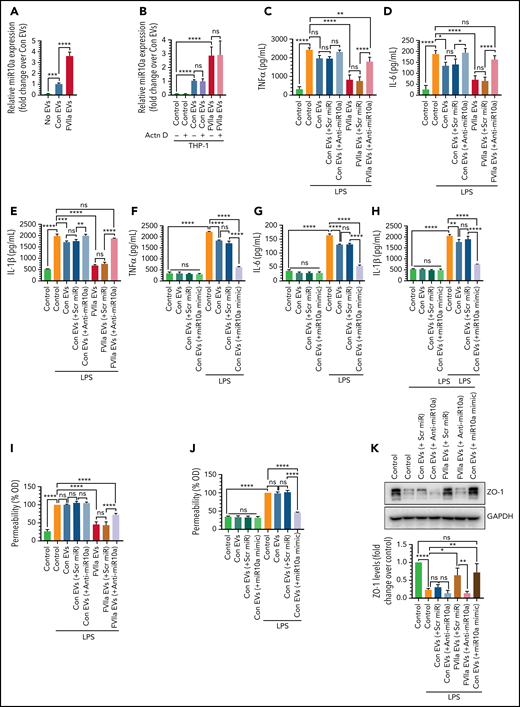
FVIIa-derived EEVs promote anti-inflammation and endothelial barrier protection via miR10a transfer. (A) miR10a expression levels in THP-1 cells after the uptake of EEVs. An equal number of EVs (2 × 108), released from endothelial cells treated with control vehicle (Con EVs) or FVIIa (FVIIa EVs), were incubated with THP-1 cells for 4 hours. Free EVs were removed, the cells were washed twice, and the miR10a level in the THP-1 cells was determined by qRT-PCR. (B) Increased miR10a level in THP-1 cells after EEV uptake resulted from the transfer of miR10a from EEVs to THP-1 cells and were not related to de novo transcription. THP-1 cells were treated with actinomycin D (Actn D; 10 µg/mL) for 8 hours before they were exposed to EEVs. The rest of the experimental procedure was the same as described in panel A. (C-E) The transfer of miR10a from EEVs to THP-1 cells confers anti-inflammatory phenotype to THP-1 cells. An equal number of EVs (2 × 108), isolated from HUVECs transfected with scrambled miR (scr miR) or anti-miR10a and then treated with a control vehicle or FVIIa, were incubated with THP-1 cells (2 × 106) for 4 hours to allow for the uptake of EEVs by THP-1 cells. Thereafter, THP-1 cells were challenged with LPS (200 ng/mL) for 12 hours, and the release of TNF-α (C), IL-6 (D), and IL-1β (E) was measured by ELISA. (F-H) Uptake by THP-1 cells of EEVs containing miR10a mimic reduced the elaboration of LPS-induced inflammatory cytokines. HUVECs were transfected with scr miR or miR10a mimic RNA (20 nM). EVs, isolated from the supernatant medium (2 × 108), were left to fuse with the THP-1 cells for 4 hours. The cells were challenged with LPS for 12 hours, and the levels of TNF-α (F), IL-6 (G), and IL-1β (H) in the supernatant medium were determined. (I-J) miR10a-dependent endothelial barrier protection in target endothelial cells after the uptake of EEVs EVs containing scr miR, anti-miR10a, or miR10a mimic were generated as described in panels C to H. They were left for 4 hours to fuse with endothelial cells grown to confluence in a transwell system. The cells were challenged with LPS (200 ng/mL), and barrier permeability was assessed 12 hours after the LPS challenge. The barrier permeability (OD readings) observed in cells treated with LPS that were not exposed to EVs were taken as 100%. (K) Naive HUVECs fused with control- or FVIIa-EEVs containing scr miR, anti-miR10a, or miR10a mimic were treated with LPS (200 ng/mL) for 6 hours, and ZO-1 levels in the cell extracts were assessed by immunoblot analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
FVIIa-derived EEVs promote anti-inflammation and endothelial barrier protection via miR10a transfer. (A) miR10a expression levels in THP-1 cells after the uptake of EEVs. An equal number of EVs (2 × 108), released from endothelial cells treated with control vehicle (Con EVs) or FVIIa (FVIIa EVs), were incubated with THP-1 cells for 4 hours. Free EVs were removed, the cells were washed twice, and the miR10a level in the THP-1 cells was determined by qRT-PCR. (B) Increased miR10a level in THP-1 cells after EEV uptake resulted from the transfer of miR10a from EEVs to THP-1 cells and were not related to de novo transcription. THP-1 cells were treated with actinomycin D (Actn D; 10 µg/mL) for 8 hours before they were exposed to EEVs. The rest of the experimental procedure was the same as described in panel A. (C-E) The transfer of miR10a from EEVs to THP-1 cells confers anti-inflammatory phenotype to THP-1 cells. An equal number of EVs (2 × 108), isolated from HUVECs transfected with scrambled miR (scr miR) or anti-miR10a and then treated with a control vehicle or FVIIa, were incubated with THP-1 cells (2 × 106) for 4 hours to allow for the uptake of EEVs by THP-1 cells. Thereafter, THP-1 cells were challenged with LPS (200 ng/mL) for 12 hours, and the release of TNF-α (C), IL-6 (D), and IL-1β (E) was measured by ELISA. (F-H) Uptake by THP-1 cells of EEVs containing miR10a mimic reduced the elaboration of LPS-induced inflammatory cytokines. HUVECs were transfected with scr miR or miR10a mimic RNA (20 nM). EVs, isolated from the supernatant medium (2 × 108), were left to fuse with the THP-1 cells for 4 hours. The cells were challenged with LPS for 12 hours, and the levels of TNF-α (F), IL-6 (G), and IL-1β (H) in the supernatant medium were determined. (I-J) miR10a-dependent endothelial barrier protection in target endothelial cells after the uptake of EEVs EVs containing scr miR, anti-miR10a, or miR10a mimic were generated as described in panels C to H. They were left for 4 hours to fuse with endothelial cells grown to confluence in a transwell system. The cells were challenged with LPS (200 ng/mL), and barrier permeability was assessed 12 hours after the LPS challenge. The barrier permeability (OD readings) observed in cells treated with LPS that were not exposed to EVs were taken as 100%. (K) Naive HUVECs fused with control- or FVIIa-EEVs containing scr miR, anti-miR10a, or miR10a mimic were treated with LPS (200 ng/mL) for 6 hours, and ZO-1 levels in the cell extracts were assessed by immunoblot analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
Next, to investigate whether EEV-mediated delivery of miR10a from endothelial cells to THP-1 is responsible for the anti-inflammatory effect of FVIIa-EEVs, EEVs were packed with anti-miR10a or scr miR by generating EEVs from HUVECs transfected with anti-miR10a or scr miR. The anti-inflammatory effect of FVIIa-EEVs was significantly attenuated if EEVs contained anti-miR10a (Figure 4C-E). To further verify the role of miR10a in exerting an anti-inflammatory effect, EEVs were generated by HUVECs transfected with miR10a mimic. In this case, the EVs generated constitutively (no FVIIa treatment) also downregulated LPS-induced expression of inflammatory cytokines in recipient THP1-cells (Figure 4F-H). Additional experiments confirmed that transfection of anti-miR10a significantly reduced (by >80%) miR10a levels in control and FVIIa-treated endothelial cells and EEVs derived from these cells (supplemental Figure 10A-B). Transfection of miR10a mimic markedly increased miR10a levels in endothelial cells. EEVs derived from miR10a mimic–transfected cells contained about threefold higher miR10a levels vs the EEVs released from nontransfected control endothelial cells and a level similar to that of FVIIa-EEVs (supplemental Figure 10A-B). In additional studies, we analyzed the role of the next abundant miRs, miR-126-3p and miR-21-5p, in FVIIa-EEV–mediated anti-inflammatory effects, using an approach similar to that used for miR10a. The data showed the anti-miR126 or anti-miR21 failed to reverse the anti-inflammatory effects of FVIIa-EEVs (supplemental Figure 11).
We next investigated whether miR10a, transported to naive endothelial cells via the EEVs, would regulate endothelial barrier permeability during inflammation. The barrier protective effect of FVIIa-EEVs was diminished significantly if FVIIa-EEVs were packed with anti-miR10a (Figure 4I). Furthermore, control-EEVs containing miR10a mimic showed a barrier protective response similar to that of FVIIa-EEVs (Figure 4J). In additional experiments, HUVECs grown in Transwells were transfected with miR10a mimic or scr miR and then challenged with LPS. Transfection of endothelial cells with miR10a mimic protected against LPS-induced barrier permeability (supplemental Figure 12).
Earlier studies showed that LPS induces vascular permeability by reducing the expression of the tight junction protein, ZO-1.40,41 Hence, we investigated whether EEV-mediated delivery of miR10a to naive HUVECs affects ZO-1 expression. LPS treatment reduced ZO-1 expression in endothelial cells, and the incorporation of FVIIa-EEVs or control-EEVs containing miR10a mimic, but not control-EEVs or FVIIa-EEVs containing anti-miR10a, diminished LPS-induced downregulation of ZO-1 (Figure 4K). Cumulatively, the data provide convincing evidence that FVIIa-EEVs promote anti-inflammatory and endothelial barrier protection in target cells via transfer of miR10a.
The transfer of miR10a from FVIIa-released EEVs promotes anti-inflammation via the downregulation of the TAK1-NF-κB signaling pathway
LPS triggers the activation of TAK1, which activates the NF-κB signaling pathway, thereby regulating the expression of various genes associated with inflammations.42 Human and murine TAK1 contain evolutionarily conserved miR10a binding sites in the 3'-UTRs (Figure 5A). Hence, we investigated whether EEV-mediated delivery of miR10a to THP-1 cells is responsible for inhibiting LPS-induced inflammatory responses via downregulation of the TAK1-NF-κB signaling pathway. Uptake of FVIIa-EEVs by THP-1 cells led to a significant reduction in TAK1 protein levels in the cells (Figure 5B). Uptake of FVIIa-EEVs containing anti-miR10a by THP-1 cells failed to reduce TAK1 levels (Figure 5B). The role of miR10a in downregulating TAK1 levels was further illustrated by the observation that uptake of control EEVs released from HUVECs transfected with miR10a mimic markedly reduced TAK1 levels in THP-1 cells (Figure 5C). Consistent with the hypothesis that TAK1 downregulation by miR10a would lead to the downregulation of the NF-κB signaling pathway, uptake of FVIIa-EEVs or EEVs released from miR10a mimic–transfected HUVECs reduced LPS-induced nuclear translocation of the NF-κB signaling protein p65 in THP-1 cells (Figure 5D-E). FVIIa-EEVs generated from HUVECs transfected with anti-miR10a failed to inhibit the nuclear translocation of p65 in THP-1 cells (Figure 5D). Overall, the data indicate that miR10a, delivered by the FVIIa-EEVs, inhibits inflammatory responses in monocytes by inhibiting the NF-κB signaling pathway through downregulation of its target TAK1 (Figure 5F).
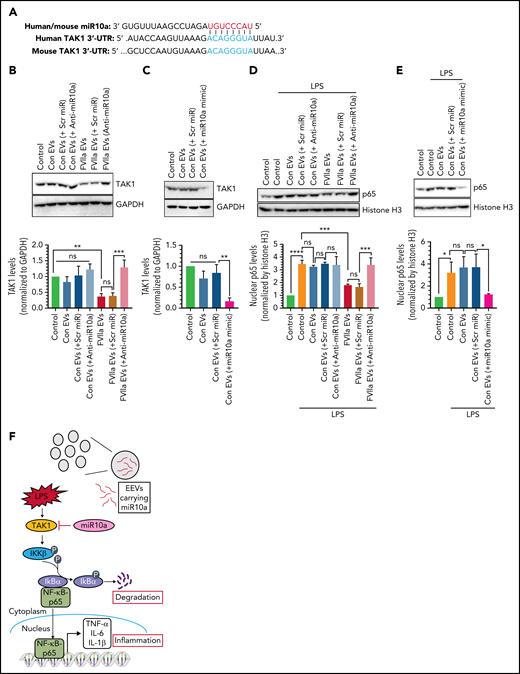
FVIIa-released EEVs prevent LPS-induced inflammation via miR10a-dependent downregulation of the TAK1-NF-ĸB signaling axis. (A) miR10a putative binding site within the 3'-UTR of TAK1. (B) An equal number of EVs (2 × 108), isolated from the conditioned medium of HUVECs that were transfected with scr miR or anti-miR10a and then treated with control vehicle or FVIIa, were left to fuse with THP-1 for 4 hours. The levels of TAK1 protein in THP-1 cells were analyzed by immunoblot analysis (top), and the band intensities were quantified by densitometric analysis (bottom). (C) EVs (2 × 108), generated from HUVECs transfected with scr miR or miR10a mimic, were added to THP-1 cells. Four hours later, the expression of TAK1 was analyzed by immunoblot analysis (top), and the band intensities were quantified by densitometric analysis (bottom). (D) THP-1 cells that incorporated control-EEVs or FVIIa-EEVs containing scr miR or anti-miR10a were treated with LPS for 30 minutes. The nuclei were isolated, and the levels p65 were determined by immunoblot analysis (top). Histone H3 was used as a control for the loading of nuclear proteins and used for normalization in densitometric analysis (bottom). (E) Control EEVs and control EEVs containing scr miR or miR10a mimic were incubated with THP-1 cells for 4 hours. The cells were treated with LPS for 30 minutes, and p65 levels in nuclei were analyzed as described in panel D. (F) Schematic representation of how miR10a, transferred via FVIIa-EEVs, downregulates the NF-ĸB–mediated inflammatory pathway in monocytes. An inflammatory stimulus, such as LPS, induces TAK1 activation, which in turn, induces IĸBα phosphorylation and its subsequent degradation to release the NF-ĸB p65 subunit to enter the nucleus to induce the expression of proinflammatory genes, such as TNF-α, IL-1β, and IL-6. miR10a, transferred from FVIIa-EEVs, regulates the TAK1-NF-ĸB signaling pathway by targeting TAK1 (F). **P < .01; ***P < .001; ****P < .0001; ns, not significant.
FVIIa-released EEVs prevent LPS-induced inflammation via miR10a-dependent downregulation of the TAK1-NF-ĸB signaling axis. (A) miR10a putative binding site within the 3'-UTR of TAK1. (B) An equal number of EVs (2 × 108), isolated from the conditioned medium of HUVECs that were transfected with scr miR or anti-miR10a and then treated with control vehicle or FVIIa, were left to fuse with THP-1 for 4 hours. The levels of TAK1 protein in THP-1 cells were analyzed by immunoblot analysis (top), and the band intensities were quantified by densitometric analysis (bottom). (C) EVs (2 × 108), generated from HUVECs transfected with scr miR or miR10a mimic, were added to THP-1 cells. Four hours later, the expression of TAK1 was analyzed by immunoblot analysis (top), and the band intensities were quantified by densitometric analysis (bottom). (D) THP-1 cells that incorporated control-EEVs or FVIIa-EEVs containing scr miR or anti-miR10a were treated with LPS for 30 minutes. The nuclei were isolated, and the levels p65 were determined by immunoblot analysis (top). Histone H3 was used as a control for the loading of nuclear proteins and used for normalization in densitometric analysis (bottom). (E) Control EEVs and control EEVs containing scr miR or miR10a mimic were incubated with THP-1 cells for 4 hours. The cells were treated with LPS for 30 minutes, and p65 levels in nuclei were analyzed as described in panel D. (F) Schematic representation of how miR10a, transferred via FVIIa-EEVs, downregulates the NF-ĸB–mediated inflammatory pathway in monocytes. An inflammatory stimulus, such as LPS, induces TAK1 activation, which in turn, induces IĸBα phosphorylation and its subsequent degradation to release the NF-ĸB p65 subunit to enter the nucleus to induce the expression of proinflammatory genes, such as TNF-α, IL-1β, and IL-6. miR10a, transferred from FVIIa-EEVs, regulates the TAK1-NF-ĸB signaling pathway by targeting TAK1 (F). **P < .01; ***P < .001; ****P < .0001; ns, not significant.
EEVs-mediated delivery of miR10a induces anti-inflammatory and endothelial barrier protection in vivo
Next, we investigated the potential of FVIIa-EEVs, through the transfer of miR10a, in preventing LPS-induced inflammation and vascular leakage in vivo. An equal number of EEVs (2 × 108), isolated from control vehicle– or FVIIa-treated murine endothelial cells that were transfected with scr miR or anti-miR10a, were injected into WT C57BL/6J mice intravenously via the tail vein. Four hours later, mice were challenged with LPS (5 mg/kg) intraperitoneally. Measurement of the inflammatory cytokines TNF-α, IL-6, and IL-1β in plasma at 12 hours after LPS showed that administration of FVIIa-EEVs, but not control-EEVs, attenuated LPS-induced increased levels of inflammatory cytokines (Figure 6A-C). More important, the protective effect of FVIIa-EEVs was significantly inhibited if FVIIa-EEVs were made to carry anti-miR10a (Figure 6A-C). Similar to those data, administration of FVIIa-EEVs significantly reduced LPS-induced vascular leakage, but this protective effect was lost if FVIIa-EEVs were engineered to contain anti-miR10a (Figure 6D-E; supplemental Figure 13). Analysis of neutrophil infiltration into the lungs showed that LPS-triggered neutrophil infiltration was significantly reduced if mice were given FVIIa-EEVs but not control-EEVs. FVIIa-EEVs containing anti-miR10a failed to suppress LPS-induced neutrophil infiltration into the lungs (Figure 6F-G). Additional experiments showed that administration of EEVs isolated from murine endothelial cells transfected with miR10a mimic but not treated with FVIIa exhibited similar anti-inflammatory and barrier protective effects as FVIIa-EEVs, suggesting that miR10a carried by the EEVs is responsible for the observed protective effects (supplemental Figure 14).
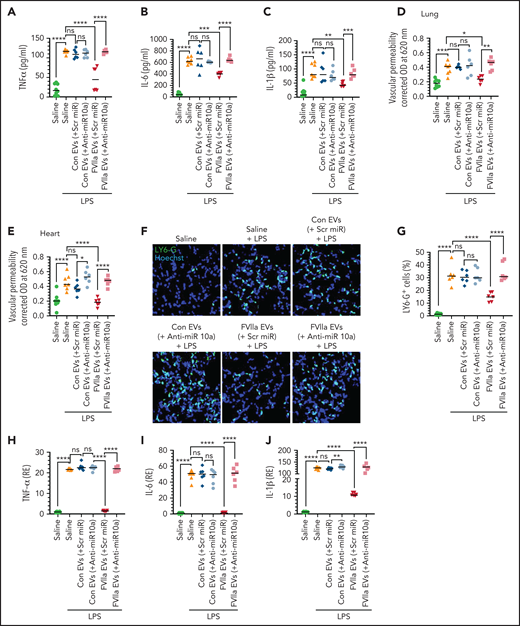
Administration of FVIIa-released EEVs to mice protects against LPS-induced inflammation and barrier disruption. (A-F) EVs were isolated from the supernatant medium of bEND.3 cells that were transfected with scr miR or anti-miR10a, and then treated with a control vehicle or FVIIa. An equal number of EVs (2 × 108) were administered to C57WT/6J mice via the tail-vein. Four hours later, mice were given an intraperitoneal injection of LPS (5 mg/kg). Twelve hours after administration of LPS, blood was obtained from the mice, and the levels of TNF-α (A), IL-6 (B), and IL-1β (C) in the plasma were measured. In a subset of the same group of mice, vascular leakage into the lung (D) and heart (E) was evaluated. In another subset, mice were euthanized 6 hours after administration of LPS, and the lungs were collected. Lung tissue sections were stained for neutrophil infiltration and imaged at 40× magnification (F), and the number of neutrophils was counted (G). (H-J) EEVs (2 × 108), isolated as described for panel A, were administered into the peritoneum of C57WT/6J mice. Four hours later, LPS (5 mg/kg) was administered to the mice IP. Two hours after administration, the mice were euthanized, and peritoneal macrophages were isolated. mRNA expression levels of TNF-α (H), IL-6 (I), and IL-1β (J) were determined by qRT-PCR. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
Administration of FVIIa-released EEVs to mice protects against LPS-induced inflammation and barrier disruption. (A-F) EVs were isolated from the supernatant medium of bEND.3 cells that were transfected with scr miR or anti-miR10a, and then treated with a control vehicle or FVIIa. An equal number of EVs (2 × 108) were administered to C57WT/6J mice via the tail-vein. Four hours later, mice were given an intraperitoneal injection of LPS (5 mg/kg). Twelve hours after administration of LPS, blood was obtained from the mice, and the levels of TNF-α (A), IL-6 (B), and IL-1β (C) in the plasma were measured. In a subset of the same group of mice, vascular leakage into the lung (D) and heart (E) was evaluated. In another subset, mice were euthanized 6 hours after administration of LPS, and the lungs were collected. Lung tissue sections were stained for neutrophil infiltration and imaged at 40× magnification (F), and the number of neutrophils was counted (G). (H-J) EEVs (2 × 108), isolated as described for panel A, were administered into the peritoneum of C57WT/6J mice. Four hours later, LPS (5 mg/kg) was administered to the mice IP. Two hours after administration, the mice were euthanized, and peritoneal macrophages were isolated. mRNA expression levels of TNF-α (H), IL-6 (I), and IL-1β (J) were determined by qRT-PCR. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
In further experiments, we evaluated the anti-inflammatory potential of FVIIa-EEVs and the role of miR10a in conferring an anti-inflammatory phenotype on macrophages, by using a murine peritonitis model.29 The data showed that LPS markedly upregulated TNF-α, IL-6, and IL-1β gene expression in murine peritoneal macrophages, and the administration of FVIIa-EEVs to the mice significantly reduced LPS-induced TNF-α, IL-6, and IL-1β expression in the macrophages (Figure 6H-J). The protective effect was dependent on miR10a, as FVIIa-EEVs containing anti-miR10a failed to exert the protective effect (Figure 6H-J).
EEVs released into the circulation in vivo in response to FVIIa treatment are enriched with miR10a, and inhibiting the biogenesis of endogenous EVs significantly diminishes the anti-inflammatory and endothelial barrier protective effects of FVIIa
When an equal number of EVs, isolated from the plasma of mice treated with saline or FVIIa, were analyzed for miR10a expression by qRT-PCR, FVIIa-released EEVs were found to contain fourfold higher miR10a levels over EEVs released from saline-treated mice (Figure 7A). When an equal number of EEVs isolated from mice treated with FVIIa or saline were added to murine peritoneal macrophages ex vivo, FVIIa-released EEVs, but not saline-released EEVs, markedly diminished LPS-induced expression of inflammatory cytokines (Figure 7B-D). Transfection of macrophages with anti-miR10a, but not scr miR, significantly diminished the anti-inflammatory effects of in vivo–derived FVIIa-EEVs (Figure 7B-D). Similar results were obtained in barrier permeability studies. The uptake of EEVs, isolated from the plasma of FVIIa-treated mice by bEND.3 cells, significantly reduced LPS-induced vascular permeability, whereas uptake of EEVs from saline-treated mice had no significant effect (Figure 7E). Transfection of bEND.3 cells with anti-miR10a, not scr miR, diminished the barrier protective effect of FVIIa-EEVs (Figure 7E).
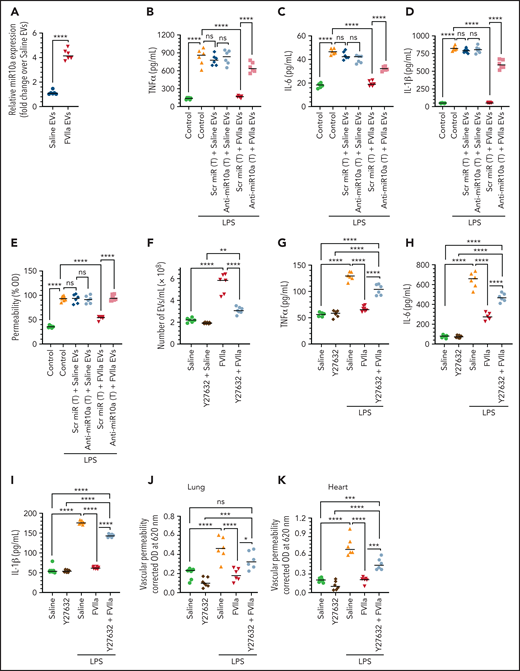
Anti-inflammatory and barrier protective effects of FVIIa-released EEVs in vivo and dependency on miR10a. (A) C57WT/6J mice were given saline or FVIIa (0.25 mg/kg) via the tail vein. Two hours later, blood was collected, and the EVs were isolated from the plasma. An equal number of EEVs (2 × 108) were used to determine miR10a expression levels by qRT-PCR. (B-D) An equal number of EVs (2 × 108), isolated from the plasma of mice treated with saline or FVIIa, as in panel A, were incubated with murine peritoneal macrophages that had been transfected (T) with scr miR or anti-miR10a. The macrophages were then challenged with LPS (200 ng/mL) for 12 hours and the release of TNF-α (B), IL-6 (C), and IL-1β (D) was measured by ELISA. (E) EEVs, isolated from mice as in panel A, were allowed to incorporate into bEND.3 cells cultured in a transwell system and transfected (T) with scr miR or anti-miR10a. The cells were challenged with LPS (200 ng/mL), and barrier permeability was measured at 12 hours after the addition of LPS. (F) C57WT/6J mice were given a ROCK inhibitor, Y27632 (1 mg/kg), via the tail vein 1 hour before injection of saline or FVIIa (0.25 mg/kg) via the same route. Two hours after administration of FVIIa, EVs in the circulating blood were isolated and quantified by NTA. (G-I) C57WT/6J mice were given Y27632 and FVIIa, as described in panel F. At 2 hours after administration of FVIIa, LPS was administered (5 mg/kg, IP). Twelve hours after LPS challenge, plasma proinflammatory cytokines, TNF-α (G), IL-6 (H), and IL-1β (I) were measured by ELISA, and vascular permeability in the lungs (J) and heart (K) were evaluated as described in “Materials and methods.” *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
Anti-inflammatory and barrier protective effects of FVIIa-released EEVs in vivo and dependency on miR10a. (A) C57WT/6J mice were given saline or FVIIa (0.25 mg/kg) via the tail vein. Two hours later, blood was collected, and the EVs were isolated from the plasma. An equal number of EEVs (2 × 108) were used to determine miR10a expression levels by qRT-PCR. (B-D) An equal number of EVs (2 × 108), isolated from the plasma of mice treated with saline or FVIIa, as in panel A, were incubated with murine peritoneal macrophages that had been transfected (T) with scr miR or anti-miR10a. The macrophages were then challenged with LPS (200 ng/mL) for 12 hours and the release of TNF-α (B), IL-6 (C), and IL-1β (D) was measured by ELISA. (E) EEVs, isolated from mice as in panel A, were allowed to incorporate into bEND.3 cells cultured in a transwell system and transfected (T) with scr miR or anti-miR10a. The cells were challenged with LPS (200 ng/mL), and barrier permeability was measured at 12 hours after the addition of LPS. (F) C57WT/6J mice were given a ROCK inhibitor, Y27632 (1 mg/kg), via the tail vein 1 hour before injection of saline or FVIIa (0.25 mg/kg) via the same route. Two hours after administration of FVIIa, EVs in the circulating blood were isolated and quantified by NTA. (G-I) C57WT/6J mice were given Y27632 and FVIIa, as described in panel F. At 2 hours after administration of FVIIa, LPS was administered (5 mg/kg, IP). Twelve hours after LPS challenge, plasma proinflammatory cytokines, TNF-α (G), IL-6 (H), and IL-1β (I) were measured by ELISA, and vascular permeability in the lungs (J) and heart (K) were evaluated as described in “Materials and methods.” *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
Finally, we determined the role of endogenously generated EEVs in FVIIa-induced anti-inflammatory and barrier protective effects. Because the ROCK inhibitor Y27632 was shown to significantly inhibit EEVs biogenesis in FVIIa-treated endothelial cells (Figure 1E), WT mice were given Y27632 via the tail vein 1 hour before administration of FVIIa (250 µg/kg) to restrict the release of EEVs. Y27632 treatment significantly reduced the FVIIa-release of EEVs in vivo (Figure 7F). Next, to determine the effect of EEVs generated endogenously in response to FVIIa on LPS-induced inflammation and vascular permeability, mice were first treated with Y27632 before FVIIa was administered, followed by LPS. FVIIa treatment, as expected, significantly reduced the LPS-induced increase of proinflammatory cytokines in the plasma (Figure 7G-I) and vascular leakage into various tissues (Figure 7J-K; supplemental Figure 15). The administration of Y27632 significantly reduced FVIIa-induced anti-inflammatory and vascular barrier protective effects (Figure 7G-K). To rule out the possibility that Y27632 directly inhibits FVIIa-induced anti-inflammatory and barrier protective effects in endothelial cells, HUVECs were pretreated with Y27632 and then exposed to FVIIa. One hour later (not sufficient time to generate EEVs in vitro13), the cells were stimulated with TNFα or LPS for 4 hours to analyze the release of proinflammatory cytokines and endothelial barrier permeability, respectively (supplemental Figure 16A-B). The data showed that the ROCK inhibitor Y27632 had no direct effect on FVIIa-induced anti-inflammatory or barrier protective effects in the cell system. Taken together, the data suggest that FVIIa may induce anti-inflammatory and vascular barrier protective effects in vivo through the release of EEVs.
Discussion
Our recent study revealed that FVIIa activation of EPCR-PAR1-mediated signaling induces EV release from endothelial cells.13 The experiments reported herein showed that FVIIa-EEVs are readily taken up by monocytes and naive endothelial cells. After the uptake of FVIIa-EEVs, monocytes assume an anti-inflammatory phenotype, whereas the endothelium was protected from barrier destabilization. Our data also showed that FVIIa-EEVs were enriched with anti-inflammatory miRs, predominantly miR10a. The transfer of miR10a from FVIIa-EEVs to target monocytes and endothelial cells results in anti-inflammatory and vascular barrier protective effects, respectively. In vivo tests showed that the administration of FVIIa-EEVs to mice protected them against LPS-induced inflammation and vascular leakage. Additional experiments showed that EEVs released into the circulation after FVIIa treatment are enriched with miR10a, and inhibition of FVIIa-induced EV biogenesis with a ROCK inhibitor diminished FVIIa-induced anti-inflammatory and barrier protective effects in mice. These findings reveal new mechanistic insights into the mode of FVIIa-induced anti-inflammatory and vascular barrier protective effects. Our present data suggest that FVIIa could influence various biological processes in different tissues via intercellular communications through the FVIIa-released EEVs.
Endothelial cells constitutively secrete a low number of EVs under physiological conditions.27 Various vascular diseases associated with systemic endothelial damage, such as atherosclerosis and diabetes, were shown to significantly increase circulating EEVs.27,43,44 EEVs are generally considered to contribute to disease development and progression, as they were found to trigger inflammation, endothelial dysfunction, and thrombosis.43,45,46 However, an increasing body of evidence indicates that, despite their detrimental effects in disease conditions, EEVs can also exert distinct beneficial effects on vascular function. EEVs have been shown to promote endothelial cell survival, regeneration, and protection against apoptosis.47-49 EEVs have also been shown to suppress endothelial cell inflammation and monocyte activation.29,50 The present data showing that FVIIa-EEVs promote vascular integrity and anti-inflammatory effects suggest that the biological functions of EEVs may depend on their origin.
In our recent study, we showed that FVIIa-mediated EV generation from the endothelium is dependent on the EPCR-PAR1 axis.13 Our earlier studies showed that FVIIa-induced PAR1 signaling in endothelial cells involves β-arrestin 1, ERK1/2, and AKT.11,12 The present study shows that FVIIa-induced EEVs biogenesis was also mediated via the β-arrestin 1–dependent pathway, but the involvement of ERK1/2 or AKT appeared to be modest. Reorganization of the cytoskeleton and cellular contractility play a crucial role in EV shedding.37,51 ROCK kinases are involved in regulating the cell shape and movement through alterations in the cytoskeleton.52,53 Our observation that Y27632, a specific ROCK inhibitor, significantly inhibited the FVIIa-induced activation of MLC2 and cofilin-1 and that the inhibition of MLC2 and cofilin-1 activation attenuated FVIIa-induced biogenesis of EEVs suggest that ROCK-dependent cytoskeletal reorganization plays a crucial role in FVIIa-induced EVs biogenesis.
EVs can transfer lipids, cytokines, proteins, mRNA, and miR to target cells to influence their biological behavior.54 Among various biological contents that can be transferred via EVs into target cells, the transfer of miR appears to play a crucial role in protein expression in recipient cells.55,56 Earlier studies of EEVs released from apoptotic human coronary artery endothelial cells showed that they predominantly contain miR126 and miR222.49,50 EEV-mediated transfer of miR126 has been shown to promote vascular repair in recipient endothelial cells,49 whereas the transfer of miR222 has been found to induce anti-inflammatory effects by reducing endothelial ICAM-1 expression.50 In our current study, the predominant miR present in EEVs upon FVIIa treatment was miR10a. The levels of miR10a are ∼20-fold higher than the next predominant miR (miR126) and >5000-fold higher than miR222. It is unclear whether differences in the abundance of specific miRs in EEVs in the present study and earlier studies reflect differences in endothelial cells originating from different vascular beds or experimental conditions used to generate EEVs.
Our data provide irrefutable evidence that the transfer of miR10a via FVIIa-EEVs functionally regulates the expression of specific proteins in target cells and alters cellular functions. The transfer of miR10a from EEVs to monocytes suppresses the expression of inflammatory cytokines in response to inflammatory stimuli, whereas miR10a transfer to target endothelial cells protects against downregulation of the tight junction protein ZO-1 in inflammation. Although uptake of EEVs by target cells potentially transfers other miRs and biomolecules present in EEVs to target cells, the observation that anti-miR10a significantly decreased the protective effects of FVIIa-EEVs in target cells clearly indicates that miR10a was primarily responsible for the cytoprotective effects of FVIIa-EEVs. Furthermore, inhibition of the next 2 abundant miRs in FVIIa-EEVs (ie, miR126-3p and miR21-5p) failed to attenuate the anti-inflammatory effects of FVIIa-EEVs. The differences between control-EEVs and FVIIa-EEVs in their ability to affect the target cell phenotype appear to be related to the variation in quantity of miR10a in their cargo. Control-EEVs, when used in high numbers, appear to exhibit protective effects similar to FVIIa-EEVs on target cells (data not shown). In an earlier study, Njock et al29 showed that EEVs, with characteristics of exosomes, secreted from unstimulated endothelial cells exhibit anti-inflammatory properties, and this is, at least in part, related to the transfer of anti-inflammatory miRs, including miR10a, to recipient monocytes/macrophages.
Endothelial microRNAs were shown to regulate the NF-κB pathway and cell adhesion molecules during inflammation.57 MAP2K7 (also referred to as TAK1) and β-transducin repeat–containing gene (βTrCP) were identified as containing an evolutionarily conserved miR10a binding site in their 3'-UTR.58 Both TAK1 and βTrCP were shown to play a key role in activating NF-κB signal transduction by promoting IκB degradation.59,60 TAK1 directly phosphorylates and activates IκB kinase β (IKKβ), which stimulates the phosphorylation of IκBs. βTrCP recognizes phosphorylated IκB and mediates ubiquitination and proteasomal degradation of IκB. Inhibition of either TAK1 or βTrCP increases the total expression of IκB and impairs NF-κB activation. It is likely that uptake of FVIIa-EEVs enriched with miR10a by target cells confers anti-inflammatory phenotype to target cells through the downregulation of TAK1 and/or βTrCP, and subsequently downregulation of the NF-κB/IκB signaling pathway. Data presented in this article demonstrate that the transfer of miR10a from FVIIa-EEVs to monocytes downregulates NF-κB signaling pathway in monocytes by suppressing TAK1.
Consistent with the data from our in vitro experiments, our in vivo results provide strong evidence of the crucial role of FVIIa-EEVs and miR10a in EEVs in mediating FVIIa-induced anti-inflammatory and barrier protective effects in vivo. The observations that administration of FVIIa-EEVs and not control EEVs attenuated LPS-induced peritonitis, systemic inflammation, and vascular leakage and that anti-miR10a reversed the FVIIa-EEV–mediated protective effects support the concept. Furthermore, our results show that FVIIa EEVs released into the circulation are enriched with miR10a and are capable of exerting anti-inflammatory and barrier protective effects in ex vivo experimental settings. More important, our results show that impeding the biogenesis of FVIIa-induced EEVs in vivo with the administration of ROCK inhibitor significantly reduced the anti-inflammatory and barrier protective effect of FVIIa. Overall, our present data suggest that FVIIa-induced cytoprotective signaling responses seen in vivo in the present study and in our earlier studies10-12,61 may have arisen from the action of FVIIa-EEVs in target cells. It may be important to point out that, although FVIIa-induced generation of EEVs and enrichment of FVIIa-EEVs with miR10a requires 4 to 8 hours in an in vitro cell model, FVIIa was shown to release EEVs in vivo in <1 hour, 13 and EEVs isolated 2 hours after FVIIa administration were found to be enriched with miR10a. FVIIa-induced anti-inflammatory and barrier protective effects in vivo, except VEGF-induced vascular permeability in the skin, were assessed at 7 to 20 hours after FVIIa administration (6-18 hours after LPS administration).10-12,61 This time frame should be sufficient for FVIIa to generate EEVs and the EEVs to act on target cells in vivo. However, our data do not preclude the possibility that FVIIa-induced cellular alterations, independent of EEVs, contribute to the cytoprotective effect of FVIIa, particularly in a shorter duration.
Our current data that suggest FVIIa-mediated activation of ROCK plays a critical role in FVIIa-induced anti-inflammatory and barrier protective effects appears, on the face of it, to be contradictory to our earlier published data,12 which showed that ROCK inhibition had no significant effect on FVIIa-induced barrier protection in endothelial cells. Differences in experimental setup may be responsible for this apparent discrepancy. Earlier experiments were conducted to investigate the direct effect of FVIIa on endothelial cells in a time frame not sufficient to generate significant amounts of EEVs and for the EEVs to exert their protective effects.
In summary, our findings provide a new paradigm for FVIIa in regulating vascular inflammation. Although EVs generated from healthy endothelial cells under basal conditions are likely to possess anti-inflammatory properties, their number or content may be below the threshold for providing a pharmacologic protective effect in response to inflammation or during disease pathogenesis. The increased release of EVs from the endothelium, selective upregulation of miR10a in the endothelium, and its packaging into EEVs in FVIIa treatment could overcome the threshold and thus provide effective protection from endothelial dysfunction. FVIIa has been widely used to treat several bleeding disorders, such as hemophilia, intracerebral hemorrhage, sepsis, and trauma.62-64 The advent of FVIIa-EEVs and understanding their role in hemostasis, anti-inflammation, and vascular barrier protection opens a new window to understanding the therapeutic potential of FVIIa and its utility in treating various diseases associated with bleeding, inflammation, and vascular dysfunction.
Acknowledgments
The authors thank Joseph Thomas Martins, a summer intern in the laboratory, for assistance with some of the experimental procedures. The authors acknowledge the service of Genomics and Microarray Core Facilities at UT Southwestern Medical Center for microRNA sequencing.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute, grants HL107483 and HL124055, and endowment funds from The Dr. and Mrs. James Vaughn Professorship in Biomedical Research (L.V.M.R.). and the Judith Graham Pool Fellowship award from the National Hemophilia Foundation (K.D.).
Authorship
Contribution: K.D. participated in the study design, performed most of the experiments, analyzed the data, and wrote the first draft of the manuscript; S.K. performed the animal experiments; U.R.P. contributed to the study design and provided technical expertise in performing the study; L.V.M.R. conceived of and designed the research, analyzed the data, and wrote the manuscript; and all authors contributed to the preparation of the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: L. Vijaya Mohan Rao, Department of Cellular and Molecular Biology, The University of Texas Health Science Center at Tyler, 11937 US Hwy 271, Tyler, TX 75708; e-mail: vijay.rao@uthct.edu.
Sequencing data have been deposited on the Gene Expression Omnibus database (accession number GSE182925).
Any data or protocols will be shared with qualified investigators upon written or e-mail request to the corresponding author.
Requests for data sharing may be submitted to L. Vijaya Mohan Rao (vijay.rao@uthct.edu.)
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal